

Abstract
One potential vaccine strategy in the fight against meningococcal disease involves the exploitation of outer-membrane components of Neisseria lactamica, a commensal bacterium closely related to the meningococcus, Neisseria meningitidis. Although N. lactamica shares many surface structures with the meningococcus, little is known about the antigenic diversity of this commensal bacterium or the antigenic relationships between N. lactamica and N. meningitidis. Here, the N. lactamica porin protein (Por) was examined and compared to the related PorB antigens of N. meningitidis, to investigate potential involvement in anti-meningococcal immunity. Relationships among porin sequences were determined using distance-based methods and FST, and maximum-likelihood analyses were used to compare the selection pressures acting on the encoded proteins. These analyses demonstrated that the N. lactamica porin was less diverse than meningococcal PorB and although it was subject to positive selection, this was not as strong as the positive selection pressures acting on the meningococcal porin. In addition, the N. lactamica porin gene sequences and the protein sequences of the loop regions predicted to be exposed to the human immune system were dissimilar to the corresponding sequences in the meningococcus. This suggests that N. lactamica Por, contrary to previous suggestions, may have limited involvement in the development of natural immunity to meningococcal disease and might not be effective as a meningococcal vaccine component.
INTRODUCTION
Meningococcal disease, caused by the Gram-negative bacterium Neisseria meningitidis, is a global health problem which cannot be comprehensively controlled by vaccination. Effective vaccines directed against the serogrouping antigen, the polysaccharide capsule, are available for disease caused by serogroups A, C, Y and W135 meningococci. However, a vaccine based on serogroup B polysaccharide has not been developed due to its poor immunological reactivity and similarity to host antigens (Finne et al., 1987). As serogroup B strains cause over 85 % of meningococcal disease in the UK (Gray et al., 2006), alternative vaccine candidates are under investigation, including outer-membrane proteins (OMPs), either purified (Boslego et al., 1995) or as part of outer-membrane vesicle (OMV) formulations (Bjune et al., 1991; van der Ley et al., 1995).
The commensal bacterium Neisseria lactamica, which is closely related to both N. meningitidis and Neisseria gonorrhoeae (Guibourdenche et al., 1986), is carried in the upper respiratory tracts of young children, in whom carriage of the meningococcus is rare. As natural immunity to meningococcal disease develops during childhood, carriage of N. lactamica may be involved in the acquisition of this immunity (Gold et al., 1978). N. lactamica is only associated with disease in exceptional circumstances (Schifman & Ryan, 1983; Wilson & Overman, 1976) and as it does not possess a polysaccharide capsule (Griffiss et al., 1987; Kim et al., 1989) or the outer-membrane protein PorA (Ward et al., 1992), this commensal or its antigens can be used in anti-meningococcal vaccines that are independent of both serogroup and serosubtype. Vaccines based on N. lactamica whole cells, OMPs, or OMVs have been proposed (Griffiss et al., 1991; Oliver et al., 2002) and research and development of a vaccine based on N. lactamica is ongoing (Finney et al., 2007; Li et al., 2006). However, the antigenic profiles of N. lactamica isolates are not well understood, and the cross-reactive epitopes that induce protection against meningococcal infection have not been defined (Tang et al., 1999). This may constitute a major obstacle to the development of vaccines based on this species.
One protein that could be involved in the induction of cross-protective immune responses is the N. lactamica porin (Troncoso et al., 2002). Porins, which are essential for growth, are pore-forming membrane proteins that create channels, allowing transport of hydrophilic molecules across lipid bilayers (Achouak et al., 2001) and are generally highly expressed. These proteins exist as trimers (Derrick et al., 1999) and consist of 16 anti-parallel β-strands connected by short periplasmic turns and eight surface-exposed loops (Maiden et al., 1991; van der Ley et al., 1991). The regions corresponding to the surface-exposed loops are less well conserved than the domains corresponding to the β-sheets, and show variation in both length and sequence, presumably as a consequence of immune selection (Maiden et al., 1991; van der Ley et al., 1991).
The N. lactamica porin is related to gonococcal PorB1a and PorB1b and to meningococcal PorB (Derrick et al., 1999; Ward et al., 1992) and is essentially identical to the Neisseria polysaccharea porin (Derrick et al., 1999). Only these members of the genus contain three conserved lysine residues, found in close proximity within the pore, which form a potential GTP-binding site implicated in pathogenesis, consistent with a role in regulating pore function when inserted into host cells (Derrick et al., 1999; Rudel et al., 1996). Meningococcal PorB proteins are divided into two distinct, mutually exclusive classes designated PorB2 and PorB3. These proteins exhibit high levels of genetic diversity, especially in the surface-exposed regions, and these regions, with the exception of putative loops II and III, are subject to the diversifying influence of immunological selection (Urwin et al., 2002). Putative loops II and III have structural roles; loop II is important in monomer–monomer interactions within the porin trimer, while loop III is sequestered in the pore of each monomer, and may be involved in regulating pore function (Derrick et al., 1999).
Here, N. lactamica por sequences, in particular the regions encoding the putative surface-exposed loops, were examined and compared to porB of N. meningitidis, to investigate the diversity of the encoded proteins and to assess whether these N. lactamica proteins could be involved in the production of cross-protective immune responses to the meningococcus. This was achieved by comparing 69 unique N. lactamica por gene sequences, obtained from isolates previously characterized by multilocus sequence typing (MLST) (Bennett et al., 2005) with a diverse collection of meningococcal porB sequences (Urwin et al., 2002). Distance-based methods and FST were used to determine relationships among the porins and maximum-likelihood analyses were used to investigate the selection pressures acting on these proteins in N. lactamica. The results indicated that the N. lactamica porin was only subject to weak positive selection pressures, had limited sequence similarity to meningococcal PorB and was less diverse than meningococcal PorB. This suggests that it might not be suitable for inclusion in anti-meningococcal vaccine formulations.
METHODS
Isolates.
