

Abstract
The Emb proteins (EmbA, EmbB, EmbC) are mycobacterial arabinosyltransferases involved in the biogenesis of the mycobacterial cell wall. EmbA and EmbB are predicted to work in unison as a heterodimer. EmbA and EmbB are involved in the formation of the crucial terminal hexaarabinoside motif [Araβ(1→2)Araα(1→5)] [Araβ(1→2)Araα(1→3)]Araα(1→5)Araα1→(Ara6) in the cell wall polysaccharide arabinogalactan. Studies conducted in Mycobacterium smegmatis revealed that mutants with disruptions in embA or embB are viable, although the growth rate was affected. In contrast, we demonstrate here that embA is an essential gene in Mycobacterium tuberculosis, since a deletion of the chromosomal gene could only be achieved when a second functional copy was provided on an integrated vector. Complementation of an embA mutant of M. smegmatis by M. tuberculosis embA confirmed that it encodes a functional arabinosyltransferase. We identified a promoter for M. tuberculosis embA located immediately upstream of the gene, indicating that it is expressed independently from the upstream gene, embC. Promoter activity from PembA(Mtb) was sevenfold lower when assayed in M. smegmatis compared to M. tuberculosis, indicating that the latter is not a good host for genetic analysis of M. tuberculosis embA expression. PembA(Mtb) activity remained constant throughout growth phases and after stress treatment, although it was reduced during hypoxia-induced non-replicating persistence. Ethambutol exposure had no effect on PembA(Mtb) activity. These data demonstrate that M. tuberculosis embA encodes a functional arabinosyltransferase which is constitutively expressed and plays a critical role in M. tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis poses a serious global health problem, causing millions of deaths and new infections every year. There is an urgent need for new antimycobacterial therapeutics, since the current treatment regimen is lengthy and ineffective against multi-drug-resistant strains. The cell wall of M. tuberculosis is an attractive drug target, since it is critical for cell survival and a number of current antitubercular agents target this structure. In mycobacteria, the peptidoglycan, located immediately outside the cytoplasmic membrane, is covalently attached to arabinogalactan (AG), which is in turn esterified to the mycolic acid layer. This structure, called the mycolyl-AG-peptidoglycan (mAGP) complex, forms the core framework of the mycobacterial cell wall. The wall provides a hydrophobic permeability barrier and is responsible for at least part of the intrinsic resistance of mycobacteria to a number of antibiotics (Brennan, 2003).
Arabinosyltransferases, encoded by emb genes, play key roles in the synthesis of mycobacterial cell wall components. Two emb genes (embA and embB) were initially identified in Mycobacterium avium (Belanger et al., 1996). Subsequently, three Emb proteins have been identified in M. tuberculosis (Cole et al., 1998) and in Mycobacterium smegmatis (Escuyer et al., 2001); no orthologues have been identified elsewhere than in the order Actinomycetales. The genomic arrangement of the emb genes in M. tuberculosis and M. smegmatis is similar, with the three genes (embCAB) being co-localized and possibly being transcribed as an operon (Telenti et al., 1997).
The Emb proteins form a family of large (>1100 residues) transmembrane proteins with a cytoplasmic N-terminal domain, 13–15 transmembrane segments and a large extracytoplasmic C-terminal domain (Berg et al., 2003; Seidel et al., 2007). Emb proteins are considered to be good drug targets, since the current antitubercular drug, ethambutol, causes cessation of the synthesis of arabinan polymers [AG and lipoarabinomannan (LAM)]. This is due to the inhibition of arabinosyltransferases (Khoo et al., 1996; Mikusova et al., 1995) and embB mutations have been associated with ethambutol resistance in M. tuberculosis (Sreevatsan et al., 1997).
Almost all of the work characterizing the role and expression of Emb proteins has been carried out in the saprophytic species M. smegmatis where the role of each protein has been determined. Biochemical analyses of deletion mutants has clearly shown that the Emb proteins are all arabinosyltransferases, but with segregated biological functions. EmbC is involved in the biosynthesis of the arabinan portion of LAM (Zhang et al., 2003), whereas EmbA and EmbB are involved in AG synthesis (Escuyer et al., 2001). Thus, the crucial hexaarabinoside terminal motif, which is the structural motif for mycolylation in AG (McNeil et al., 1994), is altered in both M. smegmatis embA and embB mutants (Escuyer et al., 2001). An EmbA deletion mutant has a number of other phenotypic changes, including altered morphology, slight loss of acid-fastness and increased susceptibility to hydrophobic antibiotics (Escuyer et al., 2001). These changes are likely to be a direct result of the defective synthesis of AG in the cell wall.
No direct information on the role of M. tuberculosis EmbA (EmbAMtb) has been obtained to date and there is no information on the expression of the embA gene or its promoter in this species. To determine the function of EmbAMtb, we attempted to construct a deletion mutant by gene replacement. We demonstrate here that embA is essential in M. tuberculosis under normal culture conditions. EmbAMtb was confirmed as a bona fide arabinosyltransferase, since it was able to complement an M. smegmatis embA mutant in an in vitro assay. We also identified the promoter of embA and demonstrated that its expression is constitutive.
METHODS
Culture.
Mycobacteria were grown in Middlebrook liquid medium (7H9-OADC) [4.7 g Middlebrook 7H9 l−1 plus 10 % (v/v) OADC (oleic acid, albumin, dextrose, catalase) supplement (Becton Dickinson)] with 0.05 % (w/v) Tween 80 (Tw) where stated, or on Middlebrook solid medium (7H10-OADC) [19 g Middlebrook 7H10 l−1 plus 10 % (v/v) OADC supplement]. Dubos medium supplemented with 5 % (w/v) glycerol and 10 % (v/v) Dubos medium albumin (Becton Dickinson) was used for hypoxic cultures. Aerobic liquid cultures of M. tuberculosis were static 10 ml cultures, inoculated 1 : 10 in 50 ml tubes. Hypoxic cultures were performed in 17 ml medium in 20 mm glass tubes with slow stirring (50 r.p.m.) from a starting OD570 of 0.004. Kanamycin was used at 20 μg ml−1, hygromycin at 100 μg ml−1, streptomycin at 20 μg ml−1, gentamicin at 10 μg ml−1, X-Gal at 50 μg ml−1 and sucrose at 5 % (w/v) unless otherwise indicated.
