

Abstract
Small GTPases of the Arf family are best known for their role in vesicular transport, wherein they nucleate the assembly of coat proteins at sites of carrier vesicle formation. However, accumulating evidence indicates that the Arfs are also important regulators of actin cytoskeleton dynamics and are involved in a variety of actin-based processes, including cell adhesion, migration and neurite outgrowth. The mechanisms of this regulation are remarkably diverse, ranging from the integration of vesicular transport with cytoskeleton assembly to the direct regulation of Rho-family GTPase function. Here, we review recent progress in our understanding of how Arfs and their interacting proteins function to integrate membrane and cytoskeletal dynamics.
Introduction
A dynamic actin cytoskeleton is essential for a wide variety of normal cellular processes, including the maintenance of cell shape and morphology, cytokinesis, adhesion, migration, neurite outgrowth, endocytosis and phagocytosis. Actin remodeling is also important under pathological conditions, such as the invasion and metastasis of cancers. It is well established that actin cytoskeleton dynamics are controlled by small GTPases of the Rho family (for review see [1]). The best characterized of these, RhoA, Rac and Cdc42, interact with distinct sets of downstream effector proteins to direct the assembly of different actin-based structures; RhoA promotes the formation of stress fibers and focal adhesions, Rac induces the formation of lamellipodia and membrane ruffles, and Cdc42 stimulates formation of filopodia. Protrusive structures such as lamellipodia might also require the vectorial addition of new membrane components, such that actin remodeling is often integrated with vesicular transport.
The ADP-ribosylation factor (Arf) proteins are small, ubiquitously expressed GTP-binding proteins best known for their role in membrane trafficking [2]. The six mammalian Arfs are divided into three classes on the basis of sequence relatedness, with class I containing Arf1, Arf2 and Arf3, class II containing Arf4 and Arf5, and class III containing only Arf6. The class I and class II Arfs are mainly concentrated in the Golgi apparatus, although they also function in endosomal compartments. By contrast, Arf6 is localized primarily in the plasma membrane and a subset of endosomes.
A large body of evidence indicates that the Arfs promote carrier vesicle biogenesis by nucleating the assembly of coat protein complexes at sites of vesicle formation. For class I and class II Arfs, these complexes include multimeric coat protein complexes such as COPI (coat protein complex I), AP-1 (adaptor protein complex 1), AP-3 and AP-4, in addition to monomeric adaptors of the GGA (Golgi-associated, gamma adaptin ear-containing and Arf binding) and Mint (Munc18-interacting protein) families [2]. These vesicle coats serve to concentrate cargo proteins in the plane of the membrane and might help to deform the membrane surface, thereby facilitating the formation of carrier vesicles.
In addition to their well characterized role in membrane trafficking, it has become apparent that the Arfs also contribute significantly to the regulation of actin cytoskeletal organization. In this review, we summarize recent progress in our understanding of how Arfs and their interacting proteins can integrate actin cytoskeleton and membrane dynamics.
Arfs, phosphoinositides and the cytoskeleton
One way in which Arfs influence actin cytoskeleton assembly is through their effects on the lipid microenvironment. There is an intimate relationship between phosphoinositides and the actin cytoskeleton. This relationship operates on several levels. First, phosphatidylinositol 4,5 bisphosphate (PtdIns(4,5)P2), binds directly to several important actin regulatory proteins and modulates their function. For example, PtdIns(4,5)P2 stimulates the activity of actin-nucleating factors, such as the WASP (Wiskott-Aldrich syndrome protein) family proteins, promotes filament growth by uncapping of barbed ends, and inhibits filament severing by proteins such as ADF (actin depolymerizing factor), cofilin and gelsolin (for review see [3]). PtdIns(4,5)P2 also enhances the association of actin with membranes by modulating the conformation of proteins such as ezrin, radixin, moesin, α-actinin and vinculin that act at the cytoskeleton–membrane interface. Second, phosphoinositides enhance the activation of Rho family GTPases by recruiting cytosolic guanine nucleotide exchange factors to the membrane surface and/or stimulating their activity.
Arf1 has been shown to recruit both PI4KIIIβ and PIP5K (phosphatidylinositol 4-phosphate 5-kinase) to the Golgi, where it also allosterically enhances the activity of both enzymes [4,5]. Similarly, Arf6 stimulates recruitment and activation of type I PIP5 kinases at the plasma membrane and endosomes [6,7], a property shared with its yeast counterpart, yArf3 [8]. At both the Golgi and the plasma membrane, the Arf-dependent recruitment of PtdIns kinases results in enhanced local production of PtdIns(4,5)P2.
In addition, all Arfs are allosteric activators of phospholipase D (PLD), which cleaves phosphatidylcholine to generate phosphatidic acid (PA). PA is a bioactive lipid that alters the physical properties of membrane bilayers, and it is thought to facilitate membrane bending at sites of vesicle formation. PA also synergizes with Arf to enhance the activity of PIP5 kinases, and the resulting PtdIns(4,5)P2 further stimulates the activity of PLD, resulting in a positive feedback loop (for review, see [9]). Thus, the ability of Arfs to alter the local lipid environment is important to their roles in both vesicular transport and in actin cytoskeleton dynamics.
