

Abstract
Charged residues play an important role in defining key mechanistic features in many biomolecules. Determining the pKa values of large, membrane or fibrillar proteins can be challenging with traditional methods. In this study we show how solid-state NMR is used to monitor chemical shift changes during a pH titration for the small soluble β1 immunoglobulin binding domain of protein G. The chemical shifts of all the amino acids with charged side-chains throughout the uniformly-13C,15N-labeled protein were monitored over several samples varying in pH; pKa values were determined from these shifts for E27, D36, and E42, and the bounds for the pKa of other acidic side-chain resonances were determined. Additionally, this study shows how the calculated pKa values give insights into the crystal packing of the protein.
Keywords: Chemical shift perturbation, GB1, Magic-angle spinning, pKa determination, Solid-state NMR
Charged residues of a protein play an important role in catalytic reactions, protein stability and substrate binding.1–3 Many enzymatic reactions are controlled by the reversible ionization of charged residues within the active site. This ionization is determined by the intrinsic pKa, which for residues involved in enzymatic mechanisms can deviate greatly from canonical values.1,4–6 Moreover, pKa values are related to the pH range over which proteins are stable, consistent with the observation of pKa differences among the unfolded and folded proteins,2,7 with aberrant pKa values often observed in the context of hydrogen bonding, through coupled protonation and deprotonation events.8 Knowledge of pKa values is useful for purposes of optimizing the absorption and stability of drugs, as well as for identifying substrate interactions in a binding pocket. The central role of ionization events in enzymology has driven the biophysical chemical community to develop methods to accurately determine pKa values across a variety of platforms.
Solution NMR and infrared (IR) spectroscopy monitor changes in spectroscopic signatures as a function of pH to determine pKa values.9 With NMR, individual protonation states can be monitored throughout the titration by tracking chemical shift changes for each resonance to yield mechanistic information10 and pH dependent protein stability.2 However, large and nonsoluble systems, such as membrane proteins, fibrils and other large crystallized proteins, are not typically amenable to these methods. For both protein fibrils and membrane proteins, X-ray structures are rare or not available to obtain pKa values from calculations either. When structures do exist, the accuracy of the calculated pKa is limited by the ability to correctly determine side-chain conformations and ionization states of nearby charged residues making moderate resolution crystal structures non-ideal candidates for this method.11,12 The importance of these systems in disease, cell signaling and ion transfer events creates a need for an alternative method for studying electrostatic interactions.
With solid-state NMR (SSNMR), large and insoluble systems can be examined to provide detailed chemical bonding and environmental information. For example, in the transition state analogue (TSA) inhibitor, carboxymethyldethia coenzyme A (CMX) bound to citrate synthase SSNMR was used to measure anisotropic chemical shifts of carboxyl groups to determine protonation states and geometry of hydrogen bonds.13 Additionally, isotropic chemical shifts were utilized to study rhodopsin and bathorhodopsin and the chromophore retinylidene to gain insights into electrostatic interactions and protonation states in ligation.14–18 However, these previous SSNMR studies were completed for a small selectively labeled cofactor in the protein systems using one- and two-dimensional experiments, and as such would require many samples in order to assess protonation states of several sites in the protein. Due to the complexity of these issues, no full titration curve to obtain exact pKa values, as routinely done now in solution NMR, has yet been completed utilizing high-resolution SSNMR. In this work, we demonstrate that SSNMR can determine both