Construction of embA deletion vector.
A deletion delivery vector for embA was constructed as follows: the upstream and downstream regions of embA were amplified using primer pairs F1 (5′-ATCGCAGTTTCCTCAACGAC-3′) and R1 (5′-CCTCGAGGGATCGAGATGTCCAG-3′), and F2 (5′-CTCGAGGTCGTCGAACCTATGGCAGT-3′) and R2 (5′-AGCGCCAGCAGGTTGTAATA-3′), respectively, and cloned into pGEM-T Easy (Promega). XhoI restriction sites (underlined) were engineered into the primers. The two fragments were excised as KpnI–XhoI and XhoI–BamHI fragments and cloned into p2NIL (Parish & Stoker, 2000). The lacZ, sacB, hyg cassette from the marker cassette vector pGOAL19 (Parish & Stoker, 2000) was excised as a PacI fragment and inserted into p2NIL to make the final vector, pEMPTY16.
Attempts to construct an embA deletion strain.
Plasmid pEMPTY16 was pre-treated with UV to promote homologous recombination and electroporated into M. tuberculosis (Hinds et al., 1999). Single cross-overs (SCOs) were isolated on hygromycin, kanamycin and X-Gal plates. One SCO was streaked onto solid medium without antibiotics to allow the second cross-over to occur. Double cross-overs (DCOs) were isolated on sucrose, X-Gal plates. White colonies were patch-tested for kanamycin and hygromycin sensitivity to confirm vector loss. A PCR screen using primers DIET1 (5′-CATCCTCACCGCCCTTAAC-3′) and DIET2 (5′-CGATTTGGGGTGCTTTTG-3′) was used to distinguish between wild-type (3.4 kb band) and deletion DCOs (0.5 kb band).
Construction of embA merodiploid strain.
The complementation vector pEMPTY22 was constructed by amplifying the embA gene with primers Empathy3 (5′-TTAATTAATGGCCAGCTACCTCAAAGAC-3′) and Empathy4 (5′-TTAATTAAACCGACAACACAAAGCCAAT-3′), cloning into pGEM-T Easy and subcloning into pAPA3 (L5 integrating vector with Ag85a promoter; unpublished) as a PacI fragment (sites underlined) in the correct orientation for expression from the Ag85a promoter. pEMPTY22 was transformed into the SCO strain to generate a merodiploid strain (Tame 68). DCOs were isolated from this strain by plating onto sucrose, X-Gal and gentamicin. Sucrose-resistant, white colonies were screened by PCR as before. ‘Del-int ’ DCO strains (one deleted copy and one integrated copy of embA) were isolated and confirmed by Southern analysis. For gene switching, two del-int strains [embAΔ (PAg85a-embA, L5 int, Gm)] were transformed with the integrating vector pUC-Hyg-Int (Mahenthiralingam et al., 1998). Transformants were plated on hygromycin±gentamicin and patch-tested for hygromycin/gentamicin resistance.
Analyses of promoter activity.
The intergenic region between embC and embA of M. tuberculosis was amplified using primers 5′-CCCAGTACTAGCGGTTGACGCCTTACTAC-3′ and 5′-CCCAGTACTAGATCGCTCATTACCGTCGT-3′, cloned as a ScaI fragment (sites underlined) into the L5-based integrating vector pSM128 (Dussurget et al., 1999) upstream of the promoterless lacZ gene to make plasmid pEMBA and the sequence verified. For site-directed mutagenesis, primers SDM1 (5′-CGC GTC GCC GAC CAG CGA GCC TCG-3′) and SDM2 (5′-CGA GGC TCG CTG GTC GGC GAC GCG-3′) (mutated nucleotides underlined) were used to generate a double mutation. The amplification reaction for site-directed mutagenesis was carried out in 50 μl total volume containing 1× Pfu Ultra reaction buffer, 0.5 mM dNTPs, 10 pmol each primer, 10 % DMSO, 10 ng template and 2.5 units Pfu Ultra (Stratagene). The thermocycling programme used was 95 °C for 1 min, followed by 18 cycles of 95 °C for 1 min, 60 °C for 1 min, 68 °C for 12 min and a final extension cycle at 68 °C for 20 min. The template was degraded using 10 units DpnI at 37 °C for 1 h. Five microlitres of the reaction was then used to transform competent Escherichia coli. Recombinant pEMBA-M plasmid was isolated and the sequence verified. Plasmids pEMBA and pEMBA-M were electroporated into M. smegmatis and M. tuberculosis and streptomycin-resistant transformants were isolated. Three independent transformants for each were selected for promoter activity determinations. Cell-free extracts were prepared (Parish & Wheeler, 1998) and β-galactosidase assays were performed as described by Miller (1972).
Isolation of RNA and identification of the embA transcript.
Total RNA was isolated from M. tuberculosis cells grown in 100 ml 7H9-OADC containing 0.05 % (w/v) Tween 80 to an OD600 of 0.8. The cells were pelleted at 3000 g for 10 min and resuspended in 10 ml Trizol (Invitrogen), vortexed and frozen at –80 °C for 1 h. The cells were broken in a FastPrep machine (Thermo Electron) at speed 6.0 for 40 s and the lysate was extracted with 2 ml chloroform. The cell lysate was centrifuged at 27 000 g at 4 °C for 30 min and the aqueous layer was precipitated with 5 ml 2-propanol at −80 °C overnight, the pellet being collected by centrifugation at 14 000 g for 10 min at 4 °C, washed with 70 % ethanol, dried and dissolved in 100 μl RNase-free water. The sample was then treated with 5 units RNase-free DNase for 1 h at 37 °C. After the removal of DNase by phenol/chloroform extraction, the RNA was precipitated with ethanol and dissolved in 50 μl deionized RNase-free water. cDNA was generated using Superscript III reverse transcriptase (RT) (Invitrogen) with the primers EMBAR (5′-CGATGGCCGAGCAGGGGATC-3′) and EMBBR (5′-GGTCGGTGACGTCCACGCG-3′). PCR was performed using primer pairs EMBAR and EMBCF2 (5′-GCGAGGTCCGGTTGCAGTGG-3′), and EMBBR and EMBAF1 (5′-CTGCCAGCGACCGTTTTCC-3′) in a 50 μl reaction using Taq DNA polymerase and 2 μl cDNA from the first strand synthesis. The amplification was performed with an initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min, followed by a final extension step at 72 °C for 7 min.