Arf regulation of actin assembly in the Golgi
It has been known for some time that actin and several actin-binding proteins, including spectrin, mAbp1 (mammalian actin-binding protein 1), drebrin, cortactin, N-WASP and the Arp2/3 complex associated with the Golgi (for review, see [10]). It is therefore not surprising that actin-depolymerizing drugs have been shown to disrupt both Golgi morphology and positioning [11,12]. Perhaps more surprising is that most of these proteins rapidly dissociate from the Golgi upon treatment of cells with brefeldin A (BFA), a fungal toxin that selectively inhibits Golgi-associated Arf nucleotide exchange factors. This indicates that the association of these proteins with Golgi membranes is dependent upon Arf activity. In the case of spectrin, this is clearly due to the Arf1-dependent synthesis of PtdIns(4,5)P2, which binds to the β-spectrin pleckstrin homology (PH) domain [13]. The mechanisms by which mAbp1, drebrin and cortactin associate with Golgi membranes are less well defined.
Cdc42 also associates strongly with Golgi membranes, where it regulates both retrograde transport from the Golgi to the ER [14] and exit from the trans-Golgi network (TGN) [15]. Early studies revealed that association of Cdc42 with the Golgi was sensitive to BFA, suggesting that Arf activation was necessary for Cdc42 recruitment [16]. Subsequent work demonstrated that Cdc42 binds to γCOP, a subunit of the COPI vesicle coat, and is brought to the Golgi, at least in part, through the Arf1-dependent assembly of the COPI complex on Golgi membranes [17]. This initiates a cascade of events in which Cdc42 activates N-WASP, which then activates the Arp2/3 complex, leading to the assembly of polymerized actin on Golgi membranes (Figure 1a). Interestingly, Cdc42 is displaced from coatomer by occupation of the cargo-binding site on γCOP by the cargo protein p23, suggesting that cargo loading is coupled to local actin polymerization. A late burst of actin assembly has been observed during formation of clathrincoated vesicles at the plasma membrane [18], leading to the hypothesis that actin polymerization either pushes nascent vesicles away from the donor membrane or contributes to their fission (for review, see [19]). Whether such a mechanism is operational in the Golgi remains to be determined.
Figure 1.

Regulation of actin assembly by Arf1. (a) Arf1 acts both upstream and downstream of Cdc42 to promote vesicle formation in the Golgi. Cdc42 interacts with the γCOP subunit of the COPI coatomer complex and is brought to Golgi membranes by Arf1-mediated coatomer recruitment. The identity of the GEF that activates Golgi-associated Cdc42 is not known. Activated Cdc42 then stimulates N-WASP, which activates the Arp2/3 complex, leading to local actin assembly, and it is thought that this promotes vesicle scission. It is not known if Arf1 remains associated with the Cdc42-N-WASP complex. In addition, Arf1 also downregulates Cdc42 activity by recruiting the Cdc42 GAP, ARHGAP21, to Golgi membranes. In the absence of ARHGAP21, unregulated actin assembly leads to disruption of the Golgi complex. How the upstream and downstream functions of Arf1 are coordinated is not currently understood. (b) Arf1, Cdc42, and ARHGAP21 also cooperate at the plasma membrane to mediate clathrin-independent endocytosis. Cdc42 activation leads to the formation of endocytic vesicles containing fluid phase markers and GPI-anchored membrane proteins (vesicle budding) [22]. It is not known if this pathway also uses N-WASP to drive actin assembly. Activation of Arf1 on the plasma membrane recruits ARHGAP21, leading to downregulation of Cdc42 activity. Depletion of either Arf1 or ARHGAP21 by RNAi results in accumulation of Cdc42 in puncta at the plasma membrane [22]. (c) In the TGN, Arf1 activity promotes the assembly of a cortactin–dynamin complex at sites of vesicle formation through a mechanism that remains unknown. In a similar fashion to N-WASP, cortactin can activate the Arp2/3 complex and presumably acts in concert with dynamin to mediate vesicle scission.
As with most GTPases, Cdc42 requires guanine nucleotide exchange factors (GEFs) to facilitate GTP loading, and GTPase-activating proteins (GAPs) to stimulate GTP hydrolysis and to terminate its interaction with downstream effectors. The identity of GEFs that might activate Cdc42 at the Golgi remains unknown. However, a recent study identified a Golgi-associated Cdc42 GAP protein, ARHGAP21, which is recruited to Golgi membranes through a direct interaction between Arf1–GTP and a region of ARHGAP21 encompassing the PH domain and an adjacent helical motif [20]. The PH domain of ARHGAP21 does not bind phospholipids, and it therefore appears that Golgi recruitment is mediated primarily through its interaction with Arf1. Depletion of endogenous ARHGAP21 by RNA interference (RNAi) resulted in fragmentation of the Golgi and an accumulation of the Arp2/3 complex in the spaces between Golgi fragments, suggesting that failure to terminate the Cdc42 signal leads to unregulated actin assembly. A similar phenotype is seen following expression of constitutively active Cdc42 [21]. Thus, it appears that Cdc42 is an important regulator of actin-dependent transport events in the Golgi, and that Arf1 acts both upstream (to recruit coatomer) and downstream (to recruit ARHGAP21) of Cdc42 in the process of vesicle formation. Intriguingly, it was shown recently that Arf1, ARHGAP21 and Cdc42 also act in concert to promote the clathrin-independent endocytosis of cholesterol-rich micro-domains at the plasma membrane (Figure 1b) [22], indicating that the Arf1–ARHGAP21 complex acts at multiple sites within the cell. It should be noted that, although originally reported as ARHGAP10, the protein described in studies [20] and [21] has been renamed ARHGAP21 (accession # NP_065875) to avoid confusion with a previously existing ARHGAP10 protein (NP_078881), also known as GRAF2 (GTPase regulator associated with focal adhesion kinase 2) (P. Chavrier, personal communication).
