

Abstract
The calpain family of cysteine proteases has a well-established causal role in neuronal cell death following acute brain injury. However, the relative contribution of calpain isoforms has not been determined in in vivo models. Identification of the calpain isoform responsible for neuronal injury is particularly important given the differential role of calpain isoforms in normal physiology. This study evaluates the role of m-calpain and μ-calpain in an in vivo model of global brain ischemia. Adeno-associated viral vectors expressing short hairpin RNAs targeting the catalytic subunits of μ- or m-calpain were used to knockdown expression of the targeted isoforms in adult rat hippocampal CA1 pyramidal neurons. Knockdown of μ-calpain, but not m-calpain, prevented calpain activity 72 hours after 6-minute transient forebrain ischemia, increased long-term survival and protected hippocampal electrophysiological function. These findings represent the first in vivo evidence that reducing expression of an individual calpain isoform can decrease post-ischemic neuronal death and preserve hippocampal function.
Keywords: calpain, ischemia, hippocampus, RNA interference, adeno-associated virus
Introduction
In the cascade of molecular events that leads to post-ischemic neuronal death a particularly attractive target for therapeutic intervention is the calpain family of calcium-activated cysteine proteases. Not only does calpain activity occur in a spatio-temporal pattern that is consistent with a causal role of post-ischemic neurodegeneration (Saido et al, 1993; Roberts-Lewis et al, 1994; Bartus et al, 1995; Yamashima et al, 1996; Neumar et al, 1996; Bartus et al, 1998; Neumar et al, 2001), but pharmacologic calpain inhibition has repeatedly been demonstrated to be neuroprotective (Frederick et al, 2008; Bartus et al, 1994a; Bartus et al, 1994b; Li et al, 1998; Markgraf et al, 1998; Lee et al, 1991; Rami and Krieglstein, 1993; Hong et al, 1994; Yokota et al, 1999). Similar results have been achieved with overexpression of calpastatin, the endogenous inhibitor of calpains, in both in vivo excitotoxicity (Takano et al, 1999) and global brain ischemia (Cao et al, 2007a) models.
A limitation of both pharmacologic inhibition and calpastatin overexpression is that neither is able to distinguish between different calpain isoforms. The two classical brain calpain isoforms are μ-calpain and m-calpain, each of which exist as heterodimers containing the distinct catalytic subunits calpain 1 and calpain 2, respectively, along with a common regulatory subunit. The major biochemical distinction between these isoforms is the level of calcium required for activation. In in vitro assays, m-calpain has been shown to require 400-800 μM Ca2+, while μ-calpain has a lower requirement of 3-50 μM Ca2+ (Goll et al, 2003). The importance of determining the contribution of these two isoforms to ischemic brain injury is best illustrated by their differential roles in physiology. While knockouts of calpain 1 are viable with the only reported physiologic defect being in platelet aggregation (Azam et al, 2001), knockout of calpain 2 is embryonic lethal (Dutt et al, 2006), and calpain 2 RNAi causes aberrant chromosome alignment during mitosis in cell culture (Honda et al, 2004). Differences also exist in the subcellular localization of calpains. While both calpain 1 and 2 are present in the cytosol, calpain 1 is also present in mitochondria (Garcia et al, 2005). However, calpain substrates in a variety of subcellular locations and functional classes have been implicated in acute neuronal injury (Bevers and Neumar, 2008). Furthermore, it is possible that the specific calpain isoform involved varies based on injury mechanism and severity. This hypothesis is supported by evidence in cell culture models, where calpain 1 RNA interference is protective following oxygen-glucose deprivation (Cao et al, 2007b) while calpain 2 knockdown prevents NMDA-induced cell death (Bevers et al, 2008).
Given the apparent physiological and pathophysiological differences between these two calpain isoforms, it is essential that their relative roles be examined in an in vivo model of brain injury. In this study we used adeno-associated viral vectors to deliver short hairpin RNAs targeting calpain 1 and 2 to CA1 sector pyramidal neurons in the adult rat hippocampus. We then evaluated the effect on post-ischemic calpain activity, neuronal survival, and hippocampal function following in vivo transient forebrain ischemia.
Materials and Methods
Materials
Antibody to calpain-cleaved spectrin (Ab38) was provided by Dr. Robert Siman (University of Pennsylvania). Alexa-568 conjugated secondary antibodies used for immunofluorescence were purchased from Invitrogen. Unless otherwise noted, all other chemicals are from Sigma-Aldrich (USA).