A total of 587 individuals, including infants and a small number of siblings and parents, were sampled in Oxfordshire, UK, during two longitudinal studies of nasopharyngeal bacterial carriage in infants, resulting in 275 N. lactamica isolates. The genetic diversity of these N. lactamica isolates was similar to that seen in isolates from other locations and likely to be representative of the global diversity of this organism. There is no evidence that the Oxfordshire isolates were a separate population and the collection contained representatives from five of the six clonal complexes currently defined for N. lactamica (Bennett, 2006). Details of these isolates are available from the Neisseria MLST database: http://pubmlst.org/neisseria/ (Jolley et al., 2004). Nucleotide sequences were determined for the entire por gene from all isolates. The majority had been collected from the same infants on successive occasions, enabling an analysis of the effect of carriage on the N. lactamica porin. The nucleotide and amino acid sequences from the N. lactamica porins were compared to the porin sequences from a diverse collection of 324 meningococcal isolates: http://neisseria.org/nm/typing/porb/ (Urwin et al., 2002) which included sequences from both carried and disease-associated isolates.
PCR amplification and sequence determination of por genes.
Amplification of the por gene was carried out by PCR with oligonucleotide primers B21 and B22 (Table 1). Amplification reaction mixtures contained reaction buffer [10 mM Tris/HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.001 % (w/v) gelatin]; 200 μM each of dATP, dCTP, dGTP, dTTP; 1 μM of each primer; 1.25 units Taq polymerase (AmpliTaq; Applied Biosystems) per 50 μl; and 0.5 μl template DNA per 50 μl (approx. 50 ng μl−1). The PCR conditions consisted of an initial denaturation step of 94 °C for 2 min, followed by 35–40 cycles of denaturation (94 °C for 1 min), annealing (50–5 °C for 1 min), extension (72 °C for 2 min), and then a final extension step of 72 °C for 2 min. The PCR products were precipitated by incubation at room temperature for 30 min with 20 % (w/v) polyethylene glycol 8000, 2.5 M NaCl. After centrifugation for 60 min at 2750 g, the precipitates were washed twice in 70 % (v/v) ethanol, dried and resuspended in 5–10 μl sterile distilled water.
Table 1.
Oligonucleotide primers used in N. lactamica por sequencing
| Primer | Sequence (5′–3′) | Function |
|---|---|---|
| B21 (forward)* | CCAAAAAAGGAATACAGC | Amplification and sequencing |
| B22 (reverse)* | GCAGATTAGAATTTGTGG | Amplification and sequencing |
| 8U (forward)† | TCCGTACGCTACGATTCTCC | Sequencing |
| 8L (reverse)† | GGAGAATCGTAGCGTACGGA | Sequencing |
| 94U (forward)† | CTCAAACCGAAGTTGCCG | Sequencing |
| 94L (reverse)† | CGGCAACTTCGGTTTGAG | Sequencing |
The nucleotide sequences of the amplified gene fragments were determined at least once on each DNA strand by cycle sequencing with BigDye Ready Reaction Mix (Applied Biosystems), used in accordance with the manufacturer's instructions. Sequencing reactions were performed with the oligonucleotide primers listed in Table 1. Unincorporated dye terminators were removed by precipitation of the termination products with 95 % (v/v) ethanol and the labelled extension products were separated and detected with either a Prism 3700 DNA analyser, or a Prism 377 XL DNA analyser (Applied Biosystems).
Analysis of sequence data.
Nucleotide sequence data from forward- and reverse-strand chromatograms were assembled into single contiguous sequences using the Staden suite of computer programs (Staden, 1996). Sequences were manually aligned using the SeqLab program, part of the GCG Wisconsin package, version 10.3 (Womble, 2000). The alignment (available on request) was based on amino acid sequence similarity, with codon integrity maintained. Each unique por allele was given a number in order of discovery, so that alleles were named por-1 to por-69. To allow comparisons of N. lactamica por with the published meningococcal porB sequences (http://neisseria.org/nm/typing/porb/) (Urwin et al., 2002), all sequences were truncated to the same length, so that the amino acid sequences began at the 32nd amino acid of the N. lactamica Por sequence (starting motif ETYRT) and ended with the 336th amino acid (motif STAST). mega version 2.1 (Kumar et al., 2001) was used to calculate genetic distances between sequences and to produce neighbour-joining trees. To construct the tree from nucleotide sequences, all three coding positions were examined and the Kimura two-parameter distance correction (Kimura, 1980) was applied. A gamma shape parameter was not calculated as the inclusion of this parameter in the analysis had a minimal effect on the phylogeny produced. To produce the trees from amino acid sequences, p-distances were used. The reliability of the inferred trees was assessed using the bootstrap test (2000 replications). FST values were calculated using Arlequin version 2.000 (Schneider et al., 2000). Alignment gaps were excluded from all analyses.
Analysis of selection pressures.
A maximum-likelihood approach was used to examine selection pressures acting on individual codon sites of the N. lactamica por gene. A phylogenetic tree of the aligned sequences was constructed using paup, version 4 (Swofford, 1998), using the HKY85 model of nucleotide substitution. Selection pressures on the aligned por sequences were examined using codeml, implemented in the paml program (Yang, 1997), in which codon substitution models were compared using the maximum-likelihood tree and the nucleotide sequence data. The dN/dS ratio (parameter ω) was calculated codon by codon using different models of codon substitution that differed in how ω varied along the sequences. Model M0 estimated a single ω parameter for all sites, whereas the M1 model divided codons into two site categories: conserved sites (p0), with ω0 set at 0, and neutral sites (p1), with ω1 set at 1. To account for positive selection the M2 model was used, with the same two classes as M1 plus a third category of sites (p2), with ω2 estimated from the data. The M3 model was a more sensitive test of selection as it estimated ω values for three classes of sites, all of which could be >1. Models M7 and M8 both used a discrete beta distribution (with ten categories described by parameters p and q) to model ω ratios among sites, although M8 also included an additional class of sites for which ω could be >1.