specific pKa values and set bounds for multiple acidic residues in a uniformly labeled protein using multidimensional homo and heteronuclear experiments. The pKa values determined in this work give insight of the protein stability and packing of β1 immunoglobulin binding domain of protein G (GB1) in a nanocrystal environment providing new and complimentary data to pKa values determined in solution. Thus, for soluble proteins which can be studied by both solution and solid-state NMR the determination of pKa values using both methods would provide exciting new details related to protein stability, crystal contacts, and salt bridges, which could in turn examine differences between the solution and solid or crystalline structures. Additionally, the methods developed within this study can be extended to larger soluble and membrane systems, such as transmembrane ion pumps and fibrillar proteins, which are uniquely suited to SSNMR studies.19–24
Seven crystalline slurries of uniformly-13C,15N labeled GB1 were prepared using pH controlled protein buffer and precipitate solutions ranging from pH 2.42 to 5.68. Crystalline material from each slurry was isolated by centrifugation and packed into a SSNMR rotor with residual mother liquor to allow proton exchange with free carboxyl groups. The complete backbone and side-chain chemical shifts were identified for every charged residue in GB1, Asp, Glu and Lys, and Asn and Gln at every titration point. 13C-13C, 15N-(13CO)-13CX and 15N-(13CA)-13CX two-dimensional dipolar correlation spectra were acquired on each sample and compared with previously published assignments for GB1 in the solid-state.25 In cases when labeled protein can be produced in large quantities, such as GB1, individual samples can be prepared at each pH value to accelerate sample preparation and data collection efforts. The methods presented in this work can be applied to larger proteins, such as membrane and fibrillar proteins where large quantities of labeled material are unavailable, by preparing a single sample at a single pH value and reconstituting the material after data acquisition and varying the pH.
The GB1 samples prepared at pH 2.53 and 2.83 were found to be unfolded with the chemical shifts predominantly consistent with a random coil secondary structure, as well as some Thr residues with secondary chemical shifts consistent with beta-sheet formation. Solution NMR studies reported that GB1 retained its structure to pH values as low as 1.8 in high and low salt solutions.7,8,26,27 The different behavior of the solid preparation reports on the energy the lattice interactions contribute to stabilizing the crystalline protein structure. The protonation of the acidic residues at low pH values, disturbs the crystal packing interactions, likely disrupting salt bridges and frustrating the formation of the crystal lattice.
For samples prepared at pH > 3.6, the protein retained its structure such that the backbone 13C resonances are conserved throughout the entire pH range for the hydrophobic residues; Val, Ile, Ala and Leu retain similar shifts in the 13C-13C spectra for samples both at the highest (5.64) and the lowest (3.63) pH values (Figure 1). The charged residues (Asp and Glu) and polar residues (Asn, Gln and Thr), display small CA-CB shift differences at the varying sample pH values (Figure 1). Small chemical shift perturbations are also observed for a small subset of the CA-C' correlations through this pH range (Figure 1). In contrast, much larger resonance changes were observed for the charged residues. For example, the chemical shifts for the Asp and Glu amino acids were shifted upfield by ~0.5 to ~3 ppm as the pH was decreased, consistent with partial to full protonation of these residues.28 The largest chemical shift differences over this pH range were observed for the acidic residues, with D46, D36, E15, E42, E27 and M1 displaying significant CA-CB perturbations due to their proximity to a titrating carboxyl group.27
Figure 1.
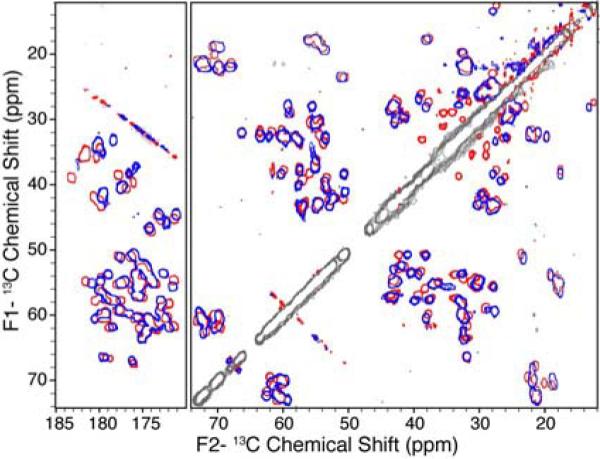
Overlay of 13C-13C 2D spectra acquired on uniformly-13C,15N labeled GB1 at pH = 3.63 (blue) and pH = 5.64 (red) using SPC53 mixing.