Viral vector generation
Recombinant adeno-associated viral (AAV) vectors were generated by the University of Pennsylvania Vector Core by triple transfection of 293 cells and purified by cesium chloride gradient sedimentation as described previously (Fisher et al, 1997). Subsequently, AAV 2/1 vectors were generated to express shRNA targeting the calpain 1 (gene name capn1) or calpain 2 (gene name capn2). These vectors have been previously demonstrated to knock down the targeted isoform in primary neuronal culture (Bevers et al, 2008). AAV 2/1 vectors expressing shRNA targeting luciferase (luc), which has no homology to any known rat gene, or scrambled shRNA were used as controls. All shRNAs were expressed under the control of the human U6 RNA polymerase III promoter. All shRNA vectors also expressed the green fluorescent marker ZsGreen under the control of the CMV promoter (Figure 1).
Figure 1.
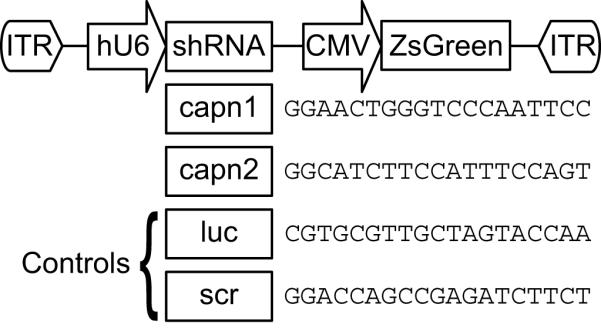
Vector design for AAV-mediated RNA interference. A plasmid containing AAV-specific inverted terminal repeats (ITRs) was designed to express short hairpin RNAs specific for the catalytic subunits of μ-calpain (capn1) or m-calpain (capn2). Two control vectors were generated, containing shRNA specific for luciferase (luc) or a scrambled shRNA (scr). Neither control sequence had any homology to any known rat gene. The sense sequences of each short hairpin are given in the figure. Expression of short hairpins was driven by the human U6 promoter (hU6). Each vector also expressed a green fluorescent marker protein (ZsGreen) under control of the CMV promoter.
Viral Vector Injections
All appropriate biosafety procedures were followed for use of AAV vectors, which are rated BSL-1 and have never been shown to cause human pathology. Male rats weighing 375-425g were anesthetized by inhalation of 4% isoflurane in 30% O2 and 70% N2O and a surgical plane of anesthesia was subsequently maintained with 2% isoflurane. The head was clipped and skin prepped with betadine. A longitudinal surgical incision was made and the skull exposed by blunt dissection. Holes were drilled bilaterally in the skull without penetration of dura 3.8mm caudal to Bregma and 1.8mm lateral to the midline. Twenty-five microliter Hamilton syringes attached to an infusion pump and equipped with 30 gauge needles with a 30° bevel were used for vector injection. The needle was inserted into brain parenchyma at a depth of 3.2mm below the dural surface and 2.0μl of AAV2/1 vector solution was infused at a rate of 0.5μl/minute. After injection, the needle was held in place for 5 minutes and then removed slowly over 2 minutes. The scalp incision was sutured closed and rats recovered from anesthesia on a heating pad.
Transient Forebrain Ischemia
Two weeks following viral vector injection, rats were again anethestized with isoflurane (4%), 70% N2O and 30% O2 by an insufflation chamber. Rats were orotracheally intubated and mechanically ventilated with 30% O2 and 70% N2O. A surgical plane of anesthesia was maintained with 2.0% isoflurane. PE 50 femoral arterial and venous catheters were placed and temperature was monitored with a needle thermocouple probe placed adjacent to the skull below the temporalis muscle. Temperature was maintained between 37.0 and 37.5 °C using a warming pad and an overhead lamp. ECG monitoring was performed with electrocardiographic limb leads. After a 10-minute stabilization period, an arterial blood sample was obtained and ventilator adjustments were made if necessary to maintain the pCO2 at 35 to 45 mm Hg.