Nested models were compared using a likelihood ratio test (LRT). Twice the difference in log-likelihood between models was compared with the value obtained under a χ2 distribution, with degrees of freedom equal to the difference in the number of parameters between models. Finally, empirical Bayesian methods were used to calculate the probability that a particular codon site belonged to a specific class.
RESULTS
Comparative diversity and relationships between N. lactamica por and meningococcal porB allele sequences
A total of 69 unique N. lactamica por alleles, obtained from 275 isolates, were aligned with 46 porB2 and 77 porB3 sequences, obtained from 324 diverse meningococcal isolates (Urwin et al., 2002). A neighbour-joining tree was constructed from these sequences (Fig. 1), grouping the porin classes into three distinct clusters, supported by bootstrap values of 99 %. The N. lactamica por sequences were more closely related to porB3 than porB2, with less diversity than both meningococcal porB classes. FST analysis of por, porB2 and porB3 was used to assess levels of gene flow among the populations and gave values >0.85 (P≤0.05) for all three comparisons, indicating separate populations with little genetic exchange among them. A neighbour-joining tree constructed from the nucleotide sequences from the β-barrel-encoding regions alone, using the same alignment, produced a tree topology almost identical to that shown in Fig. 1 (data not shown). The diversity within meningococcal porB and N. lactamica por alleles was assessed (Table 2). The number of variable sites was comparable, with 147 for porB2, 136 for porB3 and 132 for por. Diversity, however, was greater within porB2 and porB3 (4.34 % and 4.03 % respectively) than in por (2.31 %). There were 44 unique amino acid sequences for PorB2, 65 for PorB3 and 62 for Por, with more variable sites within PorB2 (70) and PorB3 (61) than in Por (49).
Fig. 1.
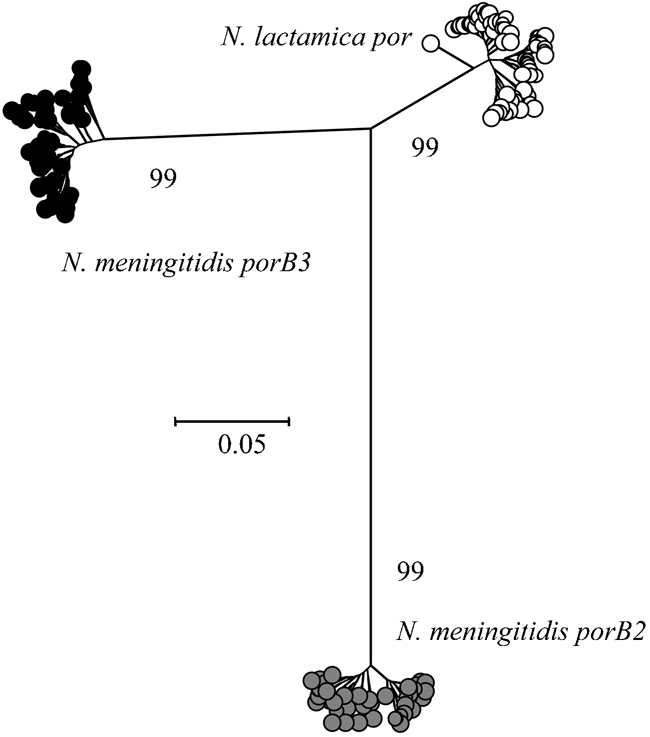
Neighbour-joining tree constructed from aligned gene sequences showing the relationship of N. lactamica por to meningococcal porB2 and porB3. Bootstrap values are shown.
Table 2.
Diversity within meningococcal porB and N. lactamica por genes
| porB2 | porB3 | por | |
|---|---|---|---|
| No. of alleles | 46 | 77 | 69 |
| Length of aligned sequences (bp) | 1053 | 894 | 1029 |
| No. of variable sites | 147 | 136 | 132 |
| Mean percentage genetic distance (Kimura's two-parameter model) | 4.34 | 4.03 | 2.31 |
| PorB2 | PorB3 | Por | |
| No. of amino acid sequences | 44 | 65 | 62 |
| No. of variable sites | 70 | 61 | 49 |
| Mean percentage p-distance | |||
| Loop I | 10.60 | 24.80 | 8.39 |
| Loop II | 6.06 | 6.06 | 12.00 |
| Loop III | 4.89 | 2.27 | 6.87 |
| Loop IV | 15.23 | 13.89 | 12.12 |
| Loop V | 24.51 | 30.67 | 13.77 |
| Loop VI | 32.78 | 28.86 | 14.72 |
| Loop VII | 15.71 | 34.84 | 7.69 |
| Loop VIII | 24.11 | 11.93 | 7.29 |
Comparative diversity and relationships between N. lactamica Por and meningococcal PorB loop regions
The amino acid sequence variants of the putative Por loop regions were named in accordance with the meningococcal PorB nomenclature used in the PorB database (http://neisseria.org/nm/typing/porb/), although the location and length of the loops were as defined previously (Derrick et al., 1999). Six variants were found in loop I, eight in loop II, 17 in loop III, four in loop IV, 10 in loop V, five in loop VI, and three in loops VII and VIII. The loops were of variable length (11 amino acids in loops II, IV and VI, 13 in loop VII, 16 in loop VIII, 18 in loop V, 23 in loop I and 41 in loop III). Some of the amino acid sequences were encoded by more than one nucleotide sequence, and one of the variants found in loop III was longer than the other variants by two dipeptides: ST and GI. The eight loop regions in N. lactamica Por were compared to the corresponding loops in meningococcal PorB (Table 2). Mean percentage p-distances within the loops of Por, PorB2 and PorB3 were determined, and for each loop, with the exception of loops II and III, divergence was greater within PorB2 and PorB3 than within Por.