Thus samples prepared at pH = 5.22 and 5.64 were in very good agreement and could be assigned from the chemical shift assignments published by Franks et al.25 De novo assignments were completed for the pH = 3.63 sample using backbone walk procedures utilizing NCACX and NCOCX three dimensional data sets.29 This assignment procedure is very well established and has been used to assign several large soluble and membrane proteins.25,29–40 The pH = 3.63 assignments were used to identify resonances for the acidic amino acids in pH = 3.89 and 4.53 samples. The complete backbone and side-chain chemical shifts were identified for every Asp, Asn, Glu, and Gln residue in GB1 at every titration point. Chemical shift assignments were confirmed in a minimum of three spectra within a standard deviation of ± 0.10 ppm (BMRB accession number 16873).
The acidic side-chain carbonyl shifts remained relatively constant at a pH of 5.64, 5.22 and 4.55 (Figure 2). At pH=3.95, all five Glu and D36 shifted upfield, due to protonation. The average determined side-chain pKa in a protein for Glu is 4.1±1.0, and for Asp it is lower at 3.1±1.0 in basic proteins to 3.9±0.9 in acidic proteins.28 Since GB1 has a pI of 4.2, we expect that the majority of Glu residues and approximately half of the Asp residues should titrate in the pH range sampled. Our results show that only one Asp, D36, has side-chain carbonyl chemical shifts consistent with a protonated acid at pH = 3.63 (Figure 2); the other residues did not titrate, displaying only slightly shifted CG resonances over the pH range.
Figure 2.

(a) Side-chain carbonyl chemical shifts over the pH titrated range. Expansion of the CG-CO and CD-CO region of the 13C-13C 2D spectra acquired with SPC53 mixing for samples at pH=3.63 (purple), pH=3.95 (blue), pH=4.55 (green), pH=5.22 (orange) and pH=5.64 (red), and pH dependence of 13C side-chain carbonyl chemical shifts in nanocrystalline GB1. (b) Aspartic acid CG resonances and (c) glutamic acid CD and C-terminus carbonyl resonances plotted against sample buffer pH showing fitted curves for D36 (blue), E27 (black) and E42 (red).
The complete chemical shifts for two acidic residues (D40 and E56) could not be assigned at the low pH values, pH = 3.63 and 3.95, since the signals from these residues were broad and weak. The D40 CB and CG resonances were not observed at pH = 3.63 and pH = 3.95, and the N-CA correlations were weak (SNR = 7); therefore the pKa of D40 could not be determined in this study. Two E56 CG-CD correlations were observed in the 13C-13C SPC5 mixing spectrum at pH = 3.63, which suggest chemical exchange or inhomogeneity at this site, and therefore its pKa was also not determined. These observations may be consistent with proton exchange at the intermediate timescale (μs to ms), which would most likely occur at pH values close to the pKa.
pKa values were determined for acidic residues that experience complete protonation over the pH range sampled. Figure 2 shows the observed chemical CG/CD chemical shifts for the Asp and Glu of GB1 plotted as a function of sample buffer pH. D36, E27 and E42 all experienced complete protonation and quantitative pKa values were determined. Although the E19 and E15 CD resonances had shifted upfield by ~1.5 ppm from pH = 5.64 to pH = 3.63, the fully protonated chemical shift could not be confirmed, thus pKa values could not be quantitatively determined. However, an upper pKa bound of 4.0 pH units can be inferred from the pH range sampled. The residues, C-terminus D22, D46, D47 and E56, which did not display significant chemical shift deviations, can be assumed to have pKa values below 3.63.