Transient forebrain ischemia was initiated by the combination of bilateral carotid occlusion and hypovolemic hypotension to a mean arterial pressure (MAP) of 30mmHg. Hypovolemic hypotension was achieved by rapidly withdrawing blood from the femoral arterial catheter and was maintained during the ischemic period by withdrawal or infusion of blood through the femoral venous catheter. Once a MAP of 30mmHg was achieved, both carotid arteries were reversibly occluded with surgical aneurysm clips. After 6 minutes of ischemia, the aneurysm clips were removed and shed blood was re-infused. Rats were maintained on mechanical ventilation for 1 hour. During this time the abdomen was surgically prepped and an intraperitoneal telemetric temperature probe was surgically implanted (Data Systems International). After one hour of reperfusion, femoral catheters were removed and all surgical wounds were closed. Rats were then extubated and observed for an additional hour for evidence of respiratory distress, which was treated with oxygen. Post-injury body temperature was regulated for 24 hours based on methods previously described (Colbourne et al., 1996; Hicks et al, 2000; and D’Cruz et al., 2002). Readings from the intraperitoneal temperature probe were transmitted to an FM receiver and recorded every 10 seconds by a PC-compatible computer using commercial software (Data Systems International). Temperature was maintained between 36.5 and 38.0 degrees celsius via software-driven relays connected to a 175-W heating lamp, water misters and a cooling fan. Sham (uninjured) animals were subjected to anesthesia and surgical preparation without bilateral carotid occlusion and hypovolemic hypotension to serve as controls.
Immunohistochemistry for calpain-cleaved spectrin
Seventy-two hours after forebrain ischemia rats were anesthetized with 4% isoflurane and transcardially perfused with cold phosphate buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde in 0.1M phosphate buffer. Brains were removed, post-fixed in 4% paraformaldehyde for 24 hours and cryoprotected by serial incubations in 0.1M phosphate buffer containing 10%, 20% and 30% sucrose. Coronal sections (20μm) were cut on a freezing sliding microtome and stored in cryoprotectant (30% sucrose, 30% glycerol in 0.1M phosphate buffer) and stored at −20°C for subsequent analysis.
Brain sections from hippocampus were washed in PBS pH 7.2 and blocked in 4% normal goat serum containing 0.5% Triton X-100 in PBS. Sections were incubated overnight in primary antibody diluted in blocking solution. Sections were then washed in PBS and incubated with Alexa 568 fluorescent secondary antibody. Finally, sections were stained with Hoescht (5μg/ml in PBS) to label nuclei. Immunostaining was examined at high power (400x and 1000x) using a Leica DM4500B microscope equipped for epifluorescence in sections transduced with luc, capn1 and capn2 shRNA.
Histopathologic Staining of CA1 Pyramidal Neurons
Two weeks following injury, rats transduced bilateral with luc, capn1 or capn2 shRNA were sacrificed and 20μm serial sections through hippocampus were mounted, dehydrated using increasing concentrations of ethanol, delipidated with 50% chloroform 50% ethanol, and then rehydrated in serial ethanol dilutions. Sections were temporarily coverslipped with water and imaged for expression of GFP. Coverslips were then removed, sections stained with hematoxylin and eosin, re-coversliped and imaged using brightfield microscopy.
Quantification of Transduced CA1 Pyramidal Neuron Survival
Two weeks following injury, rats transduced bilaterally with luc, capn1 or capn2 shRNA were sacrificed and 20μm serial sections through hippocampus were stained with Hoescht to label nuclei. Surviving cells were quantified in sections, beginning with the first section containing both blades of the dentate gyrus and in three subsequent sections spaced 240μm apart, spanning the injection site. The number of ZsGreen-expressing cells with normal nuclear morphology in these four sections was quantified using stereologic analysis using an unbiased optical fractionator technique. Briefly, this technique involved counting cells from randomly selected, systematically generated optical dissectors which represented a known fraction of the region being analyzed. Cells were counted as normal if their nuclei had diffuse chromatin and a completely rounded profile with no evidence of condensation or irregularities in the nuclear border. The number of normal cells counted was then used to generate an estimate of the total number of cells within the region of CA1 being examined. The cell number was divided by the portion of total area covered by the dissectors (0.05) and by the portion of the region represented by each section (0.083, as every twelfth section was counted). Therefore, the equation used to estimate the total surviving cells in CA1 was: Estimated total cells = (Counted cells) × (1/0.05) × (1/0.083).