Neighbour-joining trees were constructed from the amino acid sequences of the loop regions from Por, PorB2 and PorB3 (Fig. 2). For the majority of the loops, distinct clusters, corresponding to the three different classes, were formed. In loop II the groups were indistinct and not well supported by bootstrap values (not included), with two PorB2 and two PorB3 amino acid sequences identical to Por sequences from N. lactamica. One of these PorB2 sequences was encoded by a nucleotide sequence identical to that of N. lactamica. Three distinct clusters were evident for loop III, with this loop more uniform in the meningococcal porins than in the N. lactamica porin. In loop VI the PorB2 variants formed a distinct group but the PorB3 variants were more closely related to N. lactamica Por. A loop VI PorB3 amino acid sequence was identical to the amino acid sequences from three N. lactamica alleles, and clustered with the N. lactamica sequences. This PorB3 variant was encoded by a nucleotide sequence identical to one of the N. lactamica variants.
Fig. 2.

Neighbour-joining trees constructed from the aligned amino acid sequences from the loop regions of N. lactamica Por and meningococcal PorB2 and PorB3. Amino acid sequences identical in N. lactamica and N. meningitidis are indicated by dashed circles. Bootstrap values are shown.
Maximum-likelihood analysis
Maximum-likelihood analysis provided evidence for positive selection pressures acting on the por gene of N. lactamica (Table 3), as models with site classes where parameter ω was permitted to be >1 (models M2, M3 and M8) were statistically significantly better than those in which it was not (models M0, M1, M7). However, the M3 model (a sensitive test of positive selection) was not significantly better supported than the more conservative M2 model (P=0.251). Under the M2 model, 7.0 % of sites fell into a positively selected class, where ω2=4.195. Under model M3, 7.1 % of sites fell into a positively selected class, where ω2=3.936, 8.5 % of sites fell into a neutrally evolving class where ω1=1.002 and the remaining 84.4 % of sites were highly conserved (ω0=0.013). Under model M8 7.3 % of sites fell into a positively selected class, where ω1=3.886.
Table 3.
Parameter estimates for the maximum-likelihood analysis of selection pressures acting on the N. lactamica porin
| Model | Site categories (p) and dN / dS (ω) | Likelihood test | χ2 | P |
|---|---|---|---|---|
| M0 | ω=0.306 | |||
| M1 | p0=0.816, p1=0.184 | |||
| M2 | p0=0.818, p1=0.116, p2=0.070 | M0 vs M2 | 387.144 | <0.000 |
| ω2=4.195 | M1 vs M2 | 70.442 | <0.000 | |
| M3 | p0=0.844, p1=0.085, p2=0.071 | M0 vs M3 | 389.907 | <0.000 |
| ω0=0.013, ω1=1.002, ω2=3.936 | M1 vs M3 | 73.205 | <0.000 | |
| M2 vs M3 | 2.763 | 0.251 | ||
| M7 | P=0.026, q=0.118 | |||
| M8 | P=0.035, q=0.330 | M7 vs M8 | 72.475 | <0.000 |
| p1=0.073, ω1=3.886 |
Bayesian methods were used to identify the sites with the highest probability of falling into the positively selected class under models M2, M3 and M8. A dN/dS ratio (parameter ω) >1.5 was considered to be indicative of positive selection, and for model M2, there were nine selected sites with dN/dS ratios of 4.068–4.195. The same nine sites were identified using model M8, although the dN/dS ratios were 3.825–3.886. Model M3, which had previously been used to demonstrate selection in meningococcal PorB (Urwin et al., 2002) and a more sensitive test of selection than model M2, identified 16 additional selected sites with dN/dS ratios of 1.900–3.936. The majority of selected sites fell within predicted loop regions, with positive selection identified in loops I, II, IV, VI and VIII using all three models. Only model M3 identified selected sites within loops III, V and VII. All selected sites were mapped onto the translated sequence of por-2 (Fig. 3).
Fig. 3.

Positive selection acting on N. lactamica porin sequences. The translated sequence of por-2, one of the longest sequences, is used as an example. The locations of the putative loop regions (loops I–VIII) are indicated. Amino acid residues subject to positive selection under models M2 and M8 are identified with asterisks. Amino acid residues subject to positive selection under the M3 model are indicated with black boxes. Polymorphic sites in the porin alleles of the genotypically identical (ST-608) isolates carried by one infant are indicated by triangles. Figures at the end of each line of sequence denote residue number.
A N. lactamica porin structural model
A homology model for the N. lactamica porin was constructed in the same way as corresponding models for meningococcal PorB2 and PorB3 (Urwin et al., 2002). The translated sequence of N. lactamica por-2 was aligned against that of the porin Omp32 from Comamonas acidovorans (Zeth et al., 2000), which was used as a template for structural alignment and refinement, as implemented by the software package modeller (Sali et al., 1995). The three-dimensional structural model of the N. lactamica porin is shown along with the published models of meningococcal PorB2 and PorB3 (Urwin et al., 2002) in Fig. 4. Positively selected sites, as inferred from maximum-likelihood analysis using model M3, were mapped onto this model, with model M2 and model M8 positively selected sites highlighted. Whereas a number of strongly selected sites were located within the surface-exposed loops for both meningococcal PorB2 and PorB3, only weakly positively selected sites were present in the N. lactamica porin structure. In the N. lactamica porin, using model M3, weakly positively selected sites were evident within the pore. Weakly positively selected sites were also present within the pore of meningococcal PorB2, but not PorB3. The general structure of all three porins was similar, although the N. lactamica porin was closer to meningococcal PorB3, as some of the surface-exposed loops were shorter in both Por and PorB3.
Fig. 4.