The calculated pKa values for D36, E42 and E27 as well as the bounding values for other E and D residues are given in Table 1. The pH titration curves of D36, E42, and E27 have a sigmoidal shape, typical for a single titrating residue, and were fit to a modified Henderson-Hasselbalch equation. The protonated chemical shift and deprotoned chemical shifts were obtained from the pH = 3.63 and 5.64 samples, respectively. The fitted curves (Figure 2) show good agreement with the experimental data when the pKa values and the Hill coefficient are simultaneous varied during the fitting. The solid-state E42 and E27 pKa values are similar to the expected value for Glu residues (pKa= 4.2).28 The D36 determined pKa is ~0.4 pH units higher than the expected value (pKa=3.9) for an acidic protein.
Table 1.
SSNMR determined pKa values for nanocrystalline GB1 compared to values determined by solution NMR experiments and calculations from X-ray structure.
| 13C SSNMR | Hill coefficient (nH) | 1H NMR8 | Calculated (1PGB)41 | 13C NMR QQD-GB127 | 13C NMR QQD-GB1 (0.5 M salt)27 | |
|---|---|---|---|---|---|---|
| E15 | < 4.0 | 4.4 | 5.0 | 4.6 | 4.7 | |
| E19 | < 4.0 | 3.7 | 4.1 | 3.9 | 4.0 | |
| D22 | < 3.6 | 2.9 | 2.3 | 3.0 | 3.2 | |
| E27 | 4.1 ± 0.1 | 3.8 ± 0.6 | 4.5 | 4.9 | 4.8 | 4.9 |
| D36 | 4.3 ± 0.1 | 4.5 ± 0.7 | 3.8 | 4.9 | 4.2 | 4.1 |
| D40 | N.D. | 4.0 | 4.4 | 4.1 | 4.1 | |
| E42 | 4.2 ± 0.2 | 3.1 ± 0.6 | 4.4 | 4.0 | 4.9 | 4.6 |
| D46 | < 3.6 | 3.6 | 3.8 | 3.8 | 3.9 | |
| D47 | < 3.6 | 3.4 | 3.3 | 3.1 | 3.4 | |
| E56 | < 3.6 | 4.0 | 3.7 | 3.8 | 3.9 | |
| C terminus | < 3.6 | 3.1 | 3.2 |
pKa perturbations of residues can be caused by several electrostatic interactions, such as charge-charge interactions, hydrogen bonding and desolvation.1,28 In the case of solvent-exposed D36 CG, the most likely of these explanations is that a nearby negative charge shifts the pKa value up, discussed further below. The Hill coefficients, which report on cooperativity of the protonation event with other local charges, were determined for E27, D36 and E42 and they ranged from 3.1 to 4.5 units (Table 1). These values are three to four-fold larger than those typically observed for solvent exposed residues from solution NMR measurements. Hill coefficients on the order observed in our solid-state NMR measurements are the result of a highly coupled protonation event, which we attribute to the propagation of structural changes throughout the highly concentrated environment of the crystal lattice.
Table 1 also shows the pKa comparisons between the SSNMR determined values for E27, D36 and E42, as well as the other E and D bounded pKa values, and the pKa values determined by other methods. The SSNMR pKa values are in very good agreement with both those determined for wild type GB1 by solution NMR, as well as by the MCCE calculation.8,27,41 There are small differences between the SSNMR and calculated E27, D36 and E42 values. Both methods examined the trigonal crystal packing arrangement of GB1,42,43 thus similar packing interactions would be present and observed differences could be from the uncertainties of both methods. The strongest agreement was observed for the D36 pKa between SSNMR and that determined by 13C solution NMR for the QQD-GB1 (T2Q, N8D, N37D) mutant.7,26,27 The addition of a positive charge near D36 shifts the pKa up in the QQD-GB1 mutant.27 A similar event could be occurring in the SSNMR preparation from the protein crystal packing. This was confirmed in the crystal structure (2QMT), with D36 packed close to itself in the crystal lattice (Figure 3).
Figure 3.

Three GB1 molecules in trigonal crystal packing (PDB:2QMT) with chemical shift differences between low (pH = 3.63) and high (pH = 5.64) pH samples for the carboxylic acid side-chains of Asn/Asp/Gln/Glu residues colored from no change (blue) to > 2.0 ppm chemical shift differences (red). Residues with > 2.0 ppm chemical shift differences are labeled.