Recent evidence also suggests that Arf activation promotes recruitment of the actin-binding protein cortactin to sites of vesicle formation in the TGN (Figure 1c) [23]. In addition to its ability to bind F-actin and to activate the Arp2/3 complex, cortactin is also necessary for recruitment of the mechanoenzyme dynamin to the TGN. A recent study demonstrated that treatment of cells with either BFA or actin depolymerizing drugs leads to the rapid dissociation of both cortactin and dynamin from the TGN, and that Arf1–GTP stabilizes the binding of both proteins to Golgi membranes. Moreover, expression of truncated cortactin mutants that cannot couple to dynamin impaired the transport of cargo [vesticular stomatitus virus G (VSV-G) and mannose-6-phosphate receptor] from the TGN. Together, these observations suggest that Arf activation leads to the actin-dependent recruitment of a cortactin–dynamin complex that is necessary for export of at least some cargos from the TGN. It is well established that Arf1 promotes the assembly of AP1, AP3 and GGA adaptor complexes on TGN membranes, and a role for actin assembly has been suggested in the budding of clathrincoated vesicles at the TGN [24]. However, it is not yet known whether actin or associated proteins function in all Arf-dependent pathways at the TGN.
Arf regulation of actin dynamics in the cell periphery
Although both class I and class II Arfs are present in the cell periphery, the predominant Arf in this region is Arf6. Expression of dominant–negative mutants or RNAi-mediated depletion indicates that Arf6 plays an important role in a variety of actin-based processes. including phagocytosis, some forms of endocytosis, cell adhesion, migration, invasion of the extracellular matrix. and neurite outgrowth [2]. In a similar fashion to Arf1, Arf6 can activate both phospholipase D and type I PIP5 kinases, but it does so at the plasma membrane and on endosomal compartments [6]. Consistent with this function, expression of the Arf6-specific GEF, EFA6 (exchange factor for Arf6), or a constitutively active Arf6 mutant, Arf6Q67L, leads to the formation of swollen, clustered endosomes that have membranes enriched in PtdIns(4,5)P2 and surrounded by polymerized actin [25]. Lower level expression of Arf6Q67L induces the formation of actin-rich tails on intracellular particles, at least some of which contain endocytosed tracers and are presumably endosomal in nature [26]. The use of markers has revealed that these tails exist in live cells, but neither their function nor their regulation is currently understood. Another feature typical of Arf6 activation is the loss of actin stress fibers [27-29]. Again, the mechanisms through which this occurs are not well understood, but they are likely to involve crosstalk between Arf6 and Rho family GTPases.
Crosstalk between Arf6 and Rac
Arf6 can also control cytoskeletal organization through crosstalk with Rho family GTPases. Early studies demonstrated that expression of dominant inhibitory Arf6T27N inhibited membrane ruffling in response to either constitutively active Rac mutants [30] or physiological stimuli [27]. Conversely, activation of endogenous Arf6 by expression of the Arf GEF ARNO (Arf nucleotide-binding site opener) was found to trigger the activation of Rac, extension of lamellipodia and the onset of migration in normally stationary epithelial cells (Figure 2) [31]. Subsequent studies have confirmed that Arf6 activation is coupled to Rac activation in several settings, including integrin ligation [32,33] or stimulation of cells with vascular endothelial growth factor (VEGF) [34], platelet-derived growth factor (PDGF) [35] or angiotensin II [36].
Figure 2.

Induction of lamellipodia by activation of Arf in the cell periphery. MDCK cells were infected with recombinant adenoviruses encoding the Arf GEF, ARNO. Epithelial cells typically cluster together and do not form lamellipodia even at subconfluent density (a). Expression of wild type ARNO induces the formation of large lamellipodia (b), whereas a catalytically inactive mutant (E156K) does not (c), indicating that activation of Arf is sufficient to drive lamellipodium assembly. Cells were stained to identify exogenous ARNO (green), and F-actin (red). Arrows indicate sites of lamellipodia formation. Images courtesy of Lorraine Santy.
The relationship between Arf6 and Rac is complex. In epithelial cells, Rac activity is necessary for both the maintenance of cell–cell adhesions and the migration of cells induced by growth factors or wounding. In MDCK (Madin-Darby canine kidney) cells induced to scatter, there is a transient decrease in Rac activation, corresponding temporally to disassembly and internalization of cadherin-mediated junctional complexes [37]. This initial decrease is caused, at least in part, by the Arf6-dependent recruitment of NM23-H1, an allosteric inhibitor of the Rac GEF TIAM1 (T-lymphoma invasion and metastasis protein 1), to sites of cell–cell contact [38]. Subsequently, Rac1 activity increases (in parallel with Arf6) at free cell edges, triggering the assembly of lamellipodia and the onset of migration. Thus, at least in epithelia, Rac activation can be both positively and negatively regulated by Arf6, in a spatially distinct manner.