Electrophysiology
Two weeks following transient forebrain ischemia, rats were anesthetized by inhalation of 4% isoflurane in 30% O2 and 70% N2O. After confirmation of absence of response to toe pinch the rats were decapitated and the brain dissected whole and placed in ice-cold aCSF (126mM NaCl, 3.2mM KCl, 1.25mM NaHPO4, 26mM NaHCO3, 10mM glucose, 1mM MgCl2, 2mM CaCl2). The brain was placed in aCSF within 1 minute of decapitation, minimizing further ischemia, then removed from aCSF and bisected in the mid-saggital plane. The hippocampal formation was identified and removed en bloc. Time out of aCSF was minimized at all steps. The hippocampus was then sliced perpendicular to its long axis using a McIlwain Mechanical Tissue Chopper in 350μm increments. The slices were transferred to a holding chamber bathed in room temperature aCSF and oxygen to equilibrate for 30-60 minutes. Slices were transferred to the recording chamber for recording.
The Schaffer collateral afferent pathway was stimulated at 0.1 Hz with a bipolar tungsten electrode, and extracellular field potentials were recorded with a monopolar tungsten electrode. Slices were sequentially stimulated at increasing intensity (10, 20, 30, 40, 50, 60, 70, 80, 90, and 100μA) and the field excitatory postsynaptic potential (fEPSP) slope was calculated at each intensity by curve fitting between two cursors that demarcated the linear portion of the fEPSP. All field responses were normalized to the mean peak response to maximal stimulation in naïve, uninjured slices transduced with control (scrambled shRNA) vector. The fEPSP slope recorded at each intensity was compared between injured and uninjured slices transduced with scr or capn1 shRNA using One-way ANOVA with Scheffe post-hoc analysis.
Results
Effect of isoform-specific RNA-interference on calpain activity following transient forebrain ischemia
To determine if knockdown of individual calpain isoforms can prevent ischemia-induced calpain activity, adult Long-Evans rats received intra-hippocampal injections of the AAV2/1 vectors expressing luc, capn1 or capn2 shRNA that we have previously demonstrated to knock down the targeted isoform in hippocampal neuronal culture (Bevers et al, 2008). Rats received bilateral injections - the left hippocampus was injected with luc and the right with either capn1 or capn2 shRNA, allowing each animal to serve as its own control. Seventy-two hours after transient forebrain ischemia, animals were sacrificed and sections were generated through hippocampus spanning the injection site. Sections were assayed for calpain activity by immunolabeling with an antibody specific to the calpain-cleaved form of alpha-spectrin (Ab38). Hippocampal CA1 sector pyramidal neurons transduced with capn2 and luc shRNA immunolabeled for calpain-cleaved spectrin and displayed irregular nuclei indicative of early neurodegeneration. This was similar to non-transduced CA1 pyramidal neurons in the same brain sections. Hippocampal CA1 sector pyramidal neurons transduced with capn1 shRNA did not immunolabel for calpain-cleaved spectrin and had normal appearing nuclei with rounded profiles and diffuse chromatin. In the same brain sections, Ab38 labeling and irregular condensed nuclear morphology could be seen both in contralateral luc-transduced CA1 pyramidal neurons and in non-transduced neurons in the ipsilateral hippocampus both medial and lateral to the region of transduction (Figure 2).
Figure 2.

Representative images of CA1 pyramidal neurons immunolabled for calpain-cleaved spectrin (Ab38, red) and counterstained with Hoechst (blue) 72 hours after transient forebrain ischemia. Intrahippocampal injection of AAV2/1 vector expressing shRNA targeting luciferase (luc), calpain 1 (capn1) or calpain 2 (capn2) and co-expressing ZsGreen was performed 2 weeks prior to injury. Ab38 immunoreactivity and condensed, irregular nuclear profiles were observed in non-transduced CA1 neurons and those transduced with luc or capn2 shRNA. Conversely, neurons transduced with capn1 shRNA displayed normal nuclear morphology and did not label for calpain-cleaved spectrin.