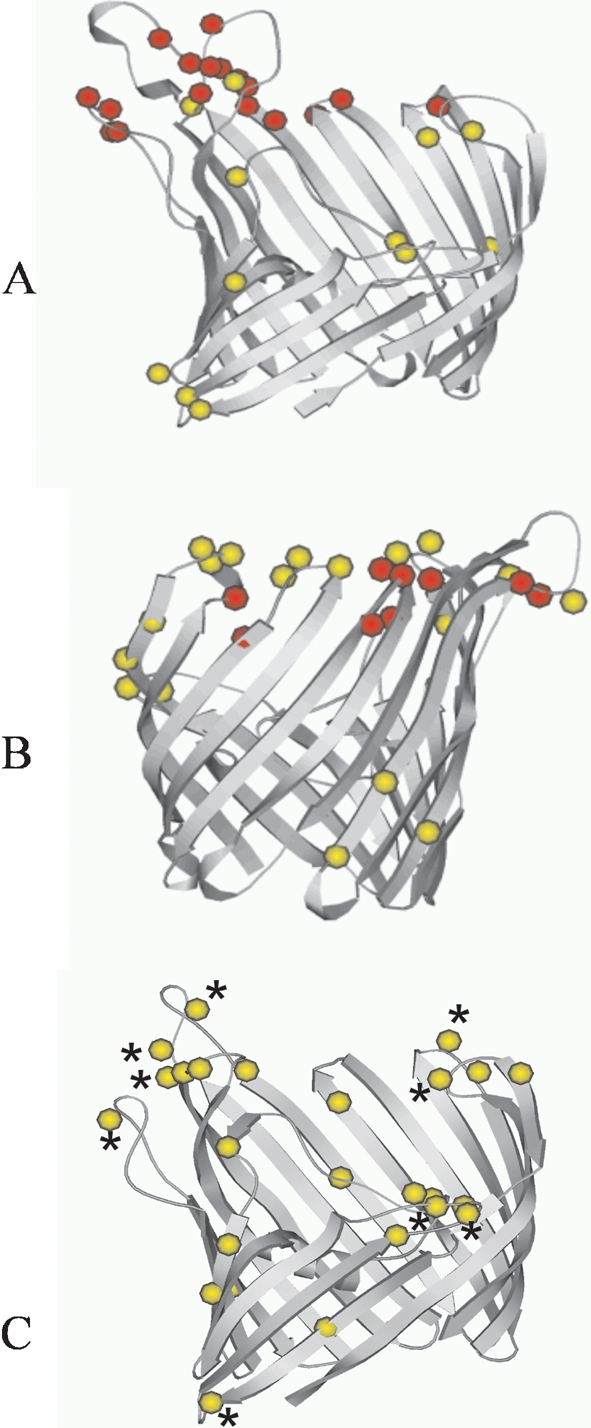
Ribbon diagrams of models for meningococcal PorB2-5 (A) PorB3-2 (B) and N. lactamica Por-2 (C), with superposition of residues subject to positive selection. Residues under strong selection are shown as red spheres and residues under weaker selection are shown as yellow spheres, under model M3. Residues subject to positive selection under models M2 and M8 are identified with asterisks. Some longer loop regions have been truncated. Diagrams produced using Molscript (Kraulis, 1991).
Selection pressures acting on N. lactamica porins during carriage
As the majority of N. lactamica isolates were collected from the same infants sampled on successive occasions, it was possible to examine the effects of carriage on the porins of these sequential isolates. There was long-term carriage of particular variants, with little evidence for replacement of variants. One child carried isolates with the MLST genotype ST-638 on seven successive samplings, between the ages of 12 weeks and 48 weeks, but at age 48 weeks the porin allele associated with these isolates altered by one nonsynonymous nucleotide substitution in loop VI. This allele was not present among the isolates obtained from any other subjects and is likely to have arisen by point mutation as a result of immune selection during carriage by this particular child. This site was also variable in other N. lactamica porin alleles (position 253, Fig. 3), with a dN/dS value >3.860 using all three models of selection.
Another infant carried ST-608 isolates on eight successive occasions, from age 8 weeks until age 96 weeks, but seven different porin alleles were identified among these isolates. There were six polymorphic sites among these alleles (Fig. 3), with each change nonsynonymous, altering the structure of the porin. Most were found within and around loop III, which potentially influences pore function. One was found within loop V, which may have occurred as a result of immune selection. These sites, except for position 95, were also variable in other N. lactamica porins. However, only model M3 detected positive selection at these sites with dN/dS values of ∼3.648, except for position 95 (dN/dS 1.487).
DISCUSSION
Establishing the diversity of antigen genes from non-pathogenic neisseriae is important, as antigens common to N. meningitidis, N. lactamica and other non-pathogenic commensals may be involved in conferring natural immunity to meningococcal disease (Troncoso et al., 2000, 2002). N. lactamica antigens are also components of anti-meningococcal vaccines currently being developed (Gorringe, 2005) and the identity and characterization of the key cross-reactive antigenic components is of great importance (Finney et al., 2007). Although the porins of N. meningitidis have been thoroughly examined (Russell et al., 2004; Urwin et al., 2002), little information was available on the diversity of the N. lactamica porin and its relationship to meningococcal porins (Derrick et al., 1999; Ward et al., 1992). Here, the diversity of 69 N. lactamica por alleles was determined and compared to the related meningococcal porB alleles to assess the potential for antigenic cross-reactivity.
The neighbour-joining tree constructed from N. lactamica por and meningococcal porB2 and porB3 alleles demonstrated the relatively low diversity among the N. lactamica por alleles and established that these alleles were distinct from porB2 and porB3. The distinct clusters observed suggest that genetic exchange between N. lactamica por and the two meningococcal porB classes is likely to be rare. This was supported by the FST results, which indicated low levels of gene flow between the three groups. The neighbour-joining tree constructed from the sequences that encode the conserved β-barrel regions alone, which were subject to stabilizing selection, produced a tree topology almost identical to the tree constructed from the sequences that encode both the β-barrel regions and the loop regions. This suggests that the porins have not diverged recently.
The amino acid sequences of the surface-exposed loop regions were analysed individually and the loops that determine antigenic variability in N. meningitidis (loops I, IV, V, VI, VII and VIII) (Derrick et al., 1999) were dissimilar to the corresponding loops in N. lactamica. Loop II sequences were the most similar between N. lactamica and N. meningitidis, and sequences from two PorB2 and two PorB3 loop II variants were identical to loop II N. lactamica variants. As structural constraints are likely to limit diversity in this region (Derrick et al., 1999), these identical sequences are probably a consequence of shared ancestry rather than recent lateral genetic transfer. This is supported by the detection of minimal gene flow between these three populations. A single example of lateral genetic transfer between species may have occurred in loop VI. This loop was subject to positive immune selection and a meningococcal PorB3 variant was encoded by a nucleotide sequence identical to a N. lactamica sequence. However, two other N. lactamica variants had amino acid sequences identical to the PorB3 variant but were encoded by different nucleotide sequences.