Arf-mediated recruitment of Rac GEFs
How does Arf6 activate Rac? In epithelial cells, Arf6 activation leads to recruitment of a bipartite Rac GEF, the Dock180–Elmo complex, to the leading edge (Figure 3a) [39]. Expression of catalytically inactive Dock180, or an Elmo mutant that cannot couple to Dock180, completely inhibits both the activation of Rac and motility downstream of Arf6. Although a direct interaction between Arf6 and Elmo or Dock180 has not yet been demonstrated, an Elmo isoform, ElmoD2, was found to act as a GAP on the Arf-like protein Arl2, and to exhibit weak activity on Arf6, suggesting that Arfs and Elmo proteins can interact directly [40]. Interestingly, the Drosophila Arf6 GEF, Loner, promotes Rac activation and localization during myoblast fusion [41], a process that also requires the Drosophila ortholog of Dock180, myoblast city [42].
Figure 3.

Models for the regulation of Rac activity by Arf6. (a) GEF recruitment. The activation of Arf6 by ARNO triggers the recruitment of the Rac GEF, Dock180–ELMO, to the plasma membrane, where it then stimulates Rac activation. In this model, cytosolic Rac reaches the membrane by diffusion. (b) Endosomal translocation. In this model, Arf6 regulates the vesicular transport of Rac from perinuclear endosomes to the plasma membrane, where it is available to be activated by local GEFs. (c) Lipid raft translocation. Arf6 can control the trafficking of lipid raft components from endosomal compartments to the plasma membrane. Incorporation of Rac into lipid rafts at the plasma membrane is thought to be important for both its activation and its coupling to downstream effector proteins. In this model, cytosolic Rac reaches the plasma membrane by diffusion, but lipid raft components are transported in vesicular carriers whose formation requires Arf6.
A second mode of Rac activation might be through the Kalirin and Trio family of Rac GEFs. Both the brain-specific Kalirin5 and the more ubiquitously expressed Trio bind Arf6 specifically through an array of spectrin repeats [43]; however, they appear to interact preferentially with Arf–GDP. Given that Arf6 remains associated with membranes in the GDP-bound state, it is possible that Arf6-GDP targets Kalirin and Trio to the membrane and releases it locally upon Arf activation.
The Rac translocation model
Early studies indicated that endogenous Rac1 is present on perinuclear endosomes, and that it can be translocated to the plasma membrane in an Arf6-dependent manner [27,30]. This led to the hypothesis that Arf6 regulates Rac activation by controlling its trafficking from endosomes to the plasma membrane in response to physiological cues (Figure 3b). However, a large cytosolic pool of Rac exists in complex with RhoGDI. In addition to preventing the dissociation of bound GDP, GDIs also enhance the solubility of Rho family GTPases by sequestering their C-terminal isoprenyl modifications, and they sterically hinder interactions with both regulatory proteins and downstream effectors [44]. Photobleaching experiments indicate that this cytosolic pool exchanges rapidly with the plasma membrane, where GDI is displaced [45]. Therefore, although recycling of endosome-associated Rac probably contributes to the pool of active Rac at the plasma membrane, the quantitative importance of this contribution remains unknown.
Accumulating evidence indicates that Rac, which is prenylated at its C terminus, needs to be partitioned into cholesterol-enriched membrane microdomains (CEMMs, more commonly called lipid rafts) to enable both its activation and its coupling to downstream effectors [46]. Therefore, Arf6 could modulate Rac activity by controlling the availability of the necessary lipids at the plasma membrane. Although some of the components of CEMMs (notably PtdIns(4,5)P2) are undoubtedly synthesized locally, recent findings suggest that the recycling of internalized rafts from endosomes also requires active Arf6, and that this has an important role in the attachment and spreading of anchorage-dependent cells [32]. Detachment of cells from the substratum induces the quantitative internalization of lipid raft components from the cell surface, and their accumulation in perinuclear recycling endosomes. Cells held in suspension exhibit low levels of active Arf6 and Rac1. However, replating onto fibronectin-coated substrates triggers the rapid activation of Arf6, the redistribution of internalized rafts to the plasma membrane, and a corresponding activation of Rac as the cells spread. Knockdown of Arf6 leads to the selective retention of raft components in recycling endosomes, a dramatic inhibition of adhesion-induced Rac activation, and impaired cell spreading [32]. These findings suggest that, in addition to its regulation of phospholipid-modifying enzymes and its interaction with Rac GEFs, Arf6 can further modulate Rac activity by controlling the availability of lipid raft components (Figure 3c).
Arf GAPs as modulators of cytoskeleton dynamics
The human genome encodes 24 proteins with recognizable Arf GAP domains, a subset of which have well-defined interactions with the actin cytoskeleton. Owing to space limitations, it is not possible to review these interactions, or all of the GAP classes, in detail. However, for an excellent and thorough review of the role of Arf GAPs in cytoskeletal dynamics, see [47]. Here, we summarize the features of this group of proteins, members of which impact directly on cytoskeleton function. Unfortunately, the nomenclature for the Arf GAPs is, to say the least, untidy. Here, we use the nomenclature adopted by Randazzo and colleagues [47], but a complete list of synonyms can be found in [48].
ARAPs
The ARAPs (Arf GAP with Rho GAP, ankyrin repeat, and pleckstrin homology domains) are characterized by the presence of both Arf GAP and Rho GAP domains, and as such represent a direct link between the two GTPase families (Figure 4). (Note that the term Rho GAP as used here connotes a domain that can stimulate GTP hydrolysis on Rho, Rac or Cdc42). There are three ARAP genes, ARAP1, ARAP2 and ARAP3. Although ARAP1 was originally reported to be primarily an Arf1 GAP in vitro [49], recent evidence suggests that, as with ARAP2 [50] and ARAP3 [35], it can act preferentially on Arf6 in the cell [51]. The Rho GAP domains of ARAP1 and ARAP3 both appear to prefer RhoA as a substrate and, consistent with this activity, ectopic expression of either protein leads to a loss of actin stress fibers. Intriguingly, the Rho GAP domain of ARAP2 contains a glutamine in place of the catalytic arginine typical of Rho GAPs, and although it does indeed bind RhoA, it lacks GAP activity [50]. In this sense, ARAP2 might actually serve as a RhoA effector rather than a GAP (see below).