Calpain 1 RNA interference increases long-term neuronal survival following transient forebrain ischemia
To determine if the reduction in calpain activity seen with μ-calpain knockdown at 72 hours after injury translated into increased neuronal survival, an additional set of animals was examined at two weeks after injury. Images from sham injured animals show abundant transduction of cells in hippocampal area CA1 by all three vectors, while H&E staining shows rounded, basophilic nuclei (Figure 3A). Following injury, brains transduced with luc or capn2 shRNA displayed green debris, qualitative CA1 pyramidal neuron loss, the appearance of irregular nuclear morphology in remaining neurons, and eosinophilic cytoplasm throughout the CA1 cell layer. Conversely, CA1 pyramidal neurons in hippocampi transduced with capn1 shRNA were much more numerous and had normal appearing basophilic nuclei (Figure 3B). Staining of adjacent sections with the fluorescent nuclear marker Hoechst shows similar results. In sham injured animals, ZsGreen expression and normal nuclear morphology was observed throughout the CA1 pyramidal layer regardless of the vector used. After TFI, sections from rats transduced with luc or capn2 shRNA had loss of ZsGreen expressing pyramidal neurons, loss of normal appearing nuclei and green debris in the neuropile. In contrast, sections from injured rats transduced with capn1 shRNA appeared similar to those from sham controls (Figure 4). Quantitative analysis confirmed the qualitative findings (Figure 5). While there was no significant difference in the estimated number of surviving transduced CA1 neurons in sham injured animals transduced with each of the vectors (capn1 = 30,602±7883; capn2 = 34,578±11,495; luc = 24,739±7382), the estimated number of surviving neurons 14 days after TFI was significantly greater in rats transduced with capn1 shRNA (34,554±6211) compared to capn2 and luc shRNA (14,217±3951 and 5,783±2484 respectively; p < 0.05, One-way ANOVA with Scheffe post-hoc analysis).
Figure 3.

Representative images of the hippocampal CA1 pyramidal layer two weeks after sham or 6 minute TFI. Intrahippocampal injection of AAV2/1 vector expressing shRNA targeting luciferase (luc), calpain 1 (capn1) or calpain 2 (capn2) and co-expressing ZsGreen was performed 2 weeks prior to injury. Sections were imaged for ZsGreen expression and then stained with hematoxylin and eosin (H&E). ZsGreen expression and rounded, basophilic nuclei were seen throughout the CA1 cell layer in sham animals regardless of the vector used, indicative of excellent transduction with all three vectors. After TFI, sections from rats transduced luc or capn2 shRNA had loss of ZsGreen expressing pyramidal neurons and green debris in the neuropile. H&E staining demonstrated nuclear loss and nuclear condensation combined with increased eosinophilic cytoplasm. In contrast, post-TFI sections from rats transduced with capn1 shRNA appeared similar to those from sham injured rats.
Figure 4.

Sections through CA1 hippocampus taken two weeks after injury, adjacent to those shown in Figure 3, now stained with the fluorescent nuclear marker Hoechst (blue). Results were similar to those seen with H&E staining. ZsGreen expression and normal nuclear morphology was observed throughout the CA1 pyramidal layer in sham injured animals regardless of the vector used. After TFI, sections from rats transduced with luc or capn2 shRNA had loss of ZsGreen expressing pyramidal neurons, loss of normal appearing nuclei and green debris in the neuropile. In contrast, post-TFI sections from rats transduced with capn1 shRNA appeared similar to those from sham injured rats. High power insets show that surviving neurons can be identified by presence of rounded nuclear profiles with diffuse cytoplasmic staining.
Figure 5.

Hippocampal CA1 sector pyramidal neurons transduced with capn1, capn2 or luc (ZsGreen positive) and with normal nuclear morphology were quantified using stereologic techniques. The estimated number of ZsGreen expressing cells with normal nuclear morphology was significantly increased after injury in animals transduced with capn1 shRNA compared to those transduced with luc or capn2 shRNA (p < 0.05, One-way ANOVA with Scheffe post-hoc analysis).
Neurons protected by transduction with capn1 shRNA retain electrophysiologic function following injury
To test whether the histological neuroprotection observed following capn1 knockdown also maintains function, electrophysiologic recordings were performed on hippocampal slices that had been transduced with control (scrambled) or capn1 shRNA prior to injury. Two weeks after transient forebrain ischemia, animals were sacrificed and acute hippocampal slices were generated for extracellular recording. A series of stimuli of increasing intensity were applied to the Schaffer collateral pathway and the field responses of CA1 neurons were recorded. Comparison of input-output curves generated from naïve and injured slices transduced with scr shRNA show that 6 minute transient forebrain ischemia produced an approximately 20% reduction in the maximal field excitatory post-synaptic potential (fEPSP) slope. The maximum fEPSP slope in injured slices transduced with capn1 shRNA was significantly greater than in those transduced with scr shRNA, and did not differ from uninjured controls (Figure 5).