For Northern analysis, 16 μg total RNA from M. tuberculosis was separated by gel electrophoresis using 1 % agarose gel and transferred to a positively charged nylon membrane (Amersham Hybond-N+) for 1.5 h using the QBIOgene vacuum blotter. The PCR-amplified embAMtb was purified and used as the hybridization probe. Labelling and detection were carried out using the AlkPhos Direct kit (Amersham), according to the manufacturer's instructions.
Arabinosyltransferase assay.
The 3285 bp embAMtb was amplified from genomic DNA using the primers TAF1 (5′-CCCCATATGGTGCCCCACGACGGTAAT-3′) and TAR1 (5′-CCCAAGCTTTCATGGCAGCGCCCTGAT-3′) and the 3279 bp embA gene from M. smegmatis (embAMsm) was amplified from genomic DNA using the primers TMF1 (5′-CCCCATATGGTGCCGGGCGATGAACAG-3′) and TMR1 (5′-CCCAAGCTTTCACGGCAGTGCCCTGAT-3′) with both primer sets having NdeI and HindIII restriction sites (underlined) in the forward and reverse primers, respectively. The amplified genes were cloned into the pVV16 expression vector to make pTA and pMA, respectively, and the sequences were verified. Plasmids pTA and pMA were transformed into an embAMsm knockout strain (ΔembAMsm) (Escuyer et al., 2001) to generate ΔembAMsm, complemented with embAMtb, and ΔembAMsm, complemented with embAMsm, respectively.
For the arabinosyltransferase assay, strains were grown to an OD600 of 0.8. Cell wall (P60) and membrane fractions were purified from the strains by standard procedures (Khasnobis et al., 2006). The arabinosyltransferase assay reaction mixture contained buffer A (50 mM MOPS, pH 8.0, 5 mM β-mercaptoethanol, 10 mM MgCl2), 1 mM ATP, 500 000 d.p.m. p[14C]Rpp (14C-labelled phosphoribosylpyrophosphate, used as a donor to form decaprenylphosphoryl-d-[14C]arabinose in situ) (Scherman et al., 1996), 300 μM acceptor [pentasaccharide, octyl-β-d-Araf-(1→2)-α-d-Araf-(1→5)-α-d-Araf-(1→5)-α-d-Araf-(1→5)-α-d-Araf, used as a substrate for arabinosyltransferase activity in a cell-free assay] (Khasnobis et al., 2006), 0.3 mg P60 and 0.8 mg membrane fractions in a total volume of 200 μl. Reactions were incubated at 37 °C for 1 h, terminated by adding 200 μl ethanol and centrifuged briefly at 14 000 g, and the supernatant was loaded onto a pre-packed strong anion exchange (SAX) column (Burdick and Jackson) pre-equilibrated with 5 ml water. Columns were eluted with 2 ml water, and the eluants were dried and partitioned between the two phases (1 : 1) of water-saturated 1-butanol and water, three times; the 1-butanol fractions collected were air-dried, reconstituted in 200 μl 1-butanol and measured by liquid scintillation spectrometry. 3000 d.p.m. were analysed by silica gel TLC developed in CHCl3/CH3OH/1 M NH4OAc/NH4OH/H2O (180 : 140 : 9 : 9 : 23), exposed for 5 days and visualized using a phosphoimager (Molecular Dynamics). The product was purified from the TLC, and approximately 5000 d.p.m. of the product was treated with endoarabinanase isolated from Cellulomonas gelida (McNeil et al., 1994) for 16 h at 37 °C (Khoo et al., 1996) and subjected to Dionex high-pH anion-exchange chromatography (HPAEC).
Preparation of LAM and soluble AG.
Cells (10 g wet wt) were delipidated with hot absolute ethanol followed by a 2 : 1 chloroform/methanol extraction (Khoo et al., 1996). Delipidated cells were resuspended in PBS and sonicated using Soniprep 150 (Sanyo, MSE Ltd). The resulting suspension was extracted three times with 50 % ethanol and the 27 000 g supernatants were combined, concentrated and digested with 1 mg proteinase K ml−1. After dialysis, aliquots of the aqueous solution containing LAM, lipomannan and phosphatidyl-myo-inositol mannosides were examined by SDS-PAGE. The remainder was applied to a Sephacryl S-200 (Pharmacia) column as described by Khoo et al. (1996). SDS-PAGE was used to monitor the elution profile, and fractions that contained LAM were individually pooled and dialysed. The cell-wall core mAGP complex was obtained from the residual pellet after LAM, lipomannan and phosphatidyl-myo-inositol mannoside extraction as described by Escuyer et al. (2001). To release soluble AG from peptidoglycans, the insoluble residue (mAGP complex) was treated with 2 M NaOH for 16 h at 80 °C and the soluble AG was purified as described by Daffe et al. (1990).
GC and GC/MS analysis of glycosyl composition.
