

Abstract
A majority of malignant melanomas harbor an oncogenic mutation in either BRAF or NRAS. If BRAF and NRAS transform melanoma cells by a similar mechanism, then additional genetic aberrations would be similar (or random). Alternatively, distinct mutation-associated changes would suggest the existence of unique cooperating requirements for each mutation group. We first analyzed a panel of 52 melanoma cell lines (n= 35, 11, 6 for BRAF*, NRAS*, and BRAF/NRASwt/wt respectively) by array-based comparative genomic hybridization for unique alterations that associate with each mutation subgroup. Subsequently, those DNA copy number changes that correlated with a mutation subgroup were used to predict the mutation status of an independent panel of 43 tumors (n=17, 13, 13 for BRAF*, NRAS*, and BRAF/NRASwt/wt respectively). BRAF mutant tumors were classified with a high rate of success (74.4%, P = 0.002), while NRAS mutants were not significantly distinguished from wild types (26/43, P = 0.12). Copy number gains of 7q32.1-36.3, 5p15.31, 8q21.11 and 8q24.11 were most strongly associated with BRAF* tumors and cell lines, as were losses of 11q24.2-24.3. BRAF* melanomas appear to be associated with a specific profile of DNA copy number aberrations that is distinct from those found in NRAS* and BRAFNRASwt/wt tumors. These findings suggest that while both BRAF and NRAS appear to function along the same signal transduction pathway, each may have different requirements for cooperating oncogenic events. The genetic loci that make up this profile may harbor therapeutic targets specific for tumors with BRAF mutations.
Keywords: DNA Copy number analysis, BRAF, melanoma, pharmacogenomics
INTRODUCTION
Melanoma, a neural-crest derived cancer, has few effective therapies that curtail its progression upon metastasis. The pathology of these heterogeneous cancers are often broadly defined by whether they are sun-induced or not and by their location (e.g. sun exposed skin vs. mucosal) (reviewed in Chudnovsky et al., 2005). Current research efforts have focused on genetic and genomic alterations associated with these pathological groupings (reviewed in Kabbarah and Chin 2005). The most common oncogenic event in melanoma is the activation of the MAPK pathway, normally caused by mutations of the BRAF or NRAS genes (Davies et al., 2002). This has been shown to increase tumor proliferation and survival (Hoeflich et al., 2006) as well as facilitate the transition to a malignancy in the stage IV vertical growth phase (Hingorani et al., 2003). The NRAS locus, although less frequently mutated in primary melanomas (Poynter et al., 2006), has been similarly shown to suppress apoptosis in knockdown experiments (Eskandarpour et al., 2005).
Early accounts of BRAF mutations in melanomas estimated their occurrence in approximately 50% of all metastatic tumors, and this frequency may be even higher in benign nevi (Poynter et al., 2006; Uribe et al., 2003) and in certain melanoma subtypes (Poynter et al., 2006). For example, non-sun induced and mucosal melanomas appear to have a lower mutation rate of BRAF than those originating in regions of intermittent sun exposure on the dermis (Maldonado et al., 2003) where mutated KIT may be a dominant oncogene (Curtin et al., 2006). Mutations of NRAS are considerably less common, and estimates of the frequency vary between ~6% (Poynter et al., 2006) and ~25% of primary sporadic melanomas (Omholt et al., 2003). Their differential occurrence in histological subtypes of melanoma is not as clear.
DNA copy number instability has been coupled with the deregulation of several key oncogenic pathways. For example, specific alterations appear to associate with somatic transformation of the RB pathway in breast cancers (Fridlyand et al., 2006). Further, particular sets of copy number alterations have been shown to be directly related to distinct expression profiles associated with breast cancer clinical subtypes (Bergamaschi et al., 2006). At the transcript level, Pavey et al., (2004) successfully classified sporadic malignant melanoma cell lines based upon activating BRAF mutations. Though not yet described in melanomas, unique NRAS* expression profiles have been noted in thyroid papillary carcinomas (Giordano et al., 2005) as well as acute myeloid leukemias (Neben et al., 2005). Previously studies of DNA copy number alterations in melanoma have indirectly demonstrated unique gains and losses associated with sporadic cases (where mutations of BRAF are most prevalent) when compared to those subject to chronic sun damage and non-dermal tumors (Curtin et al., 2005). Further, melanoma-derived cell lines broadly cluster by alterations associated with BRAF/NRAS mutations (Jonsson et al., 2007; Lin et al., 2008). However the relationship to BRAF and/or NRAS mutations in these subgroups has not been investigated in primary tumors.