The three titrating residues (E27, D36 and E42), although solvent-exposed, were not involved in salt bridges or hydrogen bonding according to the X-ray crystal structure (Figure 3).42 Three of the acidic residues that did not titrate above a pH of 3.63, E15, D47 and E56, were involved in a salt bridge with a nearby lysine.42 For example, from the GB1 structure (PDB ID: 2QMT) it can be determined that E15 is involved in an intramolecular salt bridge with K4 (3.05 Å), as D47 is in a salt bridge with K50 (3.27 Å) and E56 with K10 (3.39 Å). Additionally, by recreating the crystal packing E27 is shown to be 3.81 Å from K31, E42 is 4.81 Å from a K4 intermolecular contact, and D40 is 4.95 Å from an intermolecular contact with K10. During the titration the Lys Nζ resonances essential have no deviation of their chemical shift (shown in the SI), indicating that the salt bridges are intact during the titration. As the pH is lowered below ~3, these salt bridges and other hydrogen bonds, which are critical for protein stability and crystal packing, are broken, denaturing the protein.
In this work we have determined specific pKa values for acidic residues, E27, D36 and E42, in the nanocrystalline protein GB1 by SSNMR. This demonstrates the ability of SSNMR to study electrostatic events and determine pKa values. The determined pKa values were in good agreement with those previously determined by solution NMR,8 with one residue demonstrating a shifted pKa due to crystal packing in the solid state that was not observed in solution. These SSNMR methods for pKa determination can now be extended to large crystalline, fibrillar, and membrane proteins that other methods have traditionally experienced challenges and use this information to elucidate key mechanistic, drug targeting and interaction, and folding and unfolding information.
Supplementary Material
Acknowledgement
The authors thank Merideth Burkhart for assistance in early stages of this work and the National Institutes of Health R01GM075937 provided funding for this work.
Footnotes
Supporting Information Available: Details of the data acquisition; full bandwidth spectra at several pH values; sample preparation details; data fitting parameters; backbone chemical shift assignments at each pH value; additional chemical shift versus pH value plot of Lys and other amino acids. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Harris TK, Turner GJ. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- (2).Tollinger M, Crowhurst KA, Kay LE, Forman-Kay JD. Site-Specific Contributions to the pH Dependence of Protein Stability. Proc. Natl. Acad. Sci. USA. 2003;100:4545–4550. doi: 10.1073/pnas.0736600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cleland WW, Kreevoy MM. Low-Barrier Hydrogen-Bonds and Enzymatic Catalysis. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- (4).Das TK, Tomson FL, Gennis RB, Gordon M, Rousseau DL. pH-Dependent Structural Changes at the Heme-Copper Binuclear Center of Cytochrome c Oxidase. Biophys. J. 2001;80:2039–2045. doi: 10.1016/S0006-3495(01)76177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG. The pKa of the General Acid/Base Carboxyl Group of a Glycosidase Cycles During Catalysis: A 13C-NMR Study of Bacillus Circuluns Xylanase. Biochemistry. 1996;35:9958–9966. doi: 10.1021/bi9613234. [DOI] [PubMed] [Google Scholar]