Figure 4.
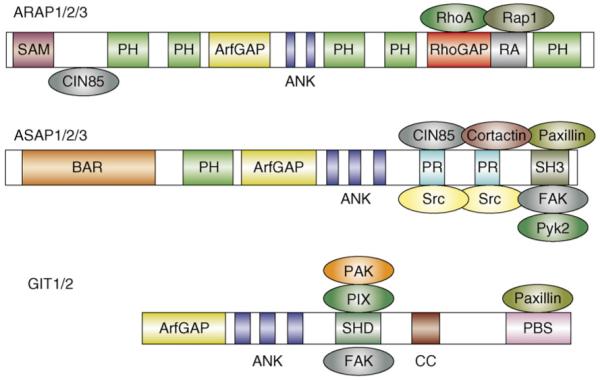
Domain organization of the ARAP, ASAP and GIT families of Arf GAPs, and their known binding partners. Abbreviations: ANK, ankyrin repeat; BAR, Bin1–amphiphysin–Rvs167 domain; CC, coiled-coil domain; PBS, paxillin-binding sequence; PH, pleckstrin homology domain; PR, proline-rich domain; RA, Ras-association domain; SAM, sterile α-motif domain; SH3, Src homology 3 domain; SHD, Spa2 homology domain.
The specific functions of ARAPs are largely unknown, although they appear to have roles in post-endocytic transport [52], cytoskeletal reorganization in response to growth factors [35], and focal adhesion dynamics [50]. Each of the ARAPs also contains a Ras-association (RA) domain, predicted to serve as a binding site for Ras or for closely related GTPases such as Rap [53]. Rap1 is best known for its role in the regulation of both integrin-mediated and cadherin-mediated adhesion [54]. In this regard, interaction of Rap1 with the RA of ARAP3 has been shown to positively regulate its Rho GAP (but not its Arf GAP) activity [55]. Although binding partners for the RA domains of ARAP1 and ARAP2 have not yet been identified, it seems likely that these proteins as a group will serve as intersection points for signaling pathways involving Arf, Rho, and Ras or Rap.
ASAPs
The three ASAPs (Arf GAP with Src homology 3, ankyrin repeat, and pleckstrin homology domains), ASAP1 (also known as AMAP1), ASAP2 (AMAP2) and ASAP3 (AMAP3), appear to prefer class I and/or class II Arfs as substrates [56]. Interestingly, ASAP1 and ASAP2 have been reported to bind Arf6 without promoting rapid GTP hydrolysis [57], and it is thought that they function as Arf6 effectors. The ASAPs interact with several focal adhesion components, including Src, FAK (focal adhesion kinase), Pyk2 (proline-rich tyrosine kinase 2) and paxillin, and localize to focal adhesions or focal complexes in many cell types (Figure 4). ASAPs are phosphorylated by Src, FAK and Pyk2, and phosphorylation by Pyk2 has been shown to inhibit Arf GAP activity [58]. ASAP1 function appears to be important for focal adhesion dynamics, because both overexpression and knockdown have been shown to affect FA assembly [59,60]. However the precise role of ASAP1 within focal adhesions remains to be determined.
ASAPs are also important for the assembly of invadopodia, dynamic actin-based structures important in the degradation and penetration of the extracellular matrix by metastatic cells. Paxillin and cortactin are key components of invadopodia, and ASAP1 (AMAP1) binds both proteins in a tripartite complex. Knockdown of ASAP1 (AMAP1) or disruption of the ASAP–paxillin–cortactin complex prevents the formation of invadopodia or related structures called podosomes [61,62]. Interestingly, formation of these structures requires the presence of ASAP1 but not its GAP activity [61], suggesting that it functions primarily as a scaffold in this context. Moreover, disruption of the ASAP–paxillin–cortactin complex inhibits the metastasis of murine breast tumor cells in an animal model [62]. These findings highlight the fact that ASAPs have both GAP-dependent and GAP-independent functions.
GITs
The two GIT proteins (G-protein-coupled receptor kinase interacting proteins), GIT1 and GIT2 (also known as PKL) associate primarily with focal adhesions or focal complexes in non-neuronal cells, and with synapses in neurons. Localization to focal adhesions is largely mediated by interaction of the C-terminal domain of GIT with paxillin [63-65]. Both GIT1 and GIT2 also bind constitutively to the Rac and Cdc42 GEF PIX (Pak-interacting exchange factor), and PIX in turn binds the serine–threonine kinase PAK (p21-activated kinase). The GIT–PIX–PAK complex appears to constitute a functional, tripartite module in which GIT targets both PIX and PAK to focal adhesions, focal complexes or synapses [64,66,67]. Early models suggested that PIX activated Rac and/or Cdc42, which then activated PAK [68]. Active PAK would then stimulate focal adhesion turnover and extension of lamellipodia, thereby promoting cell migration [67,69]. However, subsequent work determined that PAK can be activated directly by GIT in a GTPase-independent manner, calling into question whether Rac and Cdc42 activation is necessary [70]. Intriguingly, this direct activation mechanism requires the Arf GAP domain, but not GAP activity [70].