If BRAF mutations are an early event in oncogenesis of melanoma, then tumors harboring a mutant BRAF would subsequently develop additional non-random cancer promoting alterations which may include cooperating alterations. For example, this could include alterations that result in the upregulation of AKT3, a condition which promotes tumor progression in BRAF* melanomas (Cheung et al., 2008). If BRAF and NRAS mutations are similar in their transforming mechanism, then they should be associated with either the same specific aberrations or a similar random aberration profile (i.e. a copy number phenotype). To test these hypotheses we surveyed a set of melanoma cell lines and tumors for both BRAF and NRAS sequence mutations and genome-wide DNA copy number alterations using array comparative genomic hybridization (aCGH). The direct comparison between the mutation-specific profiles of cell lines and tumors both validates a putative profile as well as defines regions where cell lines may contain in vitro derived genetic aberrations.
MATERIALS AND METHODS
Melanomas
A panel of 52 unique melanoma cell lines were obtained from the lab of Mehnard Herlyn (Wistar Institute, University of Pennsylvania) and were grown in conditions similar to those described in Ji et al., (2007) (cell line details can be seen in Supplemental Table S1) . Briefly, lines were cultured in 5% CO2 atmosphere, at 37°C and in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin. All reagents for cell culture were purchased from Invitrogen (Carlsbad, CA, USA). Melanoma tumors were obtained from the University of Pennsylvania, the Karolinska Institute and Fox Chase Cancer Center under the approval of the Institutional Review Board (details in Supplementary Table S2). Only specimens containing at least 50% tumor cells (as evaluated by light microscopy) were included in the final panel. Genomic DNA was isolated from frozen tumor or cultured cells by overnight digestion, phenol-chloroform extraction and ethanol precipitation.
Mutation Detection
Using similar methods to those described in Davies et al., (2002), exons 11 and 15 of the BRAF locus and exon 2 of NRAS were screened for mutations. This involved direct sequencing of genomic DNA from PCR products using an ABI 3100 DNA Sequencer (Applied Biosystems, Foster City, CA, USA). Ultimately, every cell line and tumor was classified as being BRAF*, NRAS* or wild type for both loci.
Array CGH
Melanomas were assayed on a 1-Mb resolution BAC clone-based CGH array designed specifically for cancer analysis (Greshock et al., 2004). One μg of tumor and reference DNA (pooled lymphocyte-derived DNA from 10 individuals) were labeled with Cy3 or Cy5 flourescent dyes respectively (Amersham, Piscataway, NJ) using the BioPrime random-primed labeling kit (Invitrogen, Carlsbad, CA). In parallel experiments, melanoma DNA and reference DNA were labeled with the opposite dye (“dye swap”) in order to account for difference in dye incorporation. Labeled tumor and reference DNA were then combined and precipitated with human Cot-1 DNA to reduce non-specific binding. DNA was then resuspended and hybridized to the array for 72 h at 37°C on a rotating platform. Images were scanned with an Axon 4500 microarray scanner (Axon Instuments, Union City, CA) and analyzed with the accompanying GenePix software. Data for a subset of the samples has been previously published (Zhang et al., 2006).
Data Analysis
All BAC clones were mapped to human genome build 34 (July 2003) using data provided by the UCSC genome browser site (http://genome.ucsc.edu) or by manual alignment. For each probe on every assay, a log2 copy number ratio was measured from raw data derived from the scanned image by dividing the test channel image intensity by that of the reference channel. Ratios of duplicate clones were averaged for all assays. As a means of quality control, only clones with at least 80% foreground pixel intensity two standard deviations greater than the mean background intensity were considered for analyses. Subsequently, every assay was normalized under the assumption that median copy number was diploid resulting in the median log2 ratio being zero. Data from each dye-swap were combined and evaluated by circular binary segmentation (CBS) to estimate copy number breakpoints (Olshen et al., 2004). Global copy number assignments for each sample were made by dividing the genome into 1 Mb intervals whose copy number status was determined by its associated CBS score. For subsequent chi-squared analysis, the copy number status of each region was categorized using ratio thresholds derived from previous studies of microarray signal response (Greshock et al., 2007a). These were ± 0.25 for gains (< ~5 copies) and monosomies, and > 0.55 for, high level gains (> ~5 copies) and < -0.8 for homozygous deletions. Estimates of genome-wide aberration rates were made by simply calculating the proportion of segments gained or lost in a specific sample.