Frequent genetic recombination between species has been described for tbpB (Linz et al., 2000) and may also occur within the variable region of fetA (J. S. Bennett, E. A. L. Thompson, P. Kriz, K. A. Jolley & M. C. J. Maiden, unpublished results). However, genetic exchange frequency decreases rapidly as a function of relatedness of the donor to the recipient (Majewski, 2001). As the N. lactamica porin is distinct from the meningococcal porins, including PorA (Derrick et al., 1999), and does not experience strong selection pressures, frequent lateral genetic transfer of porin sequences between the two species is unlikely, in common with the housekeeping genes of Neisseria (Bennett et al., 2007).
Porins, including those of Neisseria, have an internal eyelet region formed by a long loop III folded into the pore, with a negatively charged cluster of side-chains facing the positive charges from R/K residues derived from β-sheets forming the β-barrel wall (Achouak et al., 2001; Schirmer, 1998). This organization produces an electrostatic field in the lumen that regulates the diffusion of molecules through the constricted area (Achouak et al., 2001). Loop III was more variable in N. lactamica Por than in meningococcal PorB. The amino acid sequences encoded by por-2 and por-8 were the longest N. lactamica sequences due to two dipeptide insertions (S T and G I), occurring at the tip of loop III. As this loop folds back into the centre of the lumen, these insertions could affect pore conductance. The sequence motif found in this region of the translated sequences of por-2 and por-8 (STKDTGI) was not present in any other N. lactamica or meningococcal sequence examined in this study and may have been transferred from another bacterial species by lateral genetic transfer.
Analysis of the selection pressures acting on the N. lactamica porin revealed codons subject to positive selection, the majority of which were located within the loop regions. However, due to the variability of these regions in both N. meningitidis and N. lactamica and the difficulty in aligning the amino acid sequences unambiguously, it was not possible to determine if the positively selected codons in N. lactamica were the same as those positively selected in N. meningitidis. Whereas some residues in meningococcal PorB2 and PorB3 were under strong positive immune selection, with dN/dS ratios (ω) of 18.553 for PorB2 and 13.923 for PorB3 (Urwin et al., 2002), residues in the N. lactamica porin experienced much weaker positive selective pressures, with dN/dS ratios that did not exceed 4.195.
Results from the three maximum-likelihood models that took account of positive selection agreed on nine positively selected codon sites in the N. lactamica porin. The M3 model identified an additional 16 positively selected sites. These additional sites were considered in this analysis as five of them were polymorphic in isolates with identical MLST genotypes that were carried by one infant on successive occasions. A sixth site was only polymorphic in the porin of the isolates carried by this particular infant, although the dN/dS value for this site (1.487) was less than the cut-off value for positive selection (>1.5) used in this analysis. Both the M2 and M8 models failed to detect positive selection at these sites which were biologically relevant as the changes were nonsynonymous, altering the structure of the porin.
Using model M3, three weakly positively selected codon sites were detected in loop II of the N. lactamica porin, whereas in the meningococcus, only one weakly positively selected site was present in loop II of PorB3 and none in loop II of PorB2 (Urwin et al., 2002). This suggests that there is less structural constraint in this region in N. lactamica than in the meningococcus. Under model M3, four weakly positively selected sites were detected in loop III of the N. lactamica porin, three of which were variable among the sequential isolates with identical MLST genotypes carried by one infant. As loop III is sequestered within the lumen, these variations may reflect changes in pore function. Three weakly positively selected sites were present in loop III of meningococcal PorB2 but none in loop III of PorB3 (Urwin et al., 2002), suggesting that N. lactamica Por is more like PorB2 in this respect. N. lactamica has a single porin as opposed to the two in most meningococci, and variations in the permeability of the pore might alter the function of this protein, improving fitness. In the gonococcus, which also has one functional porin (Feavers & Maiden, 1998), mutations in loop III were shown to reduce porin permeability to hydrophilic antibiotics and these changes may therefore have a role in resistance to penicillin and tetracycline (Gill et al., 1998). However, the genotypically identical N. lactamica isolates that expressed seven different por alleles while carried by a single infant had not been exposed to antibiotic therapy (D.W. Crook, personal communication) and therefore the reasons for the rapid amino acid variation among these isolates remain unexplained.
Although the N. lactamica porin undergoes relatively weak positive selection pressures in comparison to the meningococcus, nonsynonymous changes in the porin alleles among isolates carried by two infants showed plasticity of the porin, probably in response to selection, over a very short time period. This supports previous studies that established that meningococcal porins are subject to strong positive selection pressures and evolve rapidly (Jelfs et al., 2000; Urwin et al., 2002), hindering the design of vaccines based on these proteins.
Initially, N. lactamica por appeared to be a good candidate for involvement in the production of a cross-protective immune response to meningococci, as the related meningococcal PorB protein is subject to strong immune selection (Urwin et al., 2002). Porins are also essential for bacterial growth and are expressed at high levels, and por genes were amplified from all N. lactamica isolates examined. However, the N. lactamica porin was dissimilar to the porins of N. meningitidis, with no alleles shared, and the surface-exposed loop region amino acid sequences were rarely identical in meningococci and N. lactamica. The low sequence identity between Por and PorB, the low levels of genetic exchange, as indicated by the FST results, together with the relatively weak positive selection pressures acting on the N. lactamica porin and the lack of immunological cross-reactivity with meningococcal PorB (Finney et al., 2007; Kim et al., 1989), suggest that this protein may not be important in the induction of any cross-protective immune responses that might protect against meningococcal disease. Therefore, any protection against meningococcal disease provided by N. lactamica carriage is likely to be due to antigens other than Por.