The biology of the GITs is complex and can vary with isoform and with cellular context. In Rat2 fibroblasts, knockdown of GIT1 reduced protrusiveness and decreased the rate of focal adhesion turnover. Moreover, reconstitution experiments indicated that the catalytic activities of both β-PIX and PAK were necessary to restore normal turnover kinetics [65]. However, in HeLa cells, knockdown of GIT1 was found to have no observable effect, whereas knockdown of GIT2 actually enhanced focal adhesion turnover and lamellipodial extension [71]. This latter result is consistent with a model in which the GIT2 complex actually represses Rac activity, rather than enhancing it. In principle, this could occur if local Arf6 activity was kept low through GIT2 GAP activity, but this remains to be tested directly.
Similarly, GIT1 has been shown to repress the activation of Rac at the trailing edge of migrating cells when associated with α4β1 integrin [72]. Rac activity is typically high at the leading edge of motile cells, and lower at the rear [73]. Recent evidence indicates that GIT1 is recruited to the α4 cytoplasmic domain through its interaction with paxillin, which binds α4 directly. Given that the interaction of paxillin with α4 is negatively regulated by integrin phosphorylation (which occurs preferentially at the leading edge), the GIT1–paxillin complex binds only to dephosphorylated α4 along the sides and trailing edge. In this context, local inhibition of Rac activity requires the Arf GAP activity of GIT1; if GAP activity is counterbalanced by overexpression of an Arf6 GEF, ARNO, Arf activity remains high and directional motility is lost [72]. These data support a model in which spatially restricted inhibition of Arf6 activity by GIT1 results in local repression of Rac activation and reduced protrusiveness at the rear of migrating cells.
GIT1 has also been shown to modulate synapse formation in hippocampal neurons, through recruitment of the GIT–PIX–PAK complex to dendritic spines and synapses [74]. However, recruitment of GIT1 to spines does not require interaction with paxillin. Instead, recent work suggests that signaling through the transmembrane guidance molecule ephrinB leads to tyrosine phosphorylation of GIT1 on Y392 [75]. This creates a binding site for the adaptor molecule Grb4, which then directly couples ephrinB to GIT1. Interference with this interaction impairs spine morphogenesis and synapse formation, suggesting that ephrinB signaling promotes these processes through recruitment of the GIT–PIX–PAK complex [75].
Conclusions
In addition to their well-characterized role in vesicular transport, it is now clear that the Arfs have important functions in the regulation of actin cytoskeleton assembly. Such regulation can occur on several levels: (i) the direct, local regulation of phosphoinositide synthesis, which impacts a broad array of actin regulatory proteins; (ii) the activation of Rho family GTPases through the recruitment of GEFs (e.g. Dock180, kalirin or PIX) or trafficking of lipid raft components; (iii) the downregulation of Rho GTPases by recruitment of either GAPs (e.g. ARAPs or ARHGAP21) or inhibitors of activation (e.g. NM23-H1). Other modes of Arf-dependent regulation undoubtedly exist, and defining these will be a challenge for the future.
It is increasingly apparent that actin dynamics are intimately linked with carrier vesicle formation at many intracellular locations [10,19]. The discoveries that Cdc42 acts with Arf1 in both the cis-Golgi [17] and at the plasma membrane [22] suggest that actin remodeling is a common theme in Arf-mediated vesicle biogenesis. However, it remains unknown whether the formation of all Arf-dependent vesicle carriers require actin assembly. Conversely, how large a role does Arf-dependent membrane trafficking play in cytoskeletal dynamics at the cell periphery? It has long been hypothesized that the directed transport of membrane and adhesion components from internal pools is tightly coordinated with actin dynamics at the plasma membrane during processes like cell migration or neurite outgrowth. Thus, although many previous studies have examined the trafficking and cytoskeletal functions of the Arfs separately, hopefully future studies will focus on the role of Arfs in coordinating these two interconnected processes.
Acknowledgements
The authors wish to apologize to our colleagues whose work was not cited, owing to space constraints. We also thank Anne Allison for careful reading of the manuscript, and Lorraine Santy for providing the images used in Figure 2.
