

Abstract
Recent technological advances in flow cytometry provide a versatile platform for high throughput screening of compound libraries coupled with high-content biological testing and drug discovery. The G protein-coupled receptors (GPCRs) constitute the largest class of signaling molecules in the human genome with frequent roles in disease pathogenesis, yet many examples of orphan receptors with unknown ligands remain. The complex biology and potential for drug discovery within this class provide strong incentives for chemical biology approaches seeking to develop small molecule probes to facilitate elucidation of mechanistic pathways and enable specific manipulation of the activity of individual receptors. We have initiated small molecule probe development projects targeting two distinct families of GPCRs: the formylpeptide receptors (FPR/FPRL1) and G protein-coupled estrogen receptor (GPR30). In each case the assay for compound screening involved the development of an appropriate small molecule fluorescent probe, and the flow cytometry platform provided inherently biological rich assays that enhanced the process of identification and optimization of novel antagonists. The contributions of cheminformatics analysis tools, virtual screening, and synthetic chemistry in synergy with the biomolecular screening program have yielded valuable new chemical probes with high binding affinity, selectivity for the targeted receptor, and potent antagonist activity. This review describes the discovery of novel small molecule antagonists of FPR and FPRL1, and GPR30, and the associated characterization process involving secondary assays, cell based and in vivo studies to define the selectivity and activity of the resulting chemical probes
Keywords: flow cytometry, fluorescent, GPCR, formylpeptide receptor, inflammation, GPR30, GPER, estrogen, nongenomic, cancer, antidepressant
Flow Cytometry for Gpcr Probe Discovery
The G protein-coupled receptors (GPCRs) constitute the largest class of signaling molecules in the human genome with frequent roles in disease pathogenesis that provide targets for a large proportion of currently marketed drugs. The critical role of GPCRs in transmitting extracellular signals into cells via intracellular G protein heterotrimers, relatively few of which have been characterized, and the existence of large number of GPCRs termed “orphan receptors” for which native ligands are not known, have positioned this class prominently in the arena of drug discovery. The complex biology associated with GPCR-mediated signaling provides additional motivation for the discovery of new small molecule ligands that exhibit selective binding and functional control of specific receptors. These chemical probes can serve as valuable tools for elucidating fundamental biological mechanisms in cell-based and in vivo studies, and help to establish foundations for drug discovery initiatives that exploit the novel targets and pathways that are revealed.
Flow cytometry has matured as a platform for the quantitative multi-parameter measurement of cell fluorescence and has numerous applications including drug discovery [1-6]. The versatility of flow cytometry accommodates the display of virtually any molecular assembly or cell response. Novel bead-based assay systems allow studies of real-time interactions between solubilized receptors, ligands, protein-protein interactions and molecular signaling components that recapitulate and extend measurements in intact cells. Recent innovations allow faster serial processing of bulk cell samples. Homogeneous discrimination of free and cell-bound fluorescent probe eliminates wash steps to streamline sample processing. Flow cytometry is particularly convenient for alternately assessing both cellular and molecular activities of small molecules. By using suspension arrays of particles, which can be color-coded molecules, nanoparticles, beads or complex cell populations the resulting assays and responses can be highly multiplexed to maximize sample analysis throughput and generate multidimensional biological output. These new developments are coupled with the widespread availability of instrumentation to position flow cytometry as an attractive analysis platform for high-throughput, high-content biological testing and drug discovery.
The University of New Mexico Center for Molecular Discovery was established with support from the NIH Roadmap Molecular Libraries Screening Network (MLSCN) and subsequently through the Molecular Libraries Probe (MLP) program. The NMCMD consists of a multidisciplinary academic research team with assembled expertise from biomedical, biophysical, chemical, computational, instrumentation and engineering fields. This specialty center has established flow cytometry as a versatile platform for drug discovery, proteomics, and real-time analysis of molecular interactions while incorporating a novel sampling approach (HyperCyt®) [7], associated technology, assay design, informatics and analysis tools that support high throughput screening to identify small molecule probes in a biological information content-rich format. Small molecule probe development projects incorporating virtual screening, cheminformatics and synthetic chemistry focused on two distinct families of GPCRs have yielded valuable new chemical probes. In each case the screening application required the development of an appropriate small molecule fluorescent probe, and the flow cytometry platform provided inherently biological rich assays that facilitated the identification and optimization of novel antagonists. This review describes the discovery of novel small molecule antagonists of FPR and FPRL1, and GPR30, and the associated characterization studies that employ secondary assays, cell based and in vivo studies to define the selectivity and activity of the resulting chemical probes.
Formyl Peptide Receptors FPR/FPRL1
The G protein-coupled formylpeptide receptors (FPRs) have been implicated in a variety of inflammatory and immunological responses, and provide an important area for the application of chemical biology approaches to elucidate the complex interplay and fundamental biology of this important class of receptors, and to explore potential therapeutic applications [8-11]. FPR is expressed in a variety of cell types including astrocytes, hepatocytes, immature dendritic cells, microglial cells, neutrophils and the tunica media of coronary arteries. Two other variants have been described: FPRL1 and FPRL2 that share 69% and 56% primary sequence identity to FPR respectively, and 83% identity between the pair. FPRL1 occurs in an even greater variety of cell types than FPR including astrocytoma cells, epithelial cells, hepatocytes, microvascular endothelial cells, neuroblastoma cells, and phagocytic leukocytes. FPRL2 is expressed only in monocytes. Recent clinical interest has associated FPR with antibacterial inflammation and malignant glioma cell metastasis, while FPRL1 has been linked to chronic inflammation in systemic amyloidosis, Alzheimer's disease, and prion diseases as well as ovarian cancer metastasis. N-Formylated peptides constitute the most commonly studied class of FPR activators that trigger a variety of biological activities in myeloid cells, including chemokinesis, chemotaxis, cytokine production and superoxide generation. Many of these peptide ligands are only weak activators of FPRL1, however, a number of host-derived agonists have been identified in association with pathophysiological processes. At the time this study was undertaken there was only one report of small molecule antagonist of FPR [12], and one report of an FPRL1 antagonist was described during the course of this investigation [13].
Development of a Cross-Reactive Fluorescent Ligand for FPR/FPRL1
The conceptual approach for a screening assay to identify novel small molecule antagonists focused on achieving the simultaneous measurement of binding interactions with both FPR and FPRL1 receptors. This strategy has the potential to improve overall screening efficiency, an advantage of multiplexing, and generates data that reflects receptor specificity as an inherent feature of the screen. This approach required a fluorescent probe with high affinity for both receptors FPR and FPRL1 that could simultaneously report ligand-binding interactions in a single assay. N-Formylated peptides such as E. coli derived N-formyl-Met-Leu-Phe (fMLF) are known to be high-affinity ligands for FPR that initiate a variety of biological activities. Many potent formylpeptide FPR agonists only weakly activate FPRL1. The fMLFK-FITC has been used as a fluorescent reporter for FPR with a Kd = 3 nM, but does not bind FPRL1 [14]. Peptides possessing amino terminal tryptophan (W) residues were identified as selective high affinity ligands for FPRL1 [15], and substitutions at the second amino acid residues within the W-peptide series were tolerated without significantly compromising FPRL1 binding [16]. We investigated the hexapeptide probe H2N-Trpl-Lys(FL)-Tyr-Met-Val-(D)Met-amide (compound WK(FL)YMVm Fig (1)) incorporating a fluorescein dye attached to the lysine residue [17]. Equilibrium binding experiments with WK(FL)YMVm revealed Kd values of 1.21 ± 0.36 nM and 1.82 ± 0.78 nM for FPR and FPRL1 respectively. The affinity of the parent hexapeptide was ∼100 fold higher for FPRL1 than FPR, thus the attachment of fluorescein to the lysine residue enhanced the affinity of the resulting probe for FPR diproportionately. The observation of enhanced binding is consistent with the proposed contribution of a large hydrophobic region towards the binding interaction of FPRL1 [18]. This fluorescent probe WK(FL)YMVm represents the first cross-reactive high affinity reporter ligand, demonstrating specific ligand interactions with both FPR and FPRL1 expressing cell lines, and having satisfied the performance criteria as a fluorescent ligand probe was used to develop a flow cytometric assay.
Fig. (1).
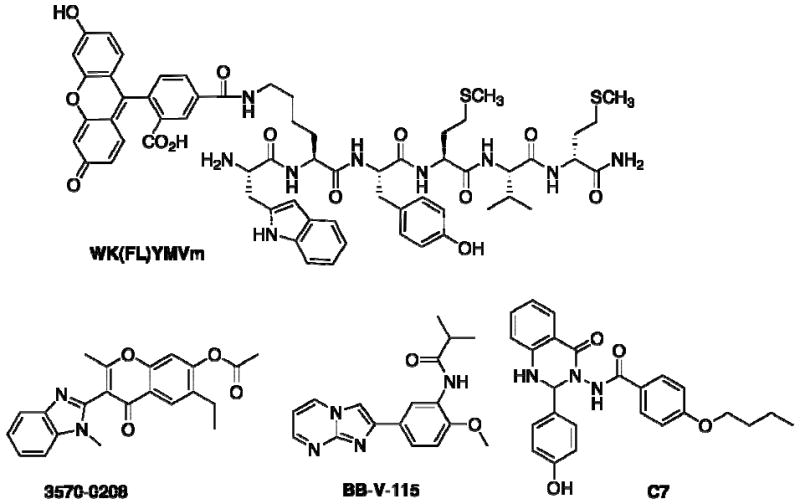
Structures of FPR/FPRL1 probes: cross-reactive fluorescent peptide ligand WK(FL)YMVm, FPR antagonist 3570-0208, FPRL1 antagonist BB-V-115, previously reported FPRL1 antagonist C7.
Identificiation of FPR/FPRL1-Selective Antagonists
A flow-cytometry-based high- throughput screening (HTS) approach was used to identify small molecules that exhibit high binding affinity with individual selectivity for FPR and FPRL1. The screening assay measured the ability of test compounds to competitively displace the fluorescent peptide WK(FL)YMVm from FPR, FPRL1, or both. In this assay U937 cells expressing FPR and rat basophil leukemia (RBL) cells expressing FPRL1 were tested together in a duplex format. The FPR-expressing U937 cells were color coded by incubation with red-fluorescent dye FuraRed™ in order to easily distinguish and separately gate these cells from the RBL/FPRL1 cells during the flow cytometric analysis. Compounds, cells, and fluorescent ligand were sequentially combined without intervening wash steps. The high-throughput HyperCyt™ flow cytometry platform was used to sample cells from 384-well microplates for introduction to the flow cytometer [6,19]. Fluorescence was excited at 488nm and detected with optical bandpass filters matched for the two reporter dyes. The time-resolved data files were analyzed with IDLeQuery software to identify active compounds in each well. The primary screen of 24,304 NIH Small Molecule Repository revealed 253 compounds with inhibition >30%, representing 181 for FPR, 72 for FPRL1 and 26 cross-reactive compounds for both receptors. Subsequent dose response studies identified a total of 40 compounds with selective binding inhibition constants Ki ≤ 4 μM, including 34 for FPR and 6 for FPRL1. No cross-reactive compounds with Ki ≤ 4 μM were found. Cheminformatic analysis classified critical chemical scaffolds or chemotype families that correlated with activity, and were compared to several chemotypes earlier reported by our group [12]. Computational screening and two-dimensional substructure searching of a commercial ChemDiv compound collection was used to identify an additional 1,446 structurally related compounds for activity optimization. The focused set of compounds selected for expanded screening included 1,276 for FPR, and a separate set of 170 for FPRL1. The single concentration HTS assay revealed strong correlation of activity for the targeted receptor with over 10-fold increase in the percentage of tested compounds with Ki ≤ 4 μM for FPR, and over 100-fold increase in the percentage of potent actives for FPRL1, providing clear confirmation of the hypothesized structure activity relationships. The most active compounds identified were resynthesized, fully characterized to confirm structure and purity, and retested to quantify activity [20]. The most potent compound against FPR was the 4H-chromen-4-one derivative 3570-0208, with Ki = 95 +/- 10 nM. A clear structure-activity relationship was observed in association with substitution of aryl and heterocyclic groups at the 3-position in this series, culminating in the maximum inhibitory activity associated with the benzimidazole derivative 3570-0208. In separate duplex competition assays this compounds affinity for FPR exceeded that for FPRL1 by more than 100-fold. The compound with the greatest activity for FPRL1 was the imidazo[1,2-a]pyrimidine derivative BB-V-115 with Ki = 270 +/- 51 nM. Structure-activity optimization focused on the carboxamide group derived from methoxyaniline in this series and achieved maximum inhibition of FPRL1 in the derivative possessing a 2-methylpropanamide group. Compound BB-V-115 exhibited receptor selectivity for FPRL1 ∼20-fold greater than for FPR. This compound was also compared with the recently reported small molecule FPRL1 antagonist C7, a 3-amino-2,3-dihydroquinazolin-4(1H)-one, which was found to exhibit ∼10-fold lower affinity for FPRL1 than BB-V-115 [13].
These compounds were evaluated in secondary assays for their ability to block the intracellular calcium response induced by high affinity peptide agonists for their respective target receptors. The calcium response elicited by WKYMVm in RBL/FPRL1 cells was blocked by the FPRL1 antagonist BB-V-115. The response induced by fMLFF in U937/FPR cells was similarly blocked by the FPR antagonist 3570-0208. Each of these antagonists demonstrated selectivity for their targeted receptor, exhibiting the inability to block the activity of the respective peptide agonists for the off-target receptor even at increased concentrations up to 100 μM. To date, these two probes represent the most potent, selective small molecule antagonist reported for their respective receptor.
The resulting selective antagonist probes developed in this study using high throughput flow cytometry to screen large compound libraries are expected to find applications in delineating pathophysiological processes associated with the FPR and FPRL1 receptors. The duplex format employing color-coded cells expressing the respective receptors reduced the overall assay time, minimized reagent requirements, and provided selectivity information at every screening stage, thus proving to be an efficient approach for identifying selective small molecule GPCR receptor ligands. The color-coding approach can be readily adapted to higher degrees of assay multiplexing to encompass even greater biological content and further enhance screening efficiency. The incorporation of in silico computational methods and substructure searching to populate an expanded set of chemotype families for characterization of structure-activity during the probe optimization stage significantly enhanced the percentage of active compounds and improved the efficiency with which highly selective antagonist probes were identified for both receptors.
Probe Development Targeting the G Protein-Coupled Estrogen Receptor GPR30
The G protein-coupled estrogen receptor GPR30 has emerged as an intriguing signaling molecule enmeshed in the complex pathways through which estrogens regulate diverse physiological processes [21-26]. Estrogen (compound E2 Fig (2)) initiates rapid biochemical and physiological responses that are not accounted for by changes in gene expression mediated by the nuclear estrogen receptors (ERα/β). Membrane-associated classical ERα has been implicated in the initiation of signaling cascades through direct interactions with membrane structures that include G proteins, caveolins, and receptor tyrosine kinases. Non-classical models attribute this activity to other membrane-associated estrogen-binding proteins, including recent studies that have identified GPR30 as a prime candidate. High affinity estrogen binding to GPR30 results in activation of stimulatory G proteins, transactivation of epidermal growth factor receptor, increased adenylyl cyclase activity, and rapid intracellular calcium mobilization, occurring within seconds to minutes, and accumulation of phosphatidylinositol 3,4,5-triphosphate in the nucleus that occurs following ∼15 minutes of ligand induced stimulation [27,28]. However, the experimental difficulty involved in distinguishing the detailed pathways through which estrogen induces these multiple cellular and physiological responses has presented a primary challenge for advancing the field. GPR30 expression in primary breast adenocarcinoma has been associated with pathological parameters commonly used to assess breast cancer progression, including the development of extramammary metastases [29]. GPR30 expression is linked to lower survival rates in endometrial and ovarian cancer [30,31]. Despite the common affinity for E2, differences in binding to synthetic estrogens, phytoestrogens and xenoestrogens have been recognized [32]. The antiestrogens tamoxifen and ICI 182,780 that have high binding affinity for GPR30 but elicit opposite activity profiles in comparison with ERα/β may have relevance for tumorigenesis and cancer therapy. No examples of GPR30-selective ligands had been reported prior to the initiation of the studies described herein. Using a combined approach integrating chemical and molecular biology we have developed the first generation of GPR30-selective probes that provide the tools for elucidating the biological roles of GPR30 from the classical nuclear estrogen receptors.
Fig. (2).
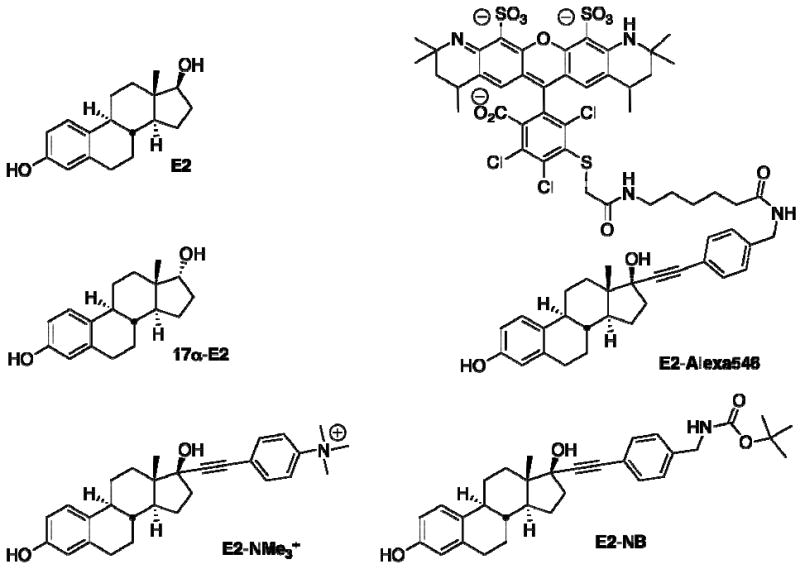
Structures of estrogen probes: 17β-estradiol E2, 17α-estradiol 17α-E2, fluorescent estrogen ligand E2-Alexa546, permeability-restricted estrogen E2-NMe3+, permeable estrogen E2-NB.
Cellular studies employing transfected COS7 monkey kidney fibroblast cells expressing GPR30 or ERα/β provide complete experimental control of receptor content that is exceptionally valuable for investigating the associated activation pathways. One particularly fascinating result from the initial study was the identification of GPR30 as an intracellular transmembrane estrogen receptor localized in the endoplasmic reticulum [28]. Identical localization patterns are revealed in cancer cell lines that endogenously express GPR30, including MCF-7, SKBr3, JEG and Hec50co. These provocative findings have broad implications towards understanding the fundamental biological function of GPCR's and for clinical applications of estrogen therapeutics. The postulate that estrogen activates GPR30 from inside the cell is consistent with the hydrophobic structure of estrogen, which contributes membrane permeability and provides ready access to the intracellular compartment. The necessary machinery for GPCR's to initiate signaling is expected to be present in the endoplasmic reticulum and the intermediate signaling molecules are present in the cytoplasm. The capacity for plasma membrane G protein-coupled receptors to translocate to other cellular structures and establish ligand-responsive signaling complexes has only been recognized relatively recently, and represents a fascinating new arena for pharmacology and drug discovery. The possibility of estrogen-stimulated GPR30 signaling from an intracellular location is consistent with reports of other functional intracellular GPCRs associated with lipophilic ligands and receptor kinases [36-39].
Development of a Fluorescent Estrogen Ligand
In order to further investigate this intriguing scenario, we designed and synthesized a series of 17α-[4-aminomethyl-phenylethynyl]-estra-1, 3, 5(10)-triene-3,17β-diol derivatives as fluorescent and functional probes to characterize the localization and functionality of GPR30 in comparison with the classical nuclear receptors ERα/β [28]. The selection of the 17α-position of substitution on the steroid scaffold combined with the rigid hydrophobic linkage were critical factors that enabled the synthesis of probes that retained high affinity for both classes of receptors. We constructed fluorescent estrogen ligands employing a pendant approach to attach a dye to a derivatized estrogen ligand, as represented by the conjugate (compound E2-Alexa546 Fig (2)). This approach provides a high degree of versatility for probe design due to the commercial availability of a wide variety of fluorescent dyes with different structural characteristics and photophysical properties that can be matched with specific excitation and emission requirements for experiments employing multiple fluorophores. The structures of most fluorescent dyes contain large hydrophobic polycyclic aromatic ring systems adorned with polar and often ionizable functional groups that provide some measure of water solubility, and this modification results in major differences in steric volume and physicochemical properties in comparison with the original receptor ligand. Interestingly, E2-Alexa546 did not exhibit observable binding to the plasma membrane in the GPR30-expressing cell lines investigated. The ionic charge of the AlexaFluor dyes rendered the corresponding estrogen conjugates membrane-impermeable over the time course of rapid signaling events studied and no staining was observed unless the cells were permeabilized with saponin,. A nuclear staining pattern with E2-Alexa546 was observed in transfected cells expressing a fusion protein of green fluorescent protein (GFP) and ERα, (ERα-GFP), as expected for the ligand binding state of nuclear receptors. Colocalization of this probe with GPR30-GFP was observed in the endoplasmic reticulum, analogous to results obtained using markers and GPR30-targeted antibodies. This binding was competitive with E2, and revealed a Ki = 6.6 nM for GPR30. The unnatural biologically inactive diastereomer 17α-estradiol (compound 17α-E2 Fig (2)) was unable to displace the fluorescent ligand, providing further verification of the selectivity of estrogen binding. These results strongly support the model for estrogen binding to GPR30 in the endoplasmic reticulum, but cannot exclude the possibility for differential activity of receptor localized in the plasma membrane.
Development of Permeability-Restricted Estrogen Probes
To further investigate the model in which GPR30 initiates signaling from an intracellular location, we synthesized a series of related small molecule probes possessing either neutral or charged appendages, thereby providing a simple parameter with which to control membrane permeability through passive diffusion [40]. We hypothesized that if GPR30 was functional at the cell surface, both charged (E2-NMe3+) and neutral (E2-NB) probes should have access to the receptor and be capable of activating the rapid signaling pathways, while differences in activation would be expected if the alternative scenario involving GPR30 activation from an intracellular site. This approach provided a complementary design for permeability-restricted estrogens that have employed macromolecular conjugates to bovine serum albumin [41], or poly(amido)amine dendrimer conjugates [42,43], with the technical advantages associated with using fully characterized, discrete small molecules. The binding properties of these probes were evaluated using transfected COS-7 cells for both ERα and GPR30 in competitive binding assays employing the fluorescent ligand E2-Alexa633. This experimental design involved membrane permeabilization with saponin to provide entry to both the charged fluorescent conjugate and the neutral or ionic estrogen probes, thereby allowing comparison of their relative binding affinities. Cells expressing ERα revealed a Ki = 0.65 nM for E2, and corresponding values for E2-NB (Ki = 2.7 nM), and E2-NMe3+ (Ki = 1.4 nM). Analogous experiments using GPR30-transfected cells revealed Ki = 9 nM for E2, and the neutral E2-NB (Ki = 16 nM), and charged compound E2-NMe3+ (Ki = 17 nM). The estrogen-probes exhibited only slightly lower affinities compared with E2 for each receptor type, associated with the strategic positioning of the arylethynyl appendages at the 17α-position, but there were no significant differences in binding affinities between the probes associated with the presence of charged or neutral hydrophobic substituents.
Two functional signaling assays were then used to evaluate the activation properties of the synthetic probes in intact, non-permeabilized cell lines. The neutral compound E2-NB initiated calcium fluxes at 10 nM concentration that were indistinguishable from those of E2 using the very rapid intracellular calcium mobilization assay in both transfected cell lines expressing either ERα or GPR30. In contrast, the charged compound E2-NMe3+ failed to stimulate measureable calcium fluxes during the time course of these experiments, even at concentrations 100-fold higher than those needed to provide maximal activity of E2. The compounds were then assessed for PIP3 production in transfected cell lines expressing either ERα or GPR30. This assay requires a slightly longer time frame (∼15 min) for stimulation, but revealed analogous results demonstrating rapid action by the neutral compound E2-NB, while the charged compound E2-NMe3+ gave virtually no response. The same activation properties were obtained using SKBr3 cells that endogenously express GPR30 but not ERα/β, providing additional confirmation for the validity of results obtained using transfected cell lines. These results are entirely consistent with the model of estrogen stimulated GPR30 signaling from an intracellular location, and the reduced ability of the cationic compounds E2-NMe3+ to enter the cell by passive diffusion. The possibility that GPR30 is capable of initiating different signaling events from distinct cell surface or intracellular locations, and the factors that regulate GPR30 localization in normal or disease states are intriguing questions with potential pharmacological implications for further investigation.
Discovery of a GPR30-Selective Agonist G-1
Distinguishing the biological roles of GPR30-mediated signaling from the classical estrogen receptors ERα/β is essential for understanding the fundamental mechanisms of estrogen action. This opportunity required the discovery of new ligands with high selectivity for GPR30, and the capacity to specifically activate or inhibit receptor-mediated signaling. The absence of detailed structural data for GPR30 detracts from the possible application of structure-guided computational approaches. The observed cross-reactivity of E2 and estrogen-derived probes for both classes of receptors provides a common scaffold for a ligand-based virtual screening approach to identify estrogen-like structures of interest for biological screening. A combination of 2D and 3D similarity approaches were used for virtual screening of a library of 10,000 molecules that was constructed using concept of GPCR-privileged substructures [44]. The combined similarity score attributed 40% weight to 2D fingerprints, 40% to the shape-based similarity and 20% to pharmacophore-based similarity to rank the top 100 molecules selected by composite score for biomolecular screening.
A competitive ligand binding assay using the fluorescent estrogen probe E2-Alexa633 was developed using a high throughput flow cytometric platform (HyperCyt®) for the primary screen of the focused library compounds selected from virtual screening. The assay used transfected COS7 cells, which do not display detectable specific binding of the fluorescent estrogen in the absence of exogenous GPR30 or ER expression, with E2 and 17a-estradiol as positive and negative controls for specific E2-Alexa633 binding. A GPR30-GFP fusion protein was expressed and cells were gated for high levels of green fluorescence, correlating with GPR30 expression, in order to maximize the bound fluorescent signal. As GPR30 expression is localized to the endoplasmic reticulum and not the plasma membrane, cells were permeabilized with saponin to permit access of the charged E2-Alexa633 to GPR30. Biomolecular screening was carried out using 17β-estradiol E2 as a positive control to block specific binding of E2-Alexa633 to GPR30. A single compound, identified as G-1 in Fig (3), consistently inhibited the binding of E2-Alexa633 to GPR30.[45] This compound possesses a tetrahydro-3H-cyclopenta[c]quinoline core structure. The compound was resynthesized using the trifluoroacetic acid catalyzed Povarov cyclization, and characterized as the racemic, but diastereomerically pure syn- or endo-isomer. The binding properties of G-1 to GPR30 were determined with Ki = 11 nM, whereas the corresponding Ki = 6 nM for E2 as previously described. The Hill coefficient for G-1 binding to GPR30 was 0.6, which suggests the possibility that there are two binding sites with similar affinity.
Fig. (3).

Structures of the GPR30-selective probes: agonist G-1, antagonist G15, I-125 radiotracer G40.
Treatment of GPR30-expressing COS7 cells with G-1 produced a comparable maximum calcium concentration (t1/2 ∼ 30 s) but was slower than the very rapid response from E2 (t1/2 < 2 s). Dose response studies revealed an EC50 for calcium mobilization of approximately 2 nM, while the corresponding EC50 for E2 was 0.3 nM. G-1 did not produce a response in non-transfected control cells, or those expressing either ERα or ERβ, both of which responded rapidly to E2. The complementary assay based on PI3K activation and determination of nuclear accumulation of PIP3 also demonstrated the capacity of G-1 to activate the same signaling pathways as estrogen through GPR30, while no response was produced in cells expressing ERα/β.
In order to evaluate the ability of G-1 to selectively target GPR30 in an environment in which both classes of receptors were present, cells were cotransfected with GPR30-monomeric red fluorescent protein-1 (GPR30-mRFP1) and ERα-GFP, which distinctly localize to the endoplasmic reticulum and nucleus respectively. The presence of G-1 selectively blocked staining of E2-Alexa633 in the endoplasmic reticulum associated with GPR30, but had no effect on the nuclear staining associated with ERα/β. The presence of G-1 initiated nuclear accumulation of PIP3 in SKBr3 breast cancer cells, which endogenously express GPR30 but not ERα/β. Similar results were obtained from G-1 treated MCF7 cells, which express GPR30, high levels of ERα and low levels of ERβ. G-1 was found to inhibit migration of SKBr3 cells with an IC50 = 0.7 nM, and MCF7 cells with IC50 = 1.6 nM. These results demonstrate the ability of G-1 to selectively bind and activate endogenous GPR30, including expression in a complex cellular environment containing classical receptors ERα/β.
Additional Biological Characterization of G-1 in Cell-Based and In Vivo Systems
Following the initial report in which G-1 was shown to activate calcium mobilization and phosphatidyl inositol-3-kinase (PI3K) in cancer cells [45], this probe has been used to examine the cellular and physiological actions of GPR30 in a variety of other published studies. Cellular effects of G-1 include activation of calcium fluxes in LHRH neurons and hypothalamic neurons [46,47], spinal neuron depolarization [48], protein kinase Ce activation [49], gene expression [50,51], proliferation [50,52], oocyte meitotic arrest [53], and primordial follicle formation [54]. The role of GPR30 in vivo has been investigated using G-1 with reported effects that include estrogen-induced thymic atrophy [55], experimental autoimmune encephalomyelitis [56], and vascular regulation [57]. In each of these animal models, the G-1-mediated effects were absent in GPR30 knockout mice, establishing the selectivity of this compound for GPR30. In one report G-1 did not show any estrogenic effects in vivo using the classical estrogen target organs, the uterus and the mammary gland [58]. However, recent studies have revealed G-1 causes a moderate increase in uterine epithelial cell proliferation [59].
Discovery of a GPR30-Selective Antagonist G15
While the GPR30 agonist G-1 provided a valuable probe for distinguishing cellular and physiological actions of GPR30 from classical nuclear receptors, further pharmacological characterization required the development of a GPR30-specific antagonist. General structural similarities between G-1 and E2 provide some degree of overlap in the respective tetrahydro-3H-cyclopenta[c]quinoline and steroid scaffolds, particularly considering the oxygen atoms that are oriented at the periphery of both polycyclic cores. Hypothesizing that the hydrogen bond acceptor properties of the ketone group in G-1 were associated with the observed agonism associated with GPR30 binding, and seeking a corresponding structural trigger to block GPR30-mediated signaling, we employed parallel virtual screening and synthetic programs to identify new compounds with altered activation profiles. The tetrahydroquinoline scaffold is synthetically accessible using the three-component imino-Diels Alder or Povarov cyclization that combines an aldehyde, aniline and electron-rich alkene components, and is subject to catalysis by a variety of Bronsted and Lewis acid catalysts. Following the precedent for lanthanide-catalyzed cyclization, we employed Sc(OTf)3 to construct a focused library of structural analogs. These conditions afforded rapid reaction times, excellent product yields and high endo-diastereoselectivity. Virtual screening was used to search the NIH Molecular Libraries Small Molecule Repository (MLSMR) in order to identify structures of interest related to the tetrahydro-3H-cyclopenta[c]quinoline scaffold of G-1. A substructure search of 144,457 molecules in the MLSMR was performed using a custom JAVA program built using the OpenEye OEJava toolkit. This virtual screen prioritized 64 molecules, of which 57 were obtained from the MLSMR. The primary biomolecular screen for GPR30 antagonism was developed using the E2-activated calcium mobilization in GPR30-expressing SKBr3 cells and evaluating compounds for the ability to block this cellular activation. Primary screening of the 57 compounds representing this scaffold from the MLSMR yielded 8 that showed some inhibition of estrogen-mediated calcium mobilization in cells expressing GPR30. Of particular interest was the compound G15 shown in Fig (3) that closely resembled G-1 but lacked the ethanone moiety of that molecule.[59]
Chemically synthesized G15 was subjected to multiple cellular and physiological assays in order to characterize its biological effects. Competitive binding assays using a radioiodinated ligand G40 demonstrated that G15 binds to GPR30 with an affinity of approximately 20 nM. This compares to the affinity for G-1, utilizing the same assay, of approximately 7 nM, similar to our previously reported affinity of G-1 for recombinant GPR30 of 11 nM and reported affinities for 17β-estradiol between 3-6 nM. Thus removal of the ethanone moiety resulted in a decrease in relative binding affinity of 2-3 fold. Additional competitive binding studies to assess interactions with ERα and ERβ revealed that similar to G-1, G15 displays little binding to ERα or ERβ at concentrations up to 10 μM, where E2 exhibits sub-nanomolar Ki values. These results reveal that G15, like G-1, displays high affinity for GPR30 with minimal binding to ERα and ERβ (Ki > 10 μM).
The receptor activity of G15 was evaluated using the functional assays employed previously. G15 did not induce the rapid calcium mobilization response in GPR30 expressing SKBr3 breast cancer cells. The pre-treatment of cells with G15 in fact caused a substantial reduction in the response produced by subsequent exposure to G-1 or E2, which confirms the ability of G15 to block the action of these GPR30 agonists. There was no inhibition of the calcium response mediated by ATP through endogenous purinergic receptors, indicating the antagonistic effect is specific to GPR30. The inhibition of agonist stimulated calcium mobilization in SKBr3 cells by G15 was dose-dependent, yielding an IC50 = 185 nM for blocking G-1, which compares closely with a similar IC50 = 190 nM for inhibiting the E2 response. Characterization of the effect of G15 on the PI3K activation pathway in the endogenous GPR30 expressing SKBr3 cells of GPR30-transfected COS7 cells revealed analogous results, producing no stimulation of this response and demonstrating the consistent ability of pre-treatment with G15 to reduce the response resulting from stimulation by G-1 and E2. In contrast, G15 had no effect on the E2-stimulated response in cells expressing ERα/β, providing confirmation of the ability of G15 to specifically block only the GPR30-mediated pathway.
The antagonist properties of G15 were then evaluated in three well-established, physiologically relevant in vivo models of estrogen action. Estrogen treatment in ovariectomized mice produces a uterine response with increasing water content (imbibition) and increased epithelial cell proliferation. Neither G-1 or G15 affected the uterine wet weight or imbibition in this model as measured by uterine weight and microscopic evaluation of histologic sections. A single injection of E2 resulted in a 17-fold increase in the proliferative index of uterine epithelia as measured by immunodetection of Ki-67 protein. The GPR30 agonist G-1 also produced a moderate (∼4-fold) increase in proliferation compared to control. The administration of G15 had no effect on proliferation. However, when these mice were treated with both G15 and E2 the relative estrogen-induced proliferation was reduced by ∼50% in a dose-dependent response. Similarly, the combination treatment with G15 blocked the G-1 stimulated proliferation in a dose-dependent manner. These results suggest that GPR30 does contribute to the proliferative response in the uterus but not towards imbibition, which appears to be mediated exclusively by ERα.
The tail suspension test serves as a convenient animal model for evaluating antidepressants by measuring their effect on the duration of immobility. Treatment with the antagonist G15 had no effect on the immobility time, while both E2 and G-1 and the tricyclic dibenzazepine antidepressant desipramine markedly reduced immobility time in comparison to control. Pretreatment of mice with G15 resulted in significant attenuation of the effect of both E2 and G-1, but had no effect on desipramine treatment. Desipramine potentiates neurotransmission by selectively blocking reuptake of norepinephrine from the neural synapse, and appears to impair serotonin transport with minor anticholinergic activity through its affinity to muscarinic receptors. These results implicate a neurological role for GPR30 in the regulation of depression that may ultimately provide a viable target for the development of new antidepressants.
Conclusion
These results demonstrate the successful application of high throughput flow cytometric screening for small molecule probe discovery. The development of the first cross reactive high affinity fluorescent probe WK(FL)YMVm was used in a duplex assay that simultaneously evaluated the binding interactions with individual cell lines expressing FPR and FPRL1. A novel 4H-chromen-4-one derivative 3570-0208 antagonist for FPR was identified with Ki = 95 +/- 10 nM. The imidazo[1,2-a]pyrimidine derivative BB-V-115 was found to be a potent antagonist for FPRL1 with Ki = 270 +/- 51 nM. Both compounds were able to block the intracellular calcium response induced by the respective high affinity peptide agonists for their target receptors, and demonstrated selectivity by their inability to block the activity of the respective peptide agonists for the off-target receptor even at increased concentrations up to 100 μM. This process was facilitated by the duplex format of the assay which provided valuable information relating to selectivity, and the application of in silico computational methods and substructure searching of chemotype families significantly enhanced the percentage of active compounds and improved the efficiency with which highly selective antagonist probes were identified for both receptors.
A pendant approach was used to develop a fluorescent estrogen ligand E2-Alexa546 which was used to confirm the intracellular localization of GPR30, corroborating results from expression of GPR30-GFP in the endoplasmic reticulum, and also served as a fluorescent ligand for a flow cytometric competitive binding assay. A series of permeability-restricted estrogens was developed to further investigate the site of functional GPR30, including the impermeant charged derivative (E2-NMe 3+) and structural related neutral (E2-NB) estrogen probes. A combined virtual and biomolecular screening approach identified the tetrahydro-3H-cyclopenta[c]quinoline compound G-1 as a potent GPR30 agonist. Additional biological characterization of G-1 in cell based and in vivo systems has demonstrated the capacity for stimulating GPR30 responses in a variety of cell types, and in animal models. A structurally related compound G15 that lacks the ethanone group, hypothesized to be associated with the agonist ligand bound conformation of GPR30, was in fact found to be an effective antagonist of GPR30 response in cell based and physiologically relevant in vivo models of estrogen action. These results suggest that GPR30 contributes to the proliferative response in the uterus but not towards imbibition, which appears to be mediated exclusively by ERα, and implicate a putative neurological role for GPR30 in the regulation of depression that may ultimately provide a viable target for the development of new antidepressants. The identification of this antagonist is expected to facilitate the further evaluation of the physiological and pharmacological roles of GPR30. In a recent evaluation of the NIH chemical probes, the following probes: the FPR antagonist 3570-0208, the FPRL1 antagonist BB-V-115, the GPR30 agonist G-1 and the GPR30 antagonist G15, respectively, were give a vote of “high confidence”, indicative of their potential usefulness in a lead and drug discovery program [60].
Acknowledgments
Grant Sponsor: NIH; Grant numbers CA127731, CA118743, R03 MH076381, R01 AI36357, U54MH074425 The New Mexico Molecular Libraries Screening Center, U54084690 The University of New Mexico Center for Molecular Discovery, The University of New Mexico Shared Flow Cytometry Resource, Cancer Research and Treatment Center (CA118100), The University of New Mexico Cancer Center Animal Models & Imaging Core, The New Mexico Tobacco Settlement fund, The Oxnard Foundation, The Stranahan Foundation, and The New Mexico Cowboys for Cancer Research.
References
- 1.Sklar LA, Carter MB, Edwards BS. Flow cytometry for drug discovery, receptor pharmacology and high-throughput screening. Curr Opin Pharmacol. 2007;7:527–34. doi: 10.1016/j.coph.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards BS, Oprea TI, Prossnitz ER, Sklar LA. Flow cytometry for high-throughput, high-content screening. Curr Opin Chem Biol. 2004;8:392–8. doi: 10.1016/j.cbpa.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Nolan JP, Sklar LA. The emergence of flow cytometry for sensitive, real-time measurements of molecular interactions. Nat Biotechnol. 1998;16:633–8. doi: 10.1038/nbt0798-633. [DOI] [PubMed] [Google Scholar]
- 4.Sklar LA, Edwards BS, Graves SW, Nolan JP, Prossnitz ER. Flow cytometric analysis of ligand-receptor interactions and molecular assemblies. Annu Rev Biophys Biomol Struct. 2002;31:97–119. doi: 10.1146/annurev.biophys.31.082901.134406. [DOI] [PubMed] [Google Scholar]
- 5.Ramirez S, Aiken CT, Andrzejewski B, Sklar LA, Edwards BS. High-throughput flow cytometry: validation in microvolume bioassays. Cytometry A. 2003;53:55–65. doi: 10.1002/cyto.a.10035. [DOI] [PubMed] [Google Scholar]
- 6.Kuckuck FW, Edwards BS, Sklar LA. High throughput flow cytometry. Cytometry. 2001;44:83–90. [PubMed] [Google Scholar]
- 7.Edwards BS, Young SM, Ivnitsky-Steele I, Ye RD, Prossnitz ER, Sklar LA. High-content screening: flow cytometry analysis. Methods Mol Biol. 2009;486:151–65. doi: 10.1007/978-1-60327-545-3_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–61. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panaro MA, Acquafredda A, Sisto M, Lisi S, Maffione AB, Mitolo V. Biological role of the N-formyl peptide receptors. Immunopharmacol Immunotoxicol. 2006;28:103–27. doi: 10.1080/08923970600625975. [DOI] [PubMed] [Google Scholar]
- 10.Blakeney JS, Fairlie DP. Nonpeptide ligands that target peptide-activated GPCRs in inflammation. Curr Med Chem. 2005;12:3027–42. doi: 10.2174/092986705774462888. [DOI] [PubMed] [Google Scholar]
- 11.Dalpiaz A, Scatturin A. Peptide derivatives as agonists or antagonists of formylpeptide receptors: analysis of their effects on neutrophils. Mini Rev Med Chem. 2003;3:167–73. doi: 10.2174/1389557033488222. [DOI] [PubMed] [Google Scholar]
- 12.Edwards BS, Bologa C, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol Pharmacol. 2005;68:1301–10. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 13.Zhou C, Zhang S, Nanamori M, Zhang Y, Liu Q, Li N, Sun M, Tian J, Ye PP, Cheng N, Ye RD, Wang MW. Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor-like 1. Mol Pharmacol. 2007;72:976–83. doi: 10.1124/mol.107.037564. [DOI] [PubMed] [Google Scholar]
- 14.Edwards BS, Young SM, Oprea TI, Bologa CG, Prossnitz ER, Sklar LA. Biomolecular screening of formylpeptide receptor ligands with a sensitive, quantitative, high-throughput flow cytometry platform. Nat Protoc. 2006;1:59–66. doi: 10.1038/nprot.2006.9. [DOI] [PubMed] [Google Scholar]
- 15.Baek SH, Seo JK, Chae CB, Suh PG, Ryu SH. Identification of the peptides that stimulate the phosphoinositide hydrolysis in lymphocyte cell lines from peptide libraries. J Biol Chem. 1996;271:8170–5. doi: 10.1074/jbc.271.14.8170. [DOI] [PubMed] [Google Scholar]
- 16.Bae YS, Song JY, Kim Y, He R, Ye RD, Kwak JY, Suh PG, Ryu SH. Differential activation of formyl peptide receptor signaling by peptide ligands. Mol Pharmacol. 2003;64:841–7. doi: 10.1124/mol.64.4.841. [DOI] [PubMed] [Google Scholar]
- 17.Strouse JJ, Young SM, Mitchell HD, Ye RD, Prossnitz ER, Sklar LA, Edwards BS. A novel fluorescent cross-reactive formylpeptide receptor/formylpeptide receptor-like 1 hexapeptide ligand. Cytometry A. 2009;75:264–70. doi: 10.1002/cyto.a.20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan HX, Zhou C, Zhang Y, Sun M, Wang X, Yu H, Yang X, Ye RD, Shen JK, Wang MW. Discovery of Trp-Nle-Tyr-Met as a novel agonist for human formyl peptide receptor-like 1. Biochem Pharmacol. 2007;74:317–26. doi: 10.1016/j.bcp.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Ramirez S, Aiken CT, Andrzejewski B, Sklar LA, Edwards BS. High-throughput flow cytometry: validation in