Discussion
While many studies have demonstrated a neuroprotective effect of pharmacologic calpain inhibition following ischemia (Bartus et al, 1994; Bartus et al, 1994; Li et al, 1998; Markgraf et al, 1998; Lee et al, 1991; Rami and Krieglstein, 1993; Hong et al, 1994; Yokota et al, 1999; Frederick et al, 2008) the compounds used often inhibit other classes of proteases, making the exact contribution of the calpains uncertain. This limitation has been overcome through the use of vector-based overexpression of the endogenous calpain inhibitor, calpastatin, which also reduces post-ischemic neurodegeneration (Cao et al, 2007). Although neuroprotection with calpastatin overexpression definitively implicates calpains, this approach does not distinguish the pathologic role of individual calpain isoforms. Furthermore, the effect of calpastatin overexpression on neuronal function after ischemia has not been examined. The findings described here represent the first demonstration of an isoform-specific role for calpain in an in vivo injury model. In this study, RNAi targeting calpain 1, but not calpain 2, prevented calpain activity and signs of early neurodegeneration in CA1 sector pyramidal neurons 72 hours after a mild ischemic insult. Importantly, knockdown of calpain 1 also produced lasting neuroprotection - increasing CA1 pyramidal neuron survival two weeks after injury and preserving electrophysiological function in hippocampal slices. Identification of an essential role for calpain 1 supports the development of isoform-specific therapeutics. Given the known importance of calpain 2 in normal cell function, specifically targeting calpain 1 could result in effective therapy while minimizing potential side effects.
It is important to recognize that while calpain 1 RNAi was neuroprotective following in vivo transient forebrain ischemia and in a cell culture model of oxygen-glucose deprivation (Cao et al, 2007), knockdown of calpain 2 was protective following in vitro excitotoxic injury (Bevers et al, 2008). This finding raises the possibility that different calpain isoforms may be involved in different model systems. A recent study of the developmental expression of calpains found that calpain 1 expression is low early in development, suggesting that cell death in culture models may disproportionally rely on calpain 2 (Li et al, 2009). Furthermore, the essential calpain isoform may differ based on the mechanism of injury.
The mechanims by which pathologic calpain activity causes neurodegeneration remains controversial. There is evidence that calpain 1 may be involved in cell death mediated by apoptosis inducing factor (AIF). Studies have shown that calpain 1 both localizes to mitochondria (Garcia et al, 2005) and is able to cleave AIF (Polster et al, 2005). Knockdown of calpain 1 also prevented nuclear translocation of AIF following oxygen glucose deprivation in neuronal culture (Cao et al, 2007). However, there is more recent evidence that calpain 1 may not in fact directly cleave AIF (Joshi et al, 2009), and that AIF can undergo nuclear translocation without being cleaved by calpain (Wang et al, 2009). Additionally, numerous extra-mitochondrial calpain substrates have been implicated in neuronal injury including calcium regulatory proteins (Bano et al, 2005), neurotransmitter receptors (Xu et al, 2007), and a wide range of signaling proteins (Wu et al, 2004; O’Hare et al, 2005; Hou et al, 2006; Jiang et al, 2007; Saito et al, 2007; Wang et al, 2007; Cao et al, 2007; Zhang et al, 2007; for review, see Bevers and Neumar, 2008), again suggesting that different calpain isoforms may be involved in different injury pathways.
The relative role of calpain isoforms in neuronal death may be influenced by the severity and mechanism of injury. The 6-minute transient forebrain ischemia used here is a relatively short-duration ischemic insult. This is reflected by the fact that histologic evidence of neurodegeneration is restricted to the rostral hippocampus and that only an approximately 20% reduction was seen in field potential slope following injury. Given the greater calcium requirement for calpain 2 activation, it is possible that a more severe injury would recruit this second calpain, and perhaps trigger alternate injury pathways. Moving forward, it will be particularly important to study isoform-specific calpain functions in more severe global ischemia models as well as other models of in vivo acute brain injury such as focal ischemia and trauma.
Supplementary Material
Figure 6.

Electrophysiologic function of post-ischemic CA1 pyramidal neurons protected by calpain 1 knockdown. Stimulus-response curves from hippocampal uninjured slices transduced with scrambled (scr, n = 9 slices from 3 animals) and injured slices transduced with scr (n = 9 slices from 3 animals) or calpain 1 (capn1, n = 8 slices from 3 animals) shRNA. The Schaffer collateral pathway was sequentially stimulated at increasing intensity and field excitatory post-synaptic potentials (fEPSPs) were recorded from CA1. An approximately 20% reduction in maximum fEPSP slope was observed in injured scr transduced slices compared to uninjured controls. This deficit was restored by transduction with capn1 shRNA. The fEPSP slope at the maximum stimulus intensity was significantly greater in injured capn1 transduced slices compared to injured scr transduced slices (p < 0.05, One-way ANOVA with Scheffe post-hoc analysis).