References
- 1.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 2.D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 3.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 4.Godi A, et al. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- 5.Jones DH, et al. Type I phosphatidylinositol 4-phosphate 5-kinase directly interacts with ADP-ribosylation factor 1 and is responsible for phosphatidylinositol 4,5-bisphosphate synthesis in the golgi compartment. J. Biol. Chem. 2000;275:13962–13966. doi: 10.1074/jbc.c901019199. [DOI] [PubMed] [Google Scholar]
- 6.Honda A, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 1999;99:521–532. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- 7.Krauss M, et al. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. J. Cell Biol. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smaczynska-de R, II, et al. Yeast Arf3p modulates plasma membrane PtdIns(4,5)P2 levels to facilitate endocytosis. Traffic. doi: 10.1111/j.1600-0854.2008.00708.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santarius M, et al. Supervised membrane swimming: small G-protein lifeguards regulate PIPK signalling and monitor intracellular PtdIns(4,5)P2 pools. Biochem. J. 2006;398:1–13. doi: 10.1042/BJ20060565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stamnes M. Regulating the actin cytoskeleton during vesicular transport. Curr. Opin. Cell Biol. 2002;14:428–433. doi: 10.1016/s0955-0674(02)00349-6. [DOI] [PubMed] [Google Scholar]
- 11.Hirschberg K, et al. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valderrama F, et al. Actin microfilaments are essential for the cytological positioning and morphology of the Golgi complex. Eur. J. Cell Biol. 1998;76:9–17. doi: 10.1016/S0171-9335(98)80012-5. [DOI] [PubMed] [Google Scholar]
- 13.Godi A, et al. ADP ribosylation factor regulates spectrin binding to the Golgi complex. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8607–8612. doi: 10.1073/pnas.95.15.8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luna A, et al. Regulation of protein transport from the Golgi complex to the endoplasmic reticulum by CDC42 and N-WASP. Mol. Biol. Cell. 2002;13:866–879. doi: 10.1091/mbc.01-12-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musch A, et al. cdc42 regulates the exit of apical and basolateral proteins from the trans-Golgi network. EMBO J. 2001;20:2171–2179. doi: 10.1093/emboj/20.9.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erickson JW, et al. Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J. Biol. Chem. 1996;271:26850–26854. doi: 10.1074/jbc.271.43.26850. [DOI] [PubMed] [Google Scholar]
- 17.Wu WJ, et al. The gamma-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature. 2000;405:800–804. doi: 10.1038/35015585. [DOI] [PubMed] [Google Scholar]
- 18.Merrifield CJ, et al. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 19.Merrifield CJ. Seeing is believing: imaging actin dynamics at single sites of endocytosis. Trends Cell Biol. 2004;14:352–358. doi: 10.1016/j.tcb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Dubois T, et al. Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat. Cell Biol. 2005;7:353–364. doi: 10.1038/ncb1244. [DOI] [PubMed] [Google Scholar]
- 21.Chen JL, et al. Coatomer-bound Cdc42 regulates dynein recruitment to COPI vesicles. J. Cell Biol. 2005;169:383–389. doi: 10.1083/jcb.200501157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumari S, Mayor S. ARF1 is directly involved in dynamin-independent endocytosis. Nat. Cell Biol. 2007;10:30–41. doi: 10.1038/ncb1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao H, et al. Actin and Arf1-dependent recruitment of a cortactin-dynamin complex to the Golgi regulates post-Golgi transport. Nat. Cell Biol. 2005;7:483–492. doi: 10.1038/ncb1246. [DOI] [PubMed] [Google Scholar]
- 24.Carreno S, et al. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J. Cell Biol. 2004;165:781–788. doi: 10.1083/jcb.200403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown FD, et al. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 2001;154:1007–1017. doi: 10.1083/jcb.200103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schafer DA, et al. Actin assembly at membranes controlled by ARF6. Traffic. 2000;1:892–903. doi: 10.1034/j.1600-0854.2000.011108.x. [DOI] [PubMed] [Google Scholar]
- 27.Boshans RL, et al. ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol. Cell. Biol. 2000;20:3685–3694. doi: 10.1128/mcb.20.10.3685-3694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunphy JL, et al. The Arf6 GEF GEP100/BRAG2 regulates cell adhesion by controlling endocytosis of beta1 integrins. Curr. Biol. 2006;16:315–320. doi: 10.1016/j.cub.2005.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank SR, et al. Remodeling of the actin cytoskeleton is coordinately regulated by protein kinase C and the ADP-ribosylation factor nucleotide exchange factor ARNO. Mol. Biol. Cell. 1998;9:3133–3146. doi: 10.1091/mbc.9.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishna H, et al. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci. 1999;112:855–866. doi: 10.1242/jcs.112.6.855. [DOI] [PubMed] [Google Scholar]
- 31.Santy LC, Casanova JE. Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J. Cell Biol. 2001;154:599–610. doi: 10.1083/jcb.200104019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balasubramanian N, et al. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 2007;9:1381–1391. doi: 10.1038/ncb1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldfinger LE, et al. RLIP76 (RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J. Cell Biol. 2006;174:877–888. doi: 10.1083/jcb.200603111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda S, et al. Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2005;96:467–475. doi: 10.1161/01.RES.0000158286.51045.16. [DOI] [PubMed] [Google Scholar]
- 35.Krugmann S, et al. ARAP3 is essential for formation of lamellipodia after growth factor stimulation. J. Cell Sci. 2006;119:425–432. doi: 10.1242/jcs.02755. [DOI] [PubMed] [Google Scholar]
- 36.Cotton M, et al. Endogenous ARF6 interacts with Rac1 upon angiotensin II stimulation to regulate membrane ruffling and cell migration. Mol. Biol. Cell. 2007;18:501–511. doi: 10.1091/mbc.E06-06-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palacios F, D’Souza-Schorey C. Modulation of Rac1 and ARF6 activation during epithelial cell scattering. J. Biol. Chem. 2003;278:17395–17400. doi: 10.1074/jbc.M300998200. [DOI] [PubMed] [Google Scholar]