Statistical Analysis
In order to identify specific DNA alterations that could distinguish between genotypes (BRAF*, NRAS*, or BRAF/NRAS wt/wt), a chi-squared test was applied to the occurrence of regional copy number changes under the null hypothesis that copy number alterations occur at equal frequencies between mutation groups. Genomic regions of 1 Mb served as units for comparison while copy number gains and losses were calculated separately under the null hypothesis that aberrations would occur at equal frequency between mutation groups. The ability to distinguish one mutation group from another was measured by constructing support vector machine models. A series of one-vs-one (OVO) models were constructed using methods previously described (Brown et al., 2000) and trained with aCGH data from cell line models in order to compare mutation groups. All models were constructed using a linear kernel. The γ parameter and cost c were optimized through leave-one-out cross-validation using the cell line data, where those yielding the highest level of success were used. Genomic regions available for inclusion in the model were those occurring on chromosomes shown to have at least a subset of regions that were significantly different between mutation groups by the chi-squared test described above. The optimal regions of alteration were then used to classify all tumors.
RESULTS
General Mutation
All the mutations at the BRAF locus were V600E. The overall frequency of this mutation in this sample set was 54.7% (52/95), though its occurrence was higher in cell lines (35/52; 67.3%) than in tumors (17/43; 39.5%) (P = 0.006). As expected, NRAS mutations were seen at lower frequencies than BRAF mutations (24/95; 25.4%), and were observed at approximately equal frequencies between cell lines (11/52; 21.2%) and tumors (13/43; 30.2%) (P = 0.22). No tissue or cell line was co-mutated for both BRAF and NRAS.
Genome-Wide CGH
Cell lines demonstrated uniformly higher rates of genome-wide chromosomal gain and loss than did tumors samples (t-test; P = 0.0004 and P = 0.0248 respectively) (Fig. 1A.). Although there was no difference in genome-wide aberration rates between mutation groups, instability of several individual chromosomes emerged as being associated with the occurrence of a mutation (Table 1). Most notably, copy number gains of chromosome 7 were seen at higher rates in BRAF* tumors and cell lines (Fig. 2B; P = 0.0199 and P = 0.0172 for tumors and cell lines respectively), while losses of chromosome 4 were associated with NRAS mutants. A survey across the genome for sub-chromosomal copy number alterations in melanoma tumors and cell lines demonstrated distinct patterns of alterations that are associated with BRAF and NRAS mutation groups (Fig. 2). When considering only tumor samples, mutations of the BRAF locus were most closely associated with the presence of copy number gains of the regions surrounding this gene, including 7q32.1-36.3 (Fig. 1C). In tumors, the regions surrounding BRAF demonstrated copy number gains in 12/17 BRAF* (71%), while only 4/26 wild types (15%) (P = 0.0004; Fisher's exact test) (Fig. 2A). Similarly, in cell lines, rates of BRAF locus gain were higher in BRAF* (23/35; 66%) than wild types (3/17; 18%) (P = 0.0012; Fisher's exact test). Other copy number alterations associated with BRAF mutations are gains of 5p15.31 and 5p13.2 as well as the frequent chromosomal losses of 11q24.2-24.3 and 17q12-21.2 (top regions differentiating mutation groups can be seen in Table 2). Expectedly, the absence of a 7q32.1-36.3 gain was associated with NRAS* tumors and BRAF/NRAS wt/wt tissues. Additionally, gains of 5p15.31 and 5p13.2 as well as losses of 22p13 occurred more frequently in NRAS* tissues when compared to BRAF/NRAS wt/wt. Similarly, 5p15.31 gains more readily distinguished BRAF* from NRAS* tumors, as did losses of 16q21.1. Losses of 11q24.2-25 occurred more frequently in NRAS* when compared to BRAF* tumors.
Figure 1.