- (6).Wang PF, McLeish MJ, Kneen MM, Lee G, Kenyon GL. An Unusually Low pKa for Cys282 in the Active Site of Human Muscle Creatine Kinase. Biochemistry. 2001;40:11698–11705. doi: 10.1021/bi011208f. [DOI] [PubMed] [Google Scholar]
- (7).Lindman S, Xue WF, Szczepankiewicz O, Bauer MC, Nilsson H, Linse S. Salting the Charged Surface: pH and Salt Dependence of Protein GB1 Stability. Biophys. J. 2006;90:2911–2921. doi: 10.1529/biophysj.105.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Khare D, Alexander P, Antosiewicz J, Bryan P, Gilson M, Orban J. pKa Measurements from Nuclear Magnetic Resonance for the B1 and B2 Immunoglobulin G-Binding Domains of Protein G: Comparison with Calculated Values for Nuclear Magnetic Resonance and X-Ray Structures. Biochemistry. 1997;36:3580–3589. doi: 10.1021/bi9630927. [DOI] [PubMed] [Google Scholar]
- (9).Onufriev A, Case DA, Ullmann GM. A Novel View of pH Titration in Biomolecules. Biochemistry. 2001;40:3413–3419. doi: 10.1021/bi002740q. [DOI] [PubMed] [Google Scholar]
- (10).Takayama SJ, Mikami S, Terui N, Mita H, Hasegawa J, Sambongi Y, Yamamoto Y. Control of the Redox Potential of Pseudomonas Aeruginosa Cytochrome C(551) through the Fe-Met Coordination Bond Strength and pKa of a Buried Heme Propionic Acid Side Chain. Biochemistry. 2005;44:5488–5494. doi: 10.1021/bi047498s. [DOI] [PubMed] [Google Scholar]
- (11).Georgescu RE, Alexov EG, Gunner MR. Combining Conformational Flexibility and Continuum Electrostatics for Calculating pKas in Proteins. Biophys. J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gilson MK, Antosiewicz J, Mccammon JA. Prediction of pKas of Ionizable Groups in Proteins. Biophys. J. 1994;66:A290–A290. [Google Scholar]
- (13).Gu ZT, Drueckhammer DG, Kurz L, Liu K, Martin DP, McDermott A. Solid State NMR Studies of Hydrogen Bonding in a Citrate Synthase Inhibitor Complex. Biochemistry. 1999;38:8022–8031. doi: 10.1021/bi9813680. [DOI] [PubMed] [Google Scholar]
- (14).Gansmuller A, Concistre M, McLean N, Johannessen OG, Marin-Montesinos I, Bovee-Geurts PHM, Verdegem P, Lugtenburg J, Brown RCD, DeGrip WJ, Levitt MH. Towards an Interpretation of 13C Chemical Shifts in Bathorhodopsin, a Functional Intermediate of a G-Protein Coupled Receptor. BBA-Biomembranes. 2009;1788:1350–1357. doi: 10.1016/j.bbamem.2009.02.018. [DOI] [PubMed] [Google Scholar]
- (15).Creemers AFL, Bovee-Geurts PHM, DeGrip WJ, Lugtenburg J, de Groot HJM. Solid-State NMR Analysis of Ligand-Receptor Interactions Reveals an Induced Misfit in the Binding Site of Isorhodopsin. Biochemistry. 2004;43:16011–16018. doi: 10.1021/bi048541e. [DOI] [PubMed] [Google Scholar]
- (16).Verhoeven MA, Creemers AFL, Bovee-Geurts PHM, De Grip WJ, Lugtenburg J, de Groot HJM. Ultra-High-Field Mas NMR Assay of a Multispin Labeled Ligand Bound to Its G-Protein Receptor Target in the Natural Membrane Environment: Electronic Structure of the Retinylidene Chromophore in Rhodopsin. Biochemistry. 2001;40:3282–3288. doi: 10.1021/bi0023798. [DOI] [PubMed] [Google Scholar]
- (17).Creemers AF, Klaassen CH, Bovee-Geurts PH, Kelle R, Kragl U, Raap J, de Grip WJ, Lugtenburg J, de Groot HJ. Solid State 15N NMR Evidence for a Complex Schiff Base Counterion in the Visual G-Protein-Coupled Receptor Rhodopsin. Biochemistry. 1999;38:7195–7199. doi: 10.1021/bi9830157. [DOI] [PubMed] [Google Scholar]