Acknowledgments
This study was funded by the Meningitis Research Foundation. M. C. J. M. is a Wellcome Trust Senior Research Fellow. The N. lactamica samples were obtained from studies carried out by the Oxford Vaccine Group supported by the Wellcome Trust, reference number 056886/2/994/Z. We are grateful to the staff of the Oxford Vaccine Group for supporting the collection of samples and the families and children who participated in the studies.
Abbreviations
MLST, multilocus sequence typing
OMP, outer-membrane protein
OMV, outer membrane vesicle
Footnotes
The GenBank accession numbers for the sequences reported in this paper are EF495264–EF495332.
References
- Achouak, W., Heulin, T. & Pages, J. M. (2001). Multiple facets of bacterial porins. FEMS Microbiol Lett 199, 1–7. [DOI] [PubMed] [Google Scholar]
- Bennett, J. S. (2006). The relationship of Neisseria lactamica to the pathogenic Neisseria: implications for vaccine development. DPhil thesis, Department of Zoology, University of Oxford, Oxford.
- Bennett, J. S., Griffiths, D. T., McCarthy, N. D., Sleeman, K. L., Jolley, K. A., Crook, D. W. & Maiden, M. C. (2005). Genetic diversity and carriage dynamics of Neisseria lactamica in infants. Infect Immun 73, 2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, J. S., Jolley, K. A., Sparling, P. F., Saunders, N. J., Hart, C. A., Feavers, I. M. & Maiden, M. C. (2007). Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from MLST. BMC Biol 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjune, G., Høiby, E. A., Grønnesby, J. K., Arnesen, O., Fredriksen, J. H., Halstensen, A., Holten, E., Lindbak, A. K., Nøkleby, H. & other authors (1991). Effect of outer membrane vesicle vaccine against group B meningococcal disease in Norway. Lancet 338, 1093–1096. [DOI] [PubMed] [Google Scholar]
- Boslego, J., Garcia, J., Cruz, C., Zollinger, W., Brandt, B., Ruiz, S., Martinez, M., Arthur, J., Underwood, P. & other authors (1995). Efficacy, safety, and immunogenicity of a meningococcal group B (15 : P1.3) outer membrane protein vaccine in Iquique, Chile. Vaccine 13, 821–829. [DOI] [PubMed] [Google Scholar]
- Derrick, J. P., Urwin, R., Suker, J., Feavers, I. M. & Maiden, M. C. J. (1999). Structural and evolutionary inference from molecular variation in Neisseria porins. Infect Immun 67, 2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feavers, I. M. & Maiden, M. C. J. (1998). A gonococcal porA pseudogene: implications for understanding the evolution and pathogenicity of Neisseria gonorrhoeae. Mol Microbiol 30, 647–656. [DOI] [PubMed] [Google Scholar]
- Finne, J., Bitter Suermann, D., Goridis, C. & Finne, U. (1987). An IgG monoclonal antibody to group B meningococci cross-reacts with developmentally regulated polysialic acid units of glycoproteins in neural and extraneural tissues. J Immunol 138, 4402–4407. [PubMed] [Google Scholar]
- Finney, M., Vaughan, T., Taylor, S., Hudson, M. J., Pratt, C., Wheeler, J. X., Vipond, C., Feavers, I., Jones, C. & other authors (2007). Characterization of the key antigenic components and pre-clinical immune responses to a meningococcal disease vaccine based on Neisseria lactamica outer membrane vesicles. Hum Vaccin 3 [DOI] [PubMed]
- Gill, M. J., Simjee, S., Al-Hattawi, K., Robertson, B. D., Easmon, C. S. & Ison, C. A. (1998). Gonococcal resistance to β-lactams and tetracycline involves mutation in loop 3 of the porin encoded at the penB locus. Antimicrob Agents Chemother 42, 2799–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold, R., Goldschneider, I., Lepow, M. L., Draper, T. F. & Randolph, M. (1978). Carriage of Neisseria meningitidis and Neisseria lactamica in infants and children. J Infect Dis 137, 112–121. [DOI] [PubMed] [Google Scholar]
- Gorringe, A. R. (2005). Can Neisseria lactamica antigens provide an effective vaccine to prevent meningococcal disease? Expert Rev Vaccines 4, 373–379. [DOI] [PubMed] [Google Scholar]
- Gray, S. J., Trotter, C. L., Ramsay, M. E., Guiver, M., Fox, A. J., Borrow, R., Mallard, R. H. & Kaczmarski, E. B. (2006). Epidemiology of meningococcal disease in England and Wales 1993/94 to 2003/04: contribution and experiences of the Meningococcal Reference Unit. J Med Microbiol 55, 887–896. [DOI] [PubMed] [Google Scholar]
- Griffiss, J. M., Brandt, B. & Jarvis, G. A. (1987). Natural immunity to Neisseria meningitidis. In Evolution of Meningococcal Disease, vol. II, pp. 99–119. Edited by N. A. Vedros. Boca Raton, FL: CRC Press.