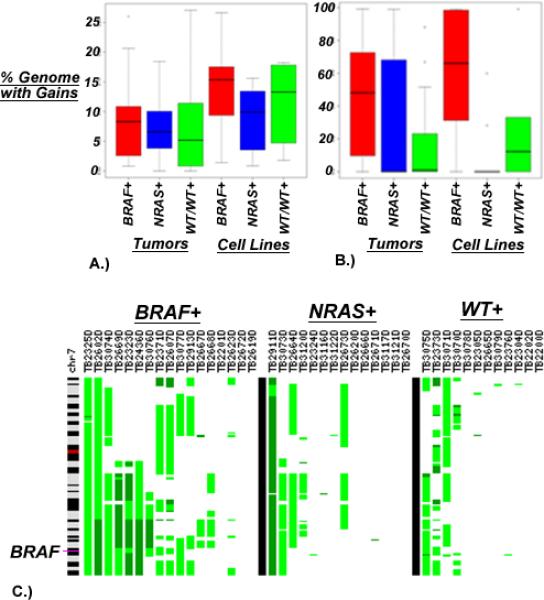
Copy number instability as measured by the proportion of each sample subject to DNA copy number gains in BRAF* tumors compared to NRAS* and BRAF/NRAS wt/wt (A) Total aberrations across the entire genome are higher in cell lines when compared to primary tumors (t-test; P = 0.0004), (B) Aberrations on chromosome 7 are more common in BRAF* tissues when compared to all BRAF wt tissues (P = 0.0199 and P = 0.0172 for tumors and cell lines respectively) (C) Specific gains of 7q32.1-36.3, represented in each sample as the green regions along the vertical ideogram of chromosome 7, strongly associate with BRAF* tissues.
TABLE 1.
Chromosomes Demonstrating Differential Aberration Rates in BRAF* or NRAS* Samples for Both Cell Lines and Tumors Include 4, 7 and 12.
| Tumors |
Cell Lines |
||||||
|---|---|---|---|---|---|---|---|
| Chrom. | Mutation association | Aberration type | Mutant (n = 17) | Wild type (n = 26) | Mutant (n = 35) | Wild type (n = 17) | P-value (tumors;cell lines) |
| 4 | NRAS+ | Loss | 1.5 (3.9) | 12.9 (22.8) | 9.4 (28.3) | 31.9 (37.1) | 0.0120; 0.0359 |
| 12 | NRAS+ | Gain | 0.8 (1.9) | 7.1 (10.0) | 0.5 (1.1) | 6.0 (10.0) | 0.0027; 0.0011 |
| 7 | BRAF+ | Gain | 48.4 (35.3) | 21.8 (34.2) | 58.2 (37.9) | 24.0 (38.0) | 0.0199; 0.0172 |
Aberration rates were quantified by the proportion of the chromosome gained or lost. Standard deviations of these proportions appear in parentheses.
Figure 2.

Genomic frequency plot depicting the copy number alteration frequency of aberrations for (A) BRAF*, (B) NRAS* and (C) BRAF/NRAS wt/wt cell lines and tumors. Cell lines, gains (plotted in green) and losses (red) generally had higher alteration frequencies than did primary tumors (dark needles). (D) A chi-squared statistic for tumors (blue) and cell lines (yellow; overlap plotted in dark shade) identifies regions differentiating mutation groups. Those P-values above the ideogram represent regions of gain, while those below represent regions of loss. The regions that demonstrate the best concordance between cell lines and tumors include gains of 7q32.1-36.3 (shaded green) and gain of 5p15.33-31 (shaded red).
TABLE 2.
Subchromosomal Loci that Best Differentiate BRAF*, NRAS* and BRAF/NRASwt/wt Melanomas as Measured by Chi-Squared Analysis.
| Region | Association | |
|---|---|---|
| BRAF* vs. WT | 7q32.1-36.3 | Gain, BRAF* |
| 5p15.31 | Gain, BRAF* | |
| 5p13.2 | Gain, BRAF* | |
| 8q21.11 | Gain, WT | |
| 8q24.11 | Gain, WT | |
| 11q24.2-24.3 | Loss, BRAF* | |
| 17q12-21.2 | Loss, BRAF* | |
| NRAS* vs. WT | 4p16.1-15.2 | Loss, WT |
| 5p15.31 | Gain, WT; Loss NRAS* | |
| 5p13.2 | Gain, WT; Loss NRAS* | |
| 7q31.11-36.3 | Gain, WT | |
| 17q.12 | Loss, WT | |
| 22p13 | Loss, NRAS* | |
| BRAF* vs. NRAS* | 5p15.31 | Gain, BRAF* |
| 7q32.1-35 | Gain, BRAF* | |
| 11q24.2-25 | Loss, NRAS* | |
| 16q21.1-qter | Loss BRAF* |
Many regions differentiating BRAF* tumors from BRAF/NRASwt/wt also distinguish BRAF* from NRAS* tissues.