- (18).Smith SO, Courtin J, Degroot H, Gebhard R, Lugtenburg J. 13C Magic-Angle Spinning NMR-Studies of Bathorhodopsin, the Primary Photoproduct of Rhodopsin. Biochemistry. 1991;30:7409–7415. doi: 10.1021/bi00244a007. [DOI] [PubMed] [Google Scholar]
- (19).Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid Fibrils of the Het-S(218–289) Prion Form a Beta Solenoid with a Triangular Hydrophobic Core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- (20).Varga K, Tian L, McDermott AE. Solid-State Nmr Study and Assignments of the KcsA Potassium Ion Channel of S. Lividans. BBA-Proteins and Proteomics. 2007;1774:1604–1613. doi: 10.1016/j.bbapap.2007.08.029. [DOI] [PubMed] [Google Scholar]
- (21).Kloepper KD, Hartman KL, Ladror DT, Rienstra CM. Solid-State Nmr Spectroscopy Reveals That Water Is Nonessential to the Core Structure of Alpha-Synuclein Fibrils. J. Phys. Chem. B. 2007;111:13353–13356. doi: 10.1021/jp077036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary Structure, Dynamics, and Topology of a Seven-Helix Receptor in Native Membranes, Studied by Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 2007;46:459–462. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- (23).Lange A, Giller K, Hornig S, Martin-Eauclaire M-F, Pongs O, Becker S, Baldus M. Toxin-Induced Conformational Changes in a Potassium Channel Revealed by Solid-State NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- (24).Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-Level Secondary Structure, Polymorphism, and Dynamics of Full-Length Alpha-Synuclein Fibrils Studied by Solid-State NMR. Proc. Natl. Acad. Sci. USA. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM. Magic-Angle Spinning Solid-State NMR Spectroscopy of the β-1 Immunoglobulin Binding Domain of Protein G (GB1): 15N and 13C Chemical Shift Assignments and Conformational Analysis. J. Am. Chem. Soc. 2005;127:12291–12305. doi: 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- (26).Lindman S, Linse S, Mulder FAA, Andre I. Electrostatic Contributions to Residue-Specific Protonation Equilibria and Proton Binding Capacitance for a Small Protein. Biochemistry. 2006;45:13993–14002. doi: 10.1021/bi061555v. [DOI] [PubMed] [Google Scholar]
- (27).Lindman S, Linse S, Mulder FAA, Andre I. pKa Values for Side-Chain Carboxyl Groups of a PGB1 Variant Explain Salt and pH-Dependent Stability. Biophys. J. 2007;92:257–266. doi: 10.1529/biophysj.106.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Forsyth WR, Antosiewiez JM, Robertson AD. Empirical Relationships between Protein Structure and Carboxyl pKa Values in Proteins. Proteins. 2002;48:388–403. doi: 10.1002/prot.10174. [DOI] [PubMed] [Google Scholar]
- (29).Baldus M, Petkova AT, Herzfeld JH, Griffin RG. Cross Polarization in the Tilted Frame: Assignment and Spectral Simplification in Heteronuclear Spin Systems. Mol. Phys. 1998;95:1197–1207. [Google Scholar]
- (30).Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and Side-Chain 13C and 15N Resonance Assignments of the Alpha-Spectrin SH3 Domain by Magic Angle Spinning Solid State NMR at 17.6 Tesla. ChemBioChem. 2001;2:101–110. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- (31).Bockmann A, Lange A, Galinier A, Luca S, Giraud N, Juy M, Heise H, Montserret R, Penin F, Baldus M. Solid State NMR Sequential Resonance Assignments and Conformational Analysis of the 2 × 10.4 Kda Dimeric Form of the Bacillus Subtilis Protein Crh. J. Biomol. NMR. 2003;27:323–339. doi: 10.1023/a:1025820611009. [DOI] [PubMed] [Google Scholar]