- 38.Palacios F, et al. ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat. Cell Biol. 2002;4:929–936. doi: 10.1038/ncb881. [DOI] [PubMed] [Google Scholar]
- 39.Santy LC, et al. The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr. Biol. 2005;15:1749–1754. doi: 10.1016/j.cub.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 40.Bowzard JB, et al. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J. Biol. Chem. 2007;282:17568–17580. doi: 10.1074/jbc.M701347200. [DOI] [PubMed] [Google Scholar]
- 41.Chen EH, et al. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell. 2003;114:751–762. doi: 10.1016/s0092-8674(03)00720-7. [DOI] [PubMed] [Google Scholar]
- 42.Rushton E, et al. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development. 1995;121:1979–1988. doi: 10.1242/dev.121.7.1979. [DOI] [PubMed] [Google Scholar]
- 43.Koo TH, et al. Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 2007;8:29. doi: 10.1186/1471-2121-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Moissoglu K, et al. In vivo dynamics of Rac-membrane interactions. Mol. Biol. Cell. 2006;17:2770–2779. doi: 10.1091/mbc.E06-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Pozo MA, et al. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- 47.Randazzo PA, et al. Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell. 2007;99:583–600. doi: 10.1042/bc20070034. [DOI] [PubMed] [Google Scholar]
- 48.Inoue H, Randazzo PA. Arf GAPs and their interacting proteins. Traffic. 2007;8:1465–1475. doi: 10.1111/j.1600-0854.2007.00624.x. [DOI] [PubMed] [Google Scholar]
- 49.Miura K, et al. ARAP1: a point of convergence for Arf and Rho signaling. Mol. Cell. 2002;9:109–119. doi: 10.1016/s1097-2765(02)00428-8. [DOI] [PubMed] [Google Scholar]
- 50.Yoon HY, et al. ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating-protein activity and binding to RhoA-GTP. J. Cell Sci. 2006;119:4650–4666. doi: 10.1242/jcs.03237. [DOI] [PubMed] [Google Scholar]
- 51.Cuthbert EJ, et al. Substrate specificities and activities of AZAP family ArfGAPs in vivo. Am. J. Physiol. Cell Physiol. 2007;294:263–270. doi: 10.1152/ajpcell.00292.2007. [DOI] [PubMed] [Google Scholar]
- 52.Kowanetz K, et al. CIN85 associates with multiple effectors controlling intracellular trafficking of epidermal growth factor receptors. Mol. Biol. Cell. 2004;15:3155–3166. doi: 10.1091/mbc.E03-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wohlgemuth S, et al. Recognizing and defining true Ras binding domains I: biochemical analysis. J. Mol. Biol. 2005;348:741–758. doi: 10.1016/j.jmb.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 54.Kooistra MR, et al. Rap1: a key regulator in cell–cell junction formation. J. Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- 55.Krugmann S, et al. ARAP3 is a PI3K- and rap-regulated GAP for RhoA. Curr. Biol. 2004;14:1380–1384. doi: 10.1016/j.cub.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 56.Kam JL, et al. Phosphoinositide-dependent activation of the ADP-ribosylation factor GTPase-activating protein ASAP1. Evidence for the pleckstrin homology domain functioning as an allosteric site. J. Biol. Chem. 2000;275:9653–9663. doi: 10.1074/jbc.275.13.9653. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto S, et al. A novel mode of action of an ArfGAP, AMAP2/PAG3/Papa lpha, in Arf6 function. J. Biol. Chem. 2004;279:37677–37684. doi: 10.1074/jbc.M404196200. [DOI] [PubMed] [Google Scholar]
- 58.Kruljac-Letunic A, et al. The tyrosine kinase Pyk2 regulates Arf1 activity by phosphorylation and inhibition of the Arf-GTPase-activating protein ASAP1. J. Biol. Chem. 2003;278:29560–29570. doi: 10.1074/jbc.M302278200. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, et al. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell. 2002;13:2147–2156. doi: 10.1091/mbc.E02-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y, et al. Mislocalization or reduced expression of Arf GTPase-activating protein ASAP1 inhibits cell spreading and migration by influencing Arf1 GTPase cycling. J. Biol. Chem. 2005;280:8884–8892. doi: 10.1074/jbc.M412200200. [DOI] [PubMed] [Google Scholar]
- 61.Bharti S, et al. Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol. Cell. Biol. 2007;27:8271–8283. doi: 10.1128/MCB.01781-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Onodera Y, et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005;24:963–973. doi: 10.1038/sj.emboj.7600588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown MC, et al. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol. Biol. Cell. 2005;16:4316–4328. doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown MC, et al. Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol. Biol. Cell. 2002;13:1550–1565. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nayal A, et al. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol. 2006;173:587–589. doi: 10.1083/jcb.200509075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manabe R, et al. GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 2002;115:1497–1510. doi: 10.1242/jcs.115.7.1497. [DOI] [PubMed] [Google Scholar]
- 67.Zhao ZS, et al. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 2000;20:6354–6363. doi: 10.1128/mcb.20.17.6354-6363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manser E, et al. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J. Cell Sci. 2005;118:2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- 70.Loo TH, et al. GIT1 activates p21-activated kinase through a mechanism independent of p21 binding. Mol. Cell. Biol. 2004;24:3849–3859. doi: 10.1128/MCB.24.9.3849-3859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frank SR, et al. GIT2 represses Crk- and Rac1-regulated cell spreading and Cdc42-mediated focal adhesion turnover. EMBO J. 2006;25:1848–1859. doi: 10.1038/sj.emboj.7601092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishiya N, et al. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat. Cell Biol. 2005;7:343–352. doi: 10.1038/ncb1234. [DOI] [PubMed] [Google Scholar]
- 73.Kraynov VS, et al. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 74.Zhang H, et al. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Segura I, et al. Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat. Neurosci. 2007;10:301–310. doi: 10.1038/nn1858. [DOI] [PubMed] [Google Scholar]