SVM modeling
A simple cell line-derived copy number alteration prediction model employing support vector machines (SVMs) for BRAF mutation status prediction was internally cross – validated in cell lines with a success rate of 86% using a leave-one-out scheme. The consensus cell line-based model (loci noted in Table 2), classified 75% of tumors correctly. Only two BRAF* tumors (n = 17) were misclassified as wild types, while 17/26 BRAFwt tumors were classified correctly, indicating a high sensitivity but reduced specificity. Based upon 1000 permutations, where mutations classes were randomly assigned, this model performed better than expected by chance given the permutated data (95% CI: 19.3-72.6%; P < 0.05). A similar model, constructed to predict NRAS* cell lines from wild types, successfully distinguished 21/30 NRAS wild types, though only correctly identified 6/13 NRAS mutants (P = 0.12).
High Level Amplifications
Although the occurrence of amplifications estimated to be > 5 copies were relatively rare compared to all copy number gains, several recurring regions of amplification appeared to be common to both tumors and cell lines. The most common high level amplification seen in the tumor population was that of 7q33 (~136-7 Mb), which was observed in 6/43 samples (Table 3). Similarly, 8/52 cell lines showed amplification of this region. This was in close proximity to the BRAF locus, though only in two case did it encompass the gene (all genes mapping to this and other regions appear in Table 3). Considering both tumors and cell lines, there was a significant tendency for this amplification to occur in BRAF* samples. Other recurring high level amplifications occurred on 1p and 20q. Recurring homozygous losses were noted on 10p14 and 16q24.3 in both tumor and cell line groups. Additionally, homozygous losses of CDKN2A were noted in 21% (11/52) of cell lines but only observed in 2.3% (1/43) primary tumors.
TABLE 3.
Multiple Genes Map to the Most Common High Level Gains and Homozygous Deletions When Considering Both Cell Lines and Tumor Groups.
| Tumors |
Cell lines |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chrom | Region (Mb) | BRAF* (n = 17) | NRAS* (n = 13) | WT/WT (n = 13) | Total | BRAF* (n = 35) | NRAS* (n = 11) | WT/WT (n = 6) | Total | P-value | Genes |
| Ampifications | |||||||||||
| 7 | 136-137 | 5 | 1 | 0 | 6 | 7 | 1 | 0 | 8 | 0.031 | CHRM2, PTN, DGKI, CREB3L2 |
| 20 | 59-60 | 3 | 0 | 1 | 4 | 5 | 2 | 0 | 7 | 0.2623 | SCAPIN1, CDH26, SYCP2, PPP1R3D |
| 20 | 32-33 | 3 | 1 | 1 | 5 | 3 | 1 | 0 | 4 | 0.4808 | DNMT3B, MAPRE1, BPIL1, SPAG4L, BASE, PLUNC, CDK5RAP1, SNTA1, E2F1 |
| 1 | 119-120 | 1 | 1 | 1 | 3 | 2 | 2 | 0 | 4 | 0.6071 | WARS2, HAO2, HSD3B1, PHGDH, ADAM30, NOTCH2 |
| | |||||||||||
| Homozygous Losses | |||||||||||
| 16 | 87-90 | 0 | 1 | 1 | 2 | 3 | 2 | 1 | 6 | 0.6298 | JPH3, CA5A, SLC7A5, ZFPM1, MVD, CYPA, IL17C, APRT, GALNS, FANCA, CDK10, DPEP1, CPNE7, SPG7, TUBB4 |
| 10 | 6.0-9.0 | 1 | 3 | 0 | 4 | 7 | 0 | 1 | 8 | 0.5123 | IL2RA, PRKCQ, PFKFB3, ITIH2, GATA3, ATP5CA |
The only high magnitude alteration that distinguished between mutation groups was gain of 7q33 (136-7 Mb).
DISCUSSION
Both BRAF and NRAS function on the same growth factor receptor tyrosine kinase pathway, and activating mutations of either causes melanomas to have an active mitogen-activated protein kinase (MAPK) cascade when compared to BRAF/NRASwt/wt cells (Shields et al., 2007). Moreover, this ongogenic convergence is supported by the ability of MEK inhibition to cause tumor regressions in either BRAF or RAS induced cancers (Ji et al., 2007). If somatic mutations of BRAF and NRAS transform melanoma cells via similar mechanisms, then the manifestation of cooperating aberrations, such as copy number aberrations, may be expected to be similar. These results indicate that BRAF* melanomas harbor a distinct set of DNA copy number alterations from those found in NRAS* and BRAF/NRASwt/wt tumors. This result suggests that while both BRAF and NRAS function along the same signaling pathway, the cooperating events required or the resultant copy number profile phenotype are distinct for different mutation groups.
This hypothesis is predicated on the observation that cell lines tend to be faithful genetic models of their parent histology both in terms of genome-wide copy number aberrations (Greshock et al., 2007b) and specific mutations (Jones et al., 2008). Previous studies of melanoma suggest this would be the case. Similar patterns of gain at 7q, and 20q as well as losses of 9p and 10q were characterized in both cell line models (Ji et al., 2007; Jonsson et al., 2007) and primary tumors (Curtin et al., 2005). Concordantly, these regions appeared as marquee alterations in both tumors and cell lines in this study. Focal amplifications also appear similar between this and previous studies. For example, the amplification of the metastatic melanoma candidate oncogene NEDD9 (6p24.2) (Kim et al., 2006) was seen in 12% (5/43) of tumors. Recurring E2F1 amplifications, thought to promote cell proliferation in melanomas (Roberts 2006), was observed in both cell lines and tumors (3/52; 6% and 5/43; 12%, respectively).
We were able to identify a DNA copy number profile that is associated with activating BRAF mutations in melanoma cell lines and tumors, where a simple copy number alteration model derived from cell line data reliably predicted the BRAF mutation status of 75% of primary tumors. This is not surprising as BRAF mutations have also been found in benign nevi, suggesting that additional cooperating events may be necessary for full transformation (Pollock et al., 2003). Two outstanding predictive features of BRAF* tumors are general instability of chromosome 7 and the specific copy number gains of 7q32.1-36.3 (the region encompassing the BRAF locus). This region also harbored the only high level copy number amplification (~> 5 copies) that was connected with a mutation group, an observation concordant with previous chromosomal CGH studies (Tanami et al., 2004). Also, Curtin et al., (2005) noted that sporadic melanomas of the skin (a subgroup with high rates of BRAF mutations) are subject to instability on 7q, though this specific association with BRAF* tumors had been previously undocumented. These data are reminiscent of previous observations that activating mutations of epidermal growth factor receptor (EGFR) in lung cancer often occur within subchromosomal amplicons, (Bell et al., 2005). Transcript abundance for genes mapping to 7q are concordant with the increased copy number alterations seen in BRAF* tumors where 15/18 differentially expressed transcripts (P < 0.05) on 7q were upregulated in BRAF* cell lines [data from Pavey et al., (2004)]. Although this observation is consistent with fluorescence in situ hybridization studies noting differential allelic imbalance and distinct amplification of mutant alleles in the region adjacent to the BRAF locus in BRAF* tumors (Willmore-Payne et al., 2006), its functional relevance remains unclear.
Although there was less consistency in DNA copy number alterations that were associated with NRAS mutations, losses of chromosome 4 and gains of chromosome 12 appeared strongly associated in both cell lines and tumors. Specific losses of 5p15.31 and 5p13.2 as well as losses of 22p13 also associate with NRAS mutations; however, we were unable to build a cell line-based predictive model for NRAS mutations that effectively characterized tumors. Under the hypothesis that associated DNA copy number changes are non-random (i.e. they have functional implications), there may be several reasons for this result: (a) the oncogenic insult conferred by NRAS* is sufficient on its own (such that additional aberrations would be random); (b) NRAS* can cooperate with a multitude of different genetic aberrations where this tumor sample size being underpowered to detect such a profile (i.e., Type II error); (c) more common cooperating genetic lesions are point mutations that would be missed by copy number analyses and (d) NRAS* occur as late in the oncogenic cascade on a background of different genetic predispositions (this explanation, however, would be contrary to numerous preclinical models whereby Ras* is sufficient for transformation). Finally, these results are consistent with the observation by Lin et al., (2008) that NRAS* cell lines inconsistently clustered based on key copy number alterations. Considering the clinical and pathological variation seen in melanomas, it is not surprising that they represent a genetically heterogeneous group of cancers. Recent data have contributed to much better understanding of the molecular classification of these tumors (Fecher et al., 2007).
In both cross validation and tumor prediction, a proportion of BRAF wild type melanomas appeared to present a DNA copy number profile similar to that of mutant tumors. This observation has several possible implications. First, this could infer that these associated alterations could be cooperating with other yet undescribed activators of RAF/MEK/ERK signaling. Secondly, this could also imply that some of the alterations compose of a copy number phenotype manifested in BRAF* melanomas that could be recapitulated in a subset of BRAF wild type melanomas. This may prove to be the case as the functional relevance of most DNA alterations associating with BRAF* and NRAS* tumors remains unclear. As a potential analogy, patterns of aneuploidy have been extensively associated with TP53* tumors (summarized in Tomasini et al., (2008), a condition that may manifest itself differently uniquely in different histologies.
This study suggests that BRAF* melanomas appear to have a more well-defined oncogenetic profile than NRAS* tumors. The more homogenous series of genetic aberrations that correlate with BRAF* melanomas may have several therapeutic implications. First, this may provide insight into specific therapeutic targets that may be relevant only in the context of an activated BRAF. For example, recently described synthetic lethality of oncrasin-1 treatment with those cells harboring KRAS mutations suggests cooperating loci that may be required for mutant tumor survival (Guo et al., 2008). Secondly, as a novel class of compounds targeting the RAF/MEK/ERK signaling pathway have proven preferentially effective for inhibiting proliferation in BRAF* cells, a more complete understanding of the genetic alterations that co-occur with mutations of BRAF* tumors could identify therapies that may prove synergistic with inhibitors of this pathway. This class of compounds includes those targeting Raf kinases directly, such as sorafenib and AZ628 (McDermott et al., 2007) as well as inhibitors targeting mitogen-activated protein kinases such as PD184352/CI-1040 (Solit et al., 2006). In this study, the cooccurring amplification of the de novo methylation regulator DNMT3B (20q11.21) in BRAF* tumors suggest synergy between existing cancer therapies such as 5-aza-2′-deoxycytidine and those targeting RAF/MEK/ERK signaling. Finally, in the context of melanoma, associated DNA amplifications may provide novel targets for preventing BRAF* nevi from progression.
Supplementary Material
Acknowledgments
Funded by:
National Cancer Institute Melanoma Specialized Program of Research Excellence (SPORE) Grant # P50 CA093372
REFERENCES
- Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB, Krebs AD, Johnson DH, Giaccone G, Herbst RS, Manegold C, Fukuoka M, Kris MG, Baselga J, Ochs JS, Haber DA. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081–8092. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- Bergamaschi A, Kim YH, Wang P, Sorlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Borresen-Dale AL, Pollack JR. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–1040. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Jr., Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–3439. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813–824. doi: 10.1172/JCI24808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Eskandarpour M, Kiaii S, Zhu C, Castro J, Sakko AJ, Hansson J. Suppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cells. Int J Cancer. 2005;115:65–73. doi: 10.1002/ijc.20873. [DOI] [PubMed] [Google Scholar]
- Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606–1620. doi: 10.1200/JCO.2006.06.0442. [DOI] [PubMed] [Google Scholar]
- Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, Segraves R, Dairkee S, Tokuyasu T, Ljung BM, Jain AN, McLennan J, Ziegler J, Chin K, Devries S, Feiler H, Gray JW, Waldman F, Pinkel D, Albertson DG. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006;6:96. doi: 10.1186/1471-2407-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano TJ, Kuick R, Thomas DG, Misek DE, Vinco M, Sanders D, Zhu Z, Ciampi R, Roh M, Shedden K, Gauger P, Doherty G, Thompson NW, Hanash S, Koenig RJ, Nikiforov YE. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24:6646–6656. doi: 10.1038/sj.onc.1208822. [DOI] [PubMed] [Google Scholar]
- Greshock J, Feng B, Nogueira C, Ivanova E, Perna I, Nathanson K, Protopopov A, Weber BL, Chin L. A comparison of DNA copy number profiling platforms. Cancer Res. 2007a;67:10173–10180. doi: 10.1158/0008-5472.CAN-07-2102. [DOI] [PubMed] [Google Scholar]
- Greshock J, Nathanson K, Martin AM, Zhang L, Coukos G, Weber BL, Zaks TZ. Cancer cell lines as genetic models of their parent histology: analyses based on array comparative genomic hybridization. Cancer Res. 2007b;67:3594–3600. doi: 10.1158/0008-5472.CAN-06-3674. [DOI] [PubMed] [Google Scholar]
- Greshock J, Naylor TL, Margolin A, Diskin S, Cleaver SH, Futreal PA, deJong PJ, Zhao S, Liebman M, Weber BL. 1-Mb resolution array-based comparative genomic hybridization using a BAC clone set optimized for cancer gene analysis. Genome Res. 2004;14:179–187. doi: 10.1101/gr.1847304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Hoeflich KP, Gray DC, Eby MT, Tien JY, Wong L, Bower J, Gogineni A, Zha J, Cole MJ, Stern HM, Murray LJ, Davis DP, Seshagiri S. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S, McNamara K, Chen L, Albert M, Sun Y, Al-Hashem R, Chirieac LR, Padera R, Bronson RT, Thomas RK, Garraway LA, Janne PA, Johnson BE, Chin L, Wong KK. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67:4933–4939. doi: 10.1158/0008-5472.CAN-06-4592. [DOI] [PubMed] [Google Scholar]
- Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, Kinzler KW, Vogelstein B, Willis J, Markowitz SD. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson G, Dahl C, Staaf J, Sandberg T, Bendahl PO, Ringner M, Guldberg P, Borg A. Genomic profiling of malignant melanoma using tiling-resolution arrayCGH. Oncogene. 2007;26:4738–4748. doi: 10.1038/sj.onc.1210252. [DOI] [PubMed] [Google Scholar]
- Kabbarah O, Chin L. Revealing the genomic heterogeneity of melanoma. Cancer Cell. 2005;8:439–441. doi: 10.1016/j.ccr.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, Flotte TJ, Duncan LM, Granter SR, Chin L. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Lin WM, Baker AC, Beroukhim R, Winckler W, Feng W, Marmion JM, Laine E, Greulich H, Tseng H, Gates C, Hodi FS, Dranoff G, Sellers WR, Thomas RK, Meyerson M, Golub TR, Dummer R, Herlyn M, Getz G, Garraway LA. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68:664–673. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, Rothenberg SM, Supko JG, Sordella R, Ulkus LE, Iafrate AJ, Maheswaran S, Njauw CN, Tsao H, Drew L, Hanke JH, Ma XJ, Erlander MG, Gray NS, Haber DA, Settleman J. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neben K, Schnittger S, Brors B, Tews B, Kokocinski F, Haferlach T, Muller J, Hahn M, Hiddemann W, Eils R, Lichter P, Schoch C. Distinct gene expression patterns associated with FLT3- and NRAS-activating mutations in acute myeloid leukemia with normal karyotype. Oncogene. 2005;24:1580–1588. doi: 10.1038/sj.onc.1208344. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483–6488. [PubMed] [Google Scholar]
- Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM, Walker GJ, Boyle GM, Harper U, Cozzi SJ, Hansen K, Yudt L, Schmidt C, Hersey P, Ellem KA, O'Rourke MG, Parsons PG, Meltzer P, Ringner M, Hayward NK. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene. 2004;23:4060–4067. doi: 10.1038/sj.onc.1207563. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, Salem G, Pohida T, Heenan P, Duray P, Kallioniemi O, Hayward NK, Trent JM, Meltzer PS. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Poynter JN, Elder JT, Fullen DR, Nair RP, Soengas MS, Johnson TM, Redman B, Thomas NE, Gruber SB. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–273. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- Roberts JD. E2F1 Amplication and Genetic Heterogeneity in Melanoma. Cancer Biol Ther. 2006;5:691–692. doi: 10.4161/cbt.5.6.2926. [DOI] [PubMed] [Google Scholar]
- Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, Hao H, Penland S, Arbiser J, Scott G, Zhou T, Bar-Eli M, Bear JE, Der CJ, Kaufmann WK, Rimm DL, Sharpless NE. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanami H, Imoto I, Hirasawa A, Yuki Y, Sonoda I, Inoue J, Yasui K, Misawa-Furihata A, Kawakami Y, Inazawa J. Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene. 2004;23:8796–8804. doi: 10.1038/sj.onc.1208152. [DOI] [PubMed] [Google Scholar]
- Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol. 2008;18:244–252. doi: 10.1016/j.tcb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Uribe P, Wistuba II, Gonzalez S. BRAF mutation: a frequent event in benign, atypical, and malignant melanocytic lesions of the skin. Am J Dermatopathol. 2003;25:365–370. doi: 10.1097/00000372-200310000-00001. [DOI] [PubMed] [Google Scholar]
- Willmore-Payne C, Holden JA, Hirschowitz S, Layfield LJ. BRAF and c-kit gene copy number in mutation-positive malignant melanoma. Hum Pathol. 2006;37:520–527. doi: 10.1016/j.humpath.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, O'Brien-Jenkins A, Katsaros D, Hatzigeorgiou A, Gimotty PA, Weber BL, Coukos G. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.