- (32).Igumenova TI, Wand AJ, McDermott AE. Assignment of the Backbone Resonances for Microcrystalline Ubiquitin. J. Am. Chem. Soc. 2004;126:5323–5331. doi: 10.1021/ja030546w. [DOI] [PubMed] [Google Scholar]
- (33).Marulanda D, Tasayco ML, McDermott A, Cataldi M, Arriaran V, Polenova T. Magic Angle Spinning Solid-State NMR Spectroscopy for Structural Studies of Protein Interfaces. Resonance Assignments of Differentially Enriched Escherichia Coli Thioredoxin Reassembled by Fragment Complementation. J. Am. Chem. Soc. 2004;126:16608–16620. doi: 10.1021/ja0464589. [DOI] [PubMed] [Google Scholar]
- (34).Marulanda D, Tasayco ML, Cataldi M, Arriaran V, Polenova T. Resonance Assignments and Secondary Structure Analysis of E. Coli Thioredoxin by Magic Angle Spinning Solid-State NMR Spectroscopy. J. Phys. Chem. B. 2005;109:18135–18145. doi: 10.1021/jp052774d. [DOI] [PubMed] [Google Scholar]
- (35).Pintacuda G, Giraud N, Pierattelli R, Bockmann A, Bertini I, Emsley L. Solid-State NMR Spectroscopy of a Paramagnetic Protein: Assignment and Study of Human Dimeric Oxidized Cuii-Znii Superoxide Dismutase (SOD) Angew. Chem. Int. Ed. 2007;46:1079–1082. doi: 10.1002/anie.200603093. [DOI] [PubMed] [Google Scholar]
- (36).Kloepper KD, Zhou DH, Li Y, Winter KA, George JM, Rienstra CM. Temperature-Dependent Sensitivity Enhancement of Solid-State NMR Spectra of Alpha-Synuclein Fibrils. J. Biomol. NMR. 2007;39:197–211. doi: 10.1007/s10858-007-9189-z. [DOI] [PubMed] [Google Scholar]
- (37).Li Y, Berthold DA, Gennis RB, Rienstra CM. Chemical Shift Assignment of the Transmembrane Helices of DsbB, a 20-kDa Integral Membrane Enzyme, by 3D Magic-Angle Spinning NMR Spectroscopy. Protein Sci. 2008;17:199–204. doi: 10.1110/ps.073225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Huang L, McDermott AE. Partial Site-Specific Assignment of a Uniformly 13C, 15N Enriched Membrane Protein, Light-Harvesting Complex 1 (LH1), by Solid State NMR. Bba-Bioenergetics. 2008;1777:1098–1108. doi: 10.1016/j.bbabio.2008.01.006. [DOI] [PubMed] [Google Scholar]
- (39).Jehle S, van Rossum B, Stout JR, Noguchi SM, Falber K, Rehbein K, Oschkinat H, Klevit RE, Rajagopal P. αB-Crystallin: A Hybrid Solid-State/Solution-State NMR Investigation Reveals Structural Aspects of the Heterogeneous Oligomer. J. Mol. Biol. 2009;385:1481–1497. doi: 10.1016/j.jmb.2008.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shi L, Ahmed MA, Zhang W, Whited G, Brown LS, Ladizhansky V. Three-Dimensional Solid-State NMR Study of a Seven-Helical Integral Membrane Proton Pump--Structural Insights. J. Mol. Biol. 2009;386:1078–1093. doi: 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- (41).Song YF, Mao JJ, Gunner MR. MCCE2: Improving Protein pKa Calculations with Extensive Side Chain Rotamer Sampling. J. Comp. Chem. 2009;30:2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Frericks Schmidt HL, Sperling LJ, Gao YG, Wylie BJ, Boettcher JM, Wilson SR, Rienstra CM. Crystal Polymorphism of Protein GB1 Examined by Solid-State NMR Spectroscopy and X-Ray Diffraction. J. Phys. Chem. B. 2007;111:14362–14369. doi: 10.1021/jp075531p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gallagher T, Alexander P, Bryan P, Gilliland GL. 2 Crystal-Structures of the B1 Immunoglobulin-Binding Domain of Streptococcal Protein-G and Comparison with Nmr. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.