- Griffiss, J. M., Yamasaki, R., Estabrook, M. & Kim, J. J. (1991). Meningococcal molecular mimicry and the search for an ideal vaccine. Trans R Soc Trop Med Hyg 85 (Suppl 1), 32–36. [DOI] [PubMed] [Google Scholar]
- Guibourdenche, M., Popoff, M. Y. & Riou, J. Y. (1986). Deoxyribonucleic acid relatedness among Neisseria gonorrhoeae, N. meningitidis, N. lactamica, N. cinerea and “Neisseria polysaccharea”. Ann Inst Pasteur Microbiol 137B, 177–185. [DOI] [PubMed] [Google Scholar]
- Jelfs, J., Munro, R., Wedege, E. & Caugant, D. A. (2000). Sequence variation in the porA gene of a clone of Neisseria meningitidis during epidemic spread. Clin Diagn Lab Immunol 7, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley, K. A., Chan, M. S. & Maiden, M. C. (2004). mlstdbNet – distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. J., Mandrell, R. E. & Griffiss, J. M. (1989). Neisseria lactamica and Neisseria meningitidis share lipooligosaccharide epitopes but lack common capsular and class 1, 2, and 3 protein epitopes. Infect Immun 57, 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16, 111–120. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. (1991). MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24, 946–950. [Google Scholar]
- Kumar, S., Tamura, K., Jakobsen, I. B. & Nei, M. (2001). mega2: Molecular Evolutionary Genetics Analysis software. Bioinformatics 17, 1244–1245. [DOI] [PubMed] [Google Scholar]
- Li, Y., Zhang, Q., Winterbotham, M., Mowe, E., Gorringe, A. & Tang, C. M. (2006). Immunization with live Neisseria lactamica protects mice against meningococcal challenge and can elicit serum bactericidal antibodies. Infect Immun 74, 6348–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz, B., Schenker, M., Zhu, P. & Achtman, M. (2000). Frequent interspecific genetic exchange between commensal neisseriae and Neisseria meningitidis. Mol Microbiol 36, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Maiden, M. C. J., Suker, J., McKenna, A. J., Bygraves, J. A. & Feavers, I. M. (1991). Comparison of the class 1 outer membrane proteins of eight serological reference strains of Neisseria meningitidis. Mol Microbiol 5, 727–736. [DOI] [PubMed] [Google Scholar]
- Majewski, J. (2001). Sexual isolation in bacteria. FEMS Microbiol Lett 199, 161–169. [DOI] [PubMed] [Google Scholar]
- Oliver, K. J., Reddin, K. M., Bracegirdle, P., Hudson, M. J., Borrow, R., Feavers, I. M., Robinson, A., Cartwright, K. & Gorringe, A. R. (2002). Neisseria lactamica protects against experimental meningococcal infection. Infect Immun 70, 3621–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel, T., Schmid, A., Benz, R., Kolb, H. A., Lang, F. & Meyer, T. F. (1996). Modulation of Neisseria porin (PorB) by cytosolic ATP/GTP of target cells: parallels between pathogen accommodation and mitochondrial endosymbiosis. Cell 85, 391–402. [DOI] [PubMed] [Google Scholar]
- Russell, J. E., Jolley, K. A., Feavers, I. M., Maiden, M. C. & Suker, J. (2004). PorA variable regions of Neisseria meningitidis. Emerg Infect Dis 10, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali, A., Potterton, L., Yuan, F., van Vlijmen, H. & Karplus, M. (1995). Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326. [DOI] [PubMed] [Google Scholar]
- Schifman, R. B. & Ryan, K. J. (1983). Neisseria lactamica septicemia in an immunocompromised patient. J Clin Microbiol 17, 934–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer, T. (1998). General and specific porins from bacterial outer membranes. J Struct Biol 121, 101–109. [DOI] [PubMed] [Google Scholar]
- Schneider, S., Roessli, D. & Excoffier, L. (2000). Arlequin version 2.000: a software for population genetic data analysis. Geneva: University of Geneva.
- Staden, R. (1996). The Staden sequence analysis package. Mol Biotechnol 5, 233–241. [DOI] [PubMed] [Google Scholar]
- Suker, J. (1997). Variation of meningococcal porin antigens. PhD thesis, Department of Biochemistry, Royal Free Hospital School of Medicine, University of London.
- Swofford, D. (1998). paup*: Phylogenetic analysis using parsimony (and other methods). Sunderland, MA: Sinauer Associates.
- Tang, C., Moxon, R. & Levine, M. M. (1999). For discussion: live attenuated vaccines for group B meningococcus. Vaccine 17, 114–117. [DOI] [PubMed] [Google Scholar]
- Troncoso, G., Sanchez, S., Moreda, M., Criado, M. T. & Ferreiros, C. M. (2000). Antigenic cross-reactivity between outer membrane proteins of Neisseria meningitidis and commensal Neisseria species. FEMS Immunol Med Microbiol 27, 103–109. [DOI] [PubMed] [Google Scholar]
- Troncoso, G., Sanchez, S., Criado, M. T. & Ferreiros, C. M. (2002). Analysis of Neisseria lactamica antigens putatively implicated in acquisition of natural immunity to Neisseria meningitidis. FEMS Immunol Med Microbiol 34, 9–15. [DOI] [PubMed] [Google Scholar]
- Urwin, R., Holmes, E. C., Fox, A. J., Derrick, J. P. & Maiden, M. C. (2002). Phylogenetic evidence for frequent positive selection and recombination in the meningococcal surface antigen PorB. Mol Biol Evol 19, 1686–1694. [DOI] [PubMed] [Google Scholar]
- van der Ley, P., Heckels, J. E., Virji, M., Hoogerhout, P. & Poolman, J. T. (1991). Topology of outer membrane porins in pathogenic Neisseria species. Infect Immun 59, 2963–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ley, P., van der Biezen, J. & Poolman, J. T. (1995). Construction of Neisseria meningitidis strains carrying multiple chromosomal copies of the porA gene for use in the production of a multivalent outer membrane vesicle vaccine. Vaccine 13, 401–407. [DOI] [PubMed] [Google Scholar]
- Ward, M. J., Lambden, P. R. & Heckels, J. E. (1992). Sequence analysis and relationships between meningococcal class 3 serotype proteins and other porins of pathogenic and non- pathogenic Neisseria species. FEMS Microbiol Lett 94, 283–290. [DOI] [PubMed] [Google Scholar]
- Wilson, H. D. & Overman, T. L. (1976). Septicemia due to Neisseria lactamica. J Clin Microbiol 4, 214–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womble, D. D. (2000). GCG: the Wisconsin Package of sequence analysis programs. Methods Mol Biol 132, 3–22. [DOI] [PubMed] [Google Scholar]
- Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Zeth, K., Diederichs, K., Welte, W. & Engelhardt, H. (2000). Crystal structure of Omp32, the anion-selective porin from Comamonas acidovorans, in complex with a periplasmic peptide at 2. Å resolution. Structure 8, 981–992. [DOI] [PubMed] [Google Scholar]