Acknowledgements
This work was supported by National Institutes of Heath Grant NS039481 (RWN) and American Heart Association Grant 0615345U (MBB). All AAV vectors used in this study were generated by the University of Pennsylvania Vector Core. Vector production was supported by National Institutes of Health Grant P30-DK-047757-14. Electrophysiology experiments were supported by National Institutes of Health Grant HD059288 (ASC). We would like to thank Dr. Robert Siman for the gift of antibody against calpain-cleaved spectrin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol. Cell. Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 3.Bartus RT, Baker KL, Heiser AD, Sawyer SD, Dean RL, Elliott PJ, Straub JA. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J. Cereb. Blood. Flow. Metab. 1994a;14:537–544. doi: 10.1038/jcbfm.1994.67. [DOI] [PubMed] [Google Scholar]
- 4.Bartus RT, Dean RL, Cavanaugh K, Eveleth D, Carriero DL, Lynch G. Time-related neuronal changes following middle cerebral artery occlusion: implications for therapeutic intervention and the role of calpain. J. Cereb. Blood. Flow. Metab. 1995;15:969–979. doi: 10.1038/jcbfm.1995.123. [DOI] [PubMed] [Google Scholar]
- 5.Bartus RT, Dean RL, Mennerick S, Eveleth D, Lynch G. Temporal ordering of pathogenic events following transient global ischemia. Brain. Res. 1998;790:1–13. doi: 10.1016/s0006-8993(97)01414-5. [DOI] [PubMed] [Google Scholar]
- 6.Bartus RT, Hayward NJ, Elliott PJ, Sawyer SD, Baker KL, Dean RL, Akiyama A, Straub JA, Harbeson SL, Li Z. Calpain inhibitor AK295 protects neurons from focal brain ischemia. Effects of postocclusion intra-arterial administration. Stroke. 1994b;25:2265–2270. doi: 10.1161/01.str.25.11.2265. [DOI] [PubMed] [Google Scholar]
- 7.Bevers MB, Lawrence E, Maronski M, Starr N, Amesquita M, Neumar RW. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J. Neurochem. 2008;108:1237–1250. doi: 10.1111/j.1471-4159.2008.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood. Flow. Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- 9.Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J. Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dutt P, Croall DE, Arthur SC, De Veyra T, Williams K, Elce JS, Greer PA. m-Calpain is required for preimplantation embryonic development in mice. BMC. Dev. Biol. 2006;6:3. doi: 10.1186/1471-213X-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher KJ, Jooss K, Alston J, Yang Y, Haecker SE, High K, Pathak R, Raper SE, Wilson JM. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 12.Frederick JR, Chen Z, Bevers MB, Ingleton LP, Ma M, Neumar RW. Neuroprotection with delayed calpain inhibition after transient forebrain ischemia. Crit. Care. Med. 2008;36:S481–S485. doi: 10.1097/ccm.0b013e31818a8ec8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia M, Bondada V, Geddes JW. Mitochondrial localization of mu-calpain. Biochem. Biophys. Res. Commun. 2005;338:1241–1247. doi: 10.1016/j.bbrc.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 14.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 15.Honda S, Marumoto T, Hirota T, Nitta M, Arima Y, Ogawa M, Saya H. Activation of m-calpain is required for chromosome alignment on the metaphase plate during mitosis. J. Biol. Chem. 2004;279:10615–10623. doi: 10.1074/jbc.M308841200. [DOI] [PubMed] [Google Scholar]
- 16.Hong SC, Lanzino G, Goto Y, Kang SK, Schottler F, Kassell NF, Lee KS. Calcium-activated proteolysis in rat neocortex induced by transient focal ischemia. Brain. Res. 1994;661:43–50. doi: 10.1016/0006-8993(94)91178-9. [DOI] [PubMed] [Google Scholar]
- 17.Hou ST, Jiang SX, Desbois A, Huang D, Kelly J, Tessier L, Karchewski L, Kappler J. Calpain-cleaved collapsin response mediator protein-3 induces neuronal death after glutamate toxicity and cerebral ischemia. J. Neurosci. 2006;26:2241–2249. doi: 10.1523/JNEUROSCI.4485-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang SX, Kappler J, Zurakowski B, Desbois A, Aylsworth A, Hou ST. Calpain cleavage of collapsin response mediator proteins in ischemic mouse brain. Eur. J. Neurosci. 2007;26:801–809. doi: 10.1111/j.1460-9568.2007.05715.x. [DOI] [PubMed] [Google Scholar]
- 19.Joshi A, Bondada V, Geddes JW. Mitochondrial mu-calpain is not involved in the processing of apoptosis-inducing factor. Exp. Neurol. 2009 doi: 10.1016/j.expneurol.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc. Natl. Acad. Sci. U. S. A. 1991;88:7233–7237. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li PA, Howlett W, He QP, Miyashita H, Siddiqui M, Shuaib A. Postischemic treatment with calpain inhibitor MDL 28170 ameliorates brain damage in a gerbil model of global ischemia. Neurosci. Lett. 1998;247:17–20. doi: 10.1016/s0304-3940(98)00266-3. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Bondada V, Joshi A, Geddes J. Calpain 1 and Calpastatin expression is developmentally regulated in rat brain. Exp. Neurol. Online. 2009 doi: 10.1016/j.expneurol.2009.09.004. DOI: 10.1016/j.expneurol.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA, Linnik MD. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke. 1998;29:152–158. doi: 10.1161/01.str.29.1.152. [DOI] [PubMed] [Google Scholar]
- 24.Neumar RW, Hagle SM, DeGracia DJ, Krause GS, White BC. Brain mu-calpain autolysis during global cerebral ischemia. J. Neurochem. 1996;66:421–424. doi: 10.1046/j.1471-4159.1996.66010421.x. [DOI] [PubMed] [Google Scholar]
- 25.Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, Siman R. Calpain activity in the rat brain after transient forebrain ischemia. Exp. Neurol. 2001;170:27–35. doi: 10.1006/exnr.2001.7708. [DOI] [PubMed] [Google Scholar]
- 26.O’Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J. Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polster BM, Basañez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 28.Rami A, Krieglstein J. Protective effects of calpain inhibitors against neuronal damage caused by cytotoxic hypoxia in vitro and ischemia in vivo. Brain. Res. 1993;609:67–70. doi: 10.1016/0006-8993(93)90856-i. [DOI] [PubMed] [Google Scholar]
- 29.Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J. Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saido TC, Yokota M, Nagao S, Yamaura I, Tani E, Tsuchiya T, Suzuki K, Kawashima S. Spatial resolution of fodrin proteolysis in postischemic brain. J. Biol. Chem. 1993;268:25239–25243. [PubMed] [Google Scholar]
- 31.Saito T, Konno T, Hosokawa T, Asada A, Ishiguro K, Hisanaga SI. p25/Cyclin-dependent kinase 5 promotes the progression of cell death in nucleus of endoplasmic reticulum-stressed neurons. J. Neurochem. 2007;102:133–140. doi: 10.1111/j.1471-4159.2007.04540.x. [DOI] [PubMed] [Google Scholar]
- 32.Takano E, Masatoshi M, Yuen P. Structure of Calpastatin and Its Inhibitory Control of Calpain. In: Wang KKW, editor. Calpain: Pharmacology and Toxicology of Calcium-Dependent Protease. Taylor &Francis; Philadelphia, PA: 1999. pp. 25–50. [Google Scholar]
- 33.Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, Dawson TM. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (Parthanatos) J. Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06167.x. Published online June 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, White MG, Akay C, et al. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J. Neurochem. 2007;103:439–455. doi: 10.1111/j.1471-4159.2007.04746.x. [DOI] [PubMed] [Google Scholar]
- 35.Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- 36.Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-Mediated mGluR1alpha Truncation: A Key Step in Excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Yamashima T, Saido TC, Takita M, et al. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur. J. Neurosci. 1996;8:1932–1944. doi: 10.1111/j.1460-9568.1996.tb01337.x. [DOI] [PubMed] [Google Scholar]
- 38.Yokota M, Tani E, Tsubuki S, Yamaura I, Nakagaki I, Hori S, Saido TC. Calpain inhibitor entrapped in liposome rescues ischemic neuronal damage. Brain. Res. 1999;819:8–14. doi: 10.1016/s0006-8993(98)01334-1. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, Wang KK. Calpain-Mediated Collapsin Response Mediator Protein-1, -2, And -4 Proteolysis after Neurotoxic And Traumatic Brain Injury. J. Neurotrauma. 2007;24:460–472. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.