Alditol acetates for GC were prepared as described by McNeil et al. (1989). GC of the alditol acetates was performed on an HP Gas Chromatograph Model 5890 fitted with an SP 2380 (30 m×0.25 mm i.d.; Supelco) column at an initial temperature of 50 °C, held for 1 min. The temperature was raised to 170 °C at 30 °C min−1 before increasing to 260 °C at 5 °C min−1. For GC/MS, partially methylated alditol acetates were dissolved in chloroform prior to injection on a DB-5 column (10 m×0.18 mm i.d.; J&W Scientific) using a ThermoQuest Trace Gas Chromatograph 2000 (ThermoQuest) connected to a GCQ/Polaris MS mass detector at an initial temperature of 50 °C held for 1 min. The temperature was raised to 150 °C at 30 °C min−1 before increasing to 260 °C at 5 °C min−1.
RESULTS
embA is an essential gene in M. tuberculosis
We were interested in exploring the role of EmbA in M. tuberculosis. Although data were available demonstrating its function in M. smegmatis (Escuyer et al., 2001), nothing was known of its function in M. tuberculosis. We first attempted to make an embA deletion in M. tuberculosis using a two-step recombination method (Parish & Stoker, 2000). The deletion vector carried an unmarked, in-frame deletion of the majority of the embA gene to ensure that any mutations did not affect expression of the downstream genes (Fig. 1a). We screened 40 DCO strains and all were wild-type. This was unexpected, since gene knockouts of embA were readily obtained in M. smegmatis (Escuyer et al., 2001). To confirm essentiality, we constructed a merodiploid strain carrying a second functional copy of embA under the control of the constitutive Ag85a promoter in an L5 integrating vector. In this background we were able to isolate DCOs with both the deletion and the wild-type allele (Fig. 1). Four out of 24 DCOs in the merodiploid background were deletions, showing that embA is an essential gene in M. tuberculosis (Fisher's exact t-test, P=0.02). The genotype of the DCO strains was confirmed by Southern blotting (Fig. 1c).
Fig. 1.

Demonstration of essentiality of embA in M. tuberculosis. (a) The genomic organization of the embA region is shown. The regions marked by black bars were amplified and cloned into p2NIL to generate the delivery vector pEMPTY16. The promoter region assayed for activity is indicated by an arrow. (b) and (c) Southern analysis of recombinant strains. (b) The maps of the wild-type and deletion mutant are shown, with the expected sizes. BamHI sites are indicated by symbols (0) and the probe is shown as a solid bar. (c) No deletion DCOs were isolated in the wild-type background. PCR was used to screen for potential deletion alleles in the merodiploid background and four potential del-int strains were identified. Genomic DNA from these four strains was digested with BamHI and hybridized to the probe. The expected sizes for the wild-type and integrated (int) copies of embA are given. Lanes: 1–4, genomic DNA from four del-int strains (deletion DCOs with integrated embA); W, wild-type genomic DNA.
Gene switching
Since the essentiality of embA was not expected, we confirmed our results using a second method (Pashley & Parish, 2003). We showed that we could not replace the integrated vector pEMPTY22 (embA, L5 int, Gm) when it carried the only functional copy of embA. Two del-int strains [Tame 91 and Tame 93: embAΔ (embA, L5 int, Gm)] were transformed with an empty integrating vector pUC-Hyg-Int (L5 int, hyg) and transformants were selected on hygromycin. If the gene was non-essential, the incoming vector would replace the resident integrated vector at a high frequency and hygromycin-resistant (gentamicin-sensitive) colonies would be isolated. If the gene was essential, then only co-integrated strains (pEMPTY22 and pUC-Hyg-Int) would be recovered; this occurs at a low frequency and strains are gentamicin- and hygromycin-resistant. We carried out this gene switching experiment with two independent del-int strains (Tame 91 and Tame 93) (Table 1). The results indicated that replacement of pEMPTY22 did not occur. The numbers of hygromycin-resistant transformants were 102- to 103-fold lower with the switching vector than with the control vector. In both cases, 24 hygromycin-resistant colonies were patch-tested and all were gentamicin-resistant, confirming that the original plasmid containing embA was still present. Since the resident vector could not be replaced, this further confirms that embA is an essential gene in M. tuberculosis.
Table 1.
Gene switching to confirm essentiality
embA del-int strains (Tame 91 and Tame 93) were electroporated with control vector (oriM, pAGAN40) or switching vector (pUC-Hyg-Int) and transformants selected on hygromycin (H) or hygromycin and gentamicin (HG). Transformation efficiencies per μg input plasmid DNA are given. nd, Not determined.
| Strain | pAGAN40 | pUC-Hyg-Int | ||
|---|---|---|---|---|
| H | HG | H | HG | |
| Tame 91 | 1.85E+08 | 2.18E+08 | 3.20E+05 | 2.15E+05 |
| Tame 93 | 1.00E+07 | nd | 1.00E+05 | nd |
embAMtb encodes an arabinosyltransferase
embAMtb has been proposed to be an arabinosyltransferase based on sequence homology with M. smegmatis embA (Cole et al., 1998; Escuyer et al., 2001), although there is no direct evidence for its biochemical activity in vitro or in vivo. We could not determine the biological consequences of embA deletion in M. tuberculosis, since we were unable to construct a deletion mutant. However, based on the premises that EmbAMsm synthesizes or transfers a disaccharide Araβ(1→2)Araα1→ to an internal 5-linked Araf of the linear glycan Araβ(1→2)Araα(1→5)Araα(1→5)Araα(1→5)Araα1→, we were able to use an enzyme assay to identify the function of EmbAMtb.
Previous work has shown that an M. smegmatis embA mutant is unable to transfer a disaccharide to the pentasaccharide acceptor, but this function is restored by complementation with functional EmbA (Khasnobis et al., 2006). We tested whether complementing this mutant strain with the embAMtb gene would restore this activity. As a control, we also complemented with the embAMsm gene. Enzyme preparations from the M. smegmatis strains were used in a cell-free assay to detect arabinosyltransferase activity. TLC analysis of the reaction products revealed a slower migrating component as compared to the starting acceptor in M. smegmatis embA strains complemented with either M. smegmatis or M. tuberculosis alleles. The product in both strains was indistinguishable (Fig. 2a), suggesting that embAMtb encodes a functional arabinosyltransferase with similar activity to EmbAMsm.
Fig. 2.

Arabinosyltransferase activity assays. Extracts from M. smegmatis strains were prepared and used in an in vitro assay for arabinosyltransferase activity. (a) TLC and (b) Dionex HPAEC was used to analyse the product formed after incubation with the pentasaccharide acceptor. (a) TLC of M. smegmatis strains. Wild-type (lanes 1 and 2), ΔembA (3 and 4), ΔembA complemented with embAMsm (5 and 6) and ΔembA complemented with embAMtb (7 and 8). Lanes 1, 3, 5 and 7 are reactions with the addition of an acceptor pentasaccharide; lanes 2, 4, 6 and 8 are reactions with no acceptor. The acceptor pentasaccharide (lane 9) was visualized on TLC with α-naphthol. (b) The enzymically formed radioactive product was purified from the TLC plate and subjected to endoarabinanase digestion followed by Dionex HPAEC analysis, resulting in a single Ara6 peak. (c) 14C-labelled AG digested with endoarabinanase showing three peaks as Ara2, Ara6 and cyclic Gal4, as established by Xin et al. (1997).
The mobility of the radioactive product does not indicate the number of arabinosyl residues incorporated on the pentasaccharide acceptor or the nature of the product. We therefore subjected the purified radioactive product excised from TLC to endoarabinanase digestion followed by Dionex HPAEC. Oligosaccharides were eluted with a gradient of sodium acetate (0–1 M) in 10 % NaOH. The digested sample yielded a single peak with a retention time comparable to the retention time of the terminal hexasaccharide Ara6 (Fig. 2b) released from the 14C-labelled AG (Fig. 2c). This result confirmed the formation of a heptamer by incorporating two Araf residues on the pentasaccharide acceptor and was in full agreement with the product formed and analysed for M. smegmatis (Khasnobis et al., 2006). In the biosynthetic product, which is a heptamer, there is only one site amenable to the Cellulomonas endoarabinanase action yielding one labelled Ara6 (Khasnobis et al., 2006) leaving behind one unlabelled Ara with the octyl aglycon. The retention time of Ara6 is close to cyclic Gal4, which will not be detected as this acquires no radioactivity in this assay.
Neutral sugar and glycosyl linkage composition
Neutral sugar composition of AG from the ΔembAMsm strain (1.2 : 1) showed diminution of the total Ara content compared to the embAMtb-complemented strain (2.2 : 1) which was in accordance with that of wild-type M. smegmatis (Escuyer et al., 2001). The sugar composition was calculated based on a single rhamnosyl (Rha) residue per AG chain. ΔembAMsm had no significant effect on the arabinosylation of LAM.
Purified LAM and soluble AG from ΔembAMsm and ΔembAMsm complemented with embAMtb were per-O-methylated and hydrolysed, reduced, acetylated and analysed by GC/MS to determine glycosyl linkage compositions. Analysis of AG yielded a molar ratio of tAraf (8.7 %), 2-Araf (13.4 %), 5-Araf (56.3 %), 3,5-Araf (21.5 %) for ΔembAMsm and a molar ratio of tAraf (13.2 %), 2-Araf (14.1 %), 5-Araf (52.6 %), 3,5-Araf (20 %) for ΔembAMsm complemented with embAMtb. Thus complementation of the mutant with embAMtb clearly resurrected the defect in tAraf, 2-Araf and reduced 5-Araf which would occur if more branching is introduced. These results are in agreement with the cell-free assay, which resulted in the branched heptamer. We did not observe any changes in the composition of LAM in both ΔembAMsm and ΔembAMsm complemented with embAMtb.
Identification of the promoter for embA
The genomic organization of embA is shown in Fig. 1. Previous work in M. smegmatis suggested that embA is expressed either from its own promoter (Escuyer et al., 2001) or from a polycistronic message encompassing the embCAB region (Telenti et al., 1997). However, the promoter(s) were never directly identified and no data are available for M. tuberculosis. Other data suggest that the EmbR regulatory protein binds to the upstream region of embAMtb and that embA is subject to regulation by EmbR in M. smegmatis (Sharma et al., 2006), although again, no data are available for M. tuberculosis. Because these experiments have not always been conducted in the native host, we investigated expression of embAMtb in its native host and in M. smegmatis.
To determine if a functional promoter for embA exists immediately upstream of the gene, we cloned the embC and embA intergenic region (Fig. 1) into the promoter-probe vector pSM128 (Dussurget et al., 1999), an integrating vector (which is therefore present in only one copy). We assayed promoter activity of this region in M. smegmatis and M. tuberculosis (Fig. 3). A functional promoter was present in this region as assayed by β-galactosidase activity. Interestingly, the activity of the promoter was significantly different in the two species (P<0.05 using Student's t-test), with a sevenfold higher expression level in its native host (M. tuberculosis). These results indicate that activity of the embAMtb promoter is not equivalent in the two organisms.
Fig. 3.

Promoter activity of the embA upstream region. (a) Sequence of the embCA intergenic region carrying PembA(Mtb). The stop and start codons of embC and embA are underlined; the predicted −10 region is in large text. The bases in bold were mutated to Gs in plasmid pEMBA-M. (b) Promoter activity from PembA(Mtb) in M. smegmatis and M. tuberculosis. M. smegmatis transformants were grown in 10 ml 7H9-OADC for 30 h. M. tuberculosis transformants were grown in 10 ml stationary cultures of 7H9-OADC-Tw for 14 days. Results are the means±sd of three individual transformants each assayed in duplicate and are expressed as Miller units. (c) RT-PCR analysis. Agarose gel analysis of RT-PCR on the embC–embA and embA–embB junctions of the embCAB locus. PCR with primers EMBCF2 and EMBAR failed to produce the expected 0.795 kb product (lane 1) while the primer pair EMBAF1 and EMBBR produced the expected 0.649 kb product (lane 2). Lane M is a 1 kb DNA ladder. (d) Genetic organization of the emb region on the M. tuberculosis chromosome and Northern analysis of total RNA from M. tuberculosis hybridized to labelled embA.
A predicted −10 promoter sequence is found upstream of embAMtb (Fig. 3a). We confirmed that this was a functional promoter region using site-directed mutagenesis. Changing the sequence TACCAT to GACCAG (pEMBA-M) resulted in abolition of promoter activity, indicating that this is a functional promoter region (Fig. 3).
embA is co-transcribed with embB in M. tuberculosis
Our data showed that embA has its own promoter and should be independent of embC. However, this did not reveal whether embA and embB are co-expressed. We used RT-PCR and Northern blotting to determine if embA was co-transcribed with embC and embB. We first utilized RT-PCR to determine if the genes were co-expressed. Primer pair EMBAF1 and EMBBR spanning the embA–embB junction was used; a 649 bp product was obtained, indicating that a co-transcript is present. In contrast, primer pair, EMBCF2 and EMBAR covering the embC–embA junction did not give rise to a product, indicating a lack of co-transcription (Fig. 3c). This result was confirmed by Northern-blot analysis (Fig. 3d). A single transcript of approximately 6.6 kb was detected using embA, corresponding to the length of both embA and embB (6590 bp). Thus our results demonstrate that embA is transcribed independently of embC but is co-transcribed with embB.
Expression of PembA(Mtb) during different growth phases
AG is a key component of the mycobacterial cell wall and it is possible that different amounts may be required during different growth phases, depending on the amount of cell wall being newly synthesized. For example, during active growth one would expect an increased requirement for new cell wall biosynthesis. Similarly, biosynthesis of AG could be reduced during stationary phase or in other non-replicating conditions where cell division has ceased. In contrast, the confirmation that embA is essential suggests that constitutive expression should be required for cell survival.
We looked at PembA(Mtb) activity during growth and under conditions of non-replicating persistence. Promoter activity was measured in M. smegmatis and M. tuberculosis to determine if there were any further differences (Fig. 4). Promoter activity was measured over 158 days in liquid and on solid media for M. tuberculosis. PembA(Mtb) was more active in the cells grown on solid medium compared to liquid medium. In both media, no significant variation in activity was observed at different growth phases, indicating that PembA(Mtb) is expressed to a constant level during growth and is not switched off during stationary phase (Fig. 4a). In contrast, PembA(Mtb) activity was markedly downregulated (tenfold) during a hypoxically induced non-replicating state (Fig. 4c). A similar pattern of expression was seen during growth in M. smegmatis, with constitutive expression throughout the growth cycle, but at a lower level (Fig. 4b). In contrast, a twofold upregulation of PembA(Mtb) was seen under hypoxic conditions (P=0.02) (Fig. 4c).
Fig. 4.
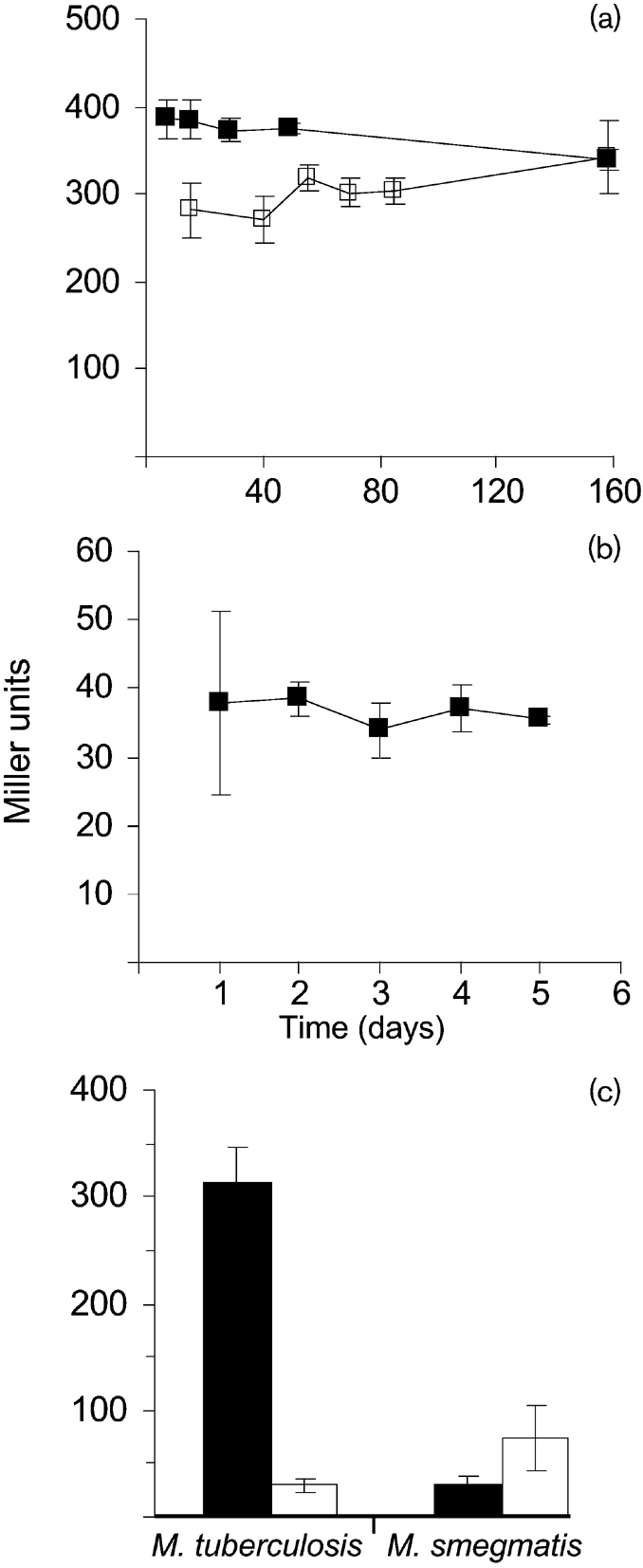
PembA(Mtb) activity during aerobic growth and under hypoxic conditions. (a) Aerobic growth. M. tuberculosis was grown in 10 ml static cultures of 7H9-OADC-Tw (open symbols) or on 7H10-OADC plates (filled symbols). (b) M. smegmatis was grown in 10 ml 7H9-OADC. (c) Hypoxic growth. PembA(Mtb) activity in hypoxic conditions (white columns) is compared to aerobic conditions (black columns). M. tuberculosis was grown under hypoxic conditions for 3 weeks and M. smegmatis for 2 weeks. Results are the means±sd of three individual transformants each assayed in duplicate and are expressed as Miller units.
Stress and drug treatments do not induce PembA(Mtb)
PembA(Mtb) activity was assayed in response to oxidative stress generated by hydrogen peroxide exposure in M. tuberculosis. For these experiments, catalase and albumin were omitted from the media. No change in promoter activity was seen (data not shown). The experiment was also conducted in M. smegmatis, where no significant changes were seen in response to 5, 10 or 15 mM hydrogen peroxide exposure (data not shown).
Previous work has suggested that both embC and embB are upregulated in response to ethambutol treatment, although embA was not measured (Papavinasasundaram et al., 2005). We looked at embA promoter activity after ethambutol or ofloxacin treatment in M. tuberculosis (data not shown). Antibiotics were added at 0.5× MIC. No induction of promoter activity was seen in response to ethambutol or ofloxacin treatment, demonstrating that expression of embA is not controlled in response to ethambutol exposure. Similarly, no induction of embA was seen in M. smegmatis (data not shown).
DISCUSSION
Essentiality of embA in M. tuberculosis
embA is a non-essential gene in M. smegmatis, although embA knockouts show an altered morphology and a slower growth rate than the wild-type, and AG synthesis and cell-wall permeability are affected (Escuyer et al., 2001). It is not immediately obvious why EmbA plays a more critical role in M. tuberculosis. One possibility is that EmbB in M. smegmatis is able to function independently of EmbA, so that deletion of one enzyme is possible. This hypothesis is supported by the fact that a double embAB mutant could not be constructed in M. smegmatis (unpublished data). In M. tuberculosis, it is clear that EmbB cannot complement the function of EmbA. We have also attempted to delete embB in M. tuberculosis, but have been unsuccessful, indicating that it may also be independently essential.
Differential expression of PembA(Mtb) in M. smegmatis
We have shown that embAMtb has its own promoter and so can be independently expressed from embC and possibly embB. The activity of the M. tuberculosis promoter was markedly different in M. smegmatis, indicating that the latter species is not a good genetic model for the study of embA. In particular, the overall level of expression of PembA(Mtb) was sevenfold lower in M. smegmatis. Recent work has suggested that PknH, a serine threonine kinase, is responsible for the phosphorylation of EmbR, which in turn is required for positive control of the embCAB genes (Sharma et al., 2006). M. smegmatis does not possess a homologue of PknH and this may explain partly why PembA(Mtb) is underexpressed in this background. The differential activity of the promoter in the two species in response to hypoxia could also be attributed to the lack of PknH, although the hypoxic response of M. smegmatis is not exactly the same as M. tuberculosis (Wayne & Sohaskey, 2001).
In conclusion, we have demonstrated that even though the gene products for the embA arabinosyltransferase in M. tuberculosis and M. smegmatis are similar, there are significant differences between the physiological requirements. In addition, the M. tuberculosis embA promoter is constitutively active, but differentially expressed in M. smegmatis. Further work using conditional expression systems as developed recently (Blokpoel et al., 2005; Carroll et al., 2005; Ehrt et al., 2005) could help to determine why this arabinosyltransferase is critical for the survival of tubercle bacilli.
Acknowledgments
A. G. A. and D. C. were funded by NIH, NIAID RO1 AI 37139; R. G. was funded by Wellcome Trust Grant 074612. The authors are grateful to Dr Michael R. McNeil for many helpful discussions.
Abbreviations
AG, arabinogalactan
DCO, SCO, double, single cross-over
del-int, one deleted copy and one integrated copy of embA
HPAEC, high-pH anion-exchange chromatography
LAM, lipoarabinomannan
mAGP, mycolyl-AG-peptidoglycan (complex)
OADC, oleic acid, albumin, dextrose, catalase
References
- Belanger, A. E., Besra, G. S., Ford, M. E., Mikusova, K., Belisle, J. T., Brennan, P. J. & Inamine, J. M. (1996). The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A 93, 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, S., Torrelles, J. B., Chatterjee, D., Crick, D. C., Escuyer, V. E. & Brennan, P. J. (2003). Point mutations in embC affect synthesis of lipoarabinomannan in Mycobacterium smegmatis. Abstract submitted for the 8th Annual Conference of the Society for Glycobiology. Glycobiology 13, 853 [Google Scholar]
- Blokpoel, M. C. J., Murphy, H. N., O'Toole, R., Wiles, S., Runn, E. S. C., Stewart, G. R., Young, D. B. & Robertson, B. D. (2005). Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res 33, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, P. J. (2003). Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 83, 91–97. [DOI] [PubMed] [Google Scholar]
- Carroll, P., Muttucumaru, D. G. N. & Parish, T. (2005). Use of a tetracycline-inducible system for conditional expression in Mycobacterium tuberculosis and Mycobacterium smegmatis. Appl Environ Microbiol 71, 3077–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S. & other authors (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- Daffe, M., Brennan, P. J. & McNeil, M. R. (1990). Predominant structural features of the cell wall arabinogalactan of Mycobacterium tuberculosis as revealed through characterization of oligoglycosyl alditol fragments by gas chromatography/mass spectrometry and by 1H and 13C NMR analysis. J Biol Chem 265, 6734–6743. [PubMed] [Google Scholar]
- Dussurget, O., Timm, J., Gomez, M., Gold, B., Yu, S. W., Sabol, S. Z., Holmes, R. K., Jacobs, W. R. & Smith, I. (1999). Transcriptional control of the iron-responsive fxbA gene by the mycobacterial regulator IdeR. J Bacteriol 181, 3402–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt, S., Guo, X. Z. V., Hickey, C. M., Ryou, M., Monteleone, M., Riley, L. W. & Schnappinger, D. (2005). Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escuyer, V. E., Lety, M. A., Torrelles, J. B., Khoo, K. H., Tang, J. B., Rithner, C. D., Frehel, C., McNeil, M. R., Brennan, P. J. & Chatterjee, D. (2001). The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J Biol Chem 276, 48854–48862. [DOI] [PubMed] [Google Scholar]
- Hinds, J., Mahenthiralingam, E., Kempsell, K. E., Duncan, K., Stokes, R. W., Parish, T. & Stoker, N. G. (1999). Enhanced gene replacement in mycobacteria. Microbiology 145, 519–527. [DOI] [PubMed] [Google Scholar]
- Khasnobis, S., Zhang, J., Angala, S. K., Amin, A. G., McNeil, M. R., Crick, D. C. & Chatterjee, D. (2006). Characterization of a specific arabinosyltransferase activity involved in mycobacterial arabinan biosynthesis. Chem Biol 13, 787–795. [DOI] [PubMed] [Google Scholar]
- Khoo, K. H., Douglas, E., Azadi, P., Inamine, J. M., Besra, G. S., Mikusova, K., Brennan, P. J. & Chatterjee, D. (1996). Truncated structural variants of lipoarabinomannan in ethambutol drug- resistant strains of Mycobacterium smegmatis: inhibition of arabinan biosynthesis by ethambutol. J Biol Chem 271, 28682–28690. [DOI] [PubMed] [Google Scholar]
- Mahenthiralingam, E., Marklund, B. I., Brooks, L. A., Smith, D. A., Bancroft, G. J. & Stokes, R. W. (1998). Site-directed mutagenesis of the 19-kilodalton lipoprotein antigen reveals no essential role for the protein in the growth and virulence of Mycobacterium intracellulare. Infect Immun 66, 3626–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil, M. R., Chatterjee, D., Hunter, S. W. & Brennan, P. J. (1989). Mycobacterial glycolipids: isolation, structures, antigenicity, and synthesis of neoantigens. Methods Enzymol 179, 215–242. [DOI] [PubMed] [Google Scholar]
- McNeil, M. R., Robuck, K. G., Harter, M. & Brennan, P. J. (1994). Enzymatic evidence for the presence of a critical hexa-arabinoside in the cell walls of Mycobacterium tuberculosis. Glycobiology 4, 165–173. [DOI] [PubMed] [Google Scholar]
- Mikusova, K., Slayden, R. A., Besra, G. S. & Brennan, P. J. (1995). Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob Agents Chemother 39, 2484–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Papavinasasundaram, K. G., Chan, B., Chung, J. H., Colston, M. J., Davis, E. O. & Av-Gay, Y. (2005). Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J Bacteriol 187, 5751–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish, T. & Stoker, N. G. (2000). Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975. [DOI] [PubMed] [Google Scholar]
- Parish, T. & Wheeler, P. R. (1998). Preparation of cell-free extracts from mycobacteria. In Mycobacteria Protocols, pp. 77–90. Edited by T. Parish & N. G. Stoker. Totowa: Humana Press. [DOI] [PubMed]
- Pashley, C. A. & Parish, T. (2003). Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett 229, 211–215. [DOI] [PubMed] [Google Scholar]
- Scherman, M. S., Kalbe-Bournonville, L., Bush, D., Xin, Y., Deng, L. & McNeil, M. (1996). Polyprenylphosphate-pentoses in mycobacteria are synthesized from 5-phosphoribose pyrophosphate. J Biol Chem 271, 29652–29658. [DOI] [PubMed] [Google Scholar]
- Seidel, M., Alderwick, L. J., Sahm, H., Besra, G. S. & Eggeling, L. (2007). Topology and mutational analysis of the single Emb arabinofuranosyltransferase of Corynebacterium glutamicum as a model of Emb proteins of Mycobacterium tuberculosis. Glycobiology 17, 210–219. [DOI] [PubMed] [Google Scholar]
- Sharma, K., Gupta, M., Pathak, M., Gupta, N., Koul, A., Sarangi, S., Baweja, R. & Singh, Y. (2006). Transcriptional control of the mycobacterial embCAB operon by PknH through a regulatory protein, EmbR, in vivo. J Bacteriol 188, 2936–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevatsan, S., Pan, X., Zhang, Y., Kreiswirth, B. N. & Musser, J. M. (1997). Mutations associated with pyrazinamide resistance in pncA of Mycobacterium tuberculosis complex organisms. Antimicrob Agents Chemother 41, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti, A., Philipp, W. J., Sreevatsan, S., Bernasconi, C., Stockbauer, K. E., Wieles, B., Musser, J. M. & Jacobs, W. R., Jr (1997). The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat Med 3, 567–570. [DOI] [PubMed] [Google Scholar]
- Wayne, L. G. & Sohaskey, C. D. (2001). Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol 55, 139–163. [DOI] [PubMed] [Google Scholar]
- Xin, Y., Lee, R. E., Scherman, M. S., Khoo, K. H., Besra, G. S., Brennan, P. J. & McNeil, M. (1997). Characterization of the in vitro synthesized arabinan of mycobacterial cell walls. Biochim Biophys Acta 1335, 231–234. [DOI] [PubMed] [Google Scholar]
- Zhang, N., Torrelles, J., McNeil, M., Escuyer, V. E., Khoo, K. H., Brennan, P. J. & Chatterjee, D. (2003). The Emb proteins of mycobacteria direct arabinosylation of lipoarabinomannan and arabinogalactan via an N-terminal recognition region and a C-terminal synthetic region. Mol Microbiol 50, 69–76. [DOI] [PubMed] [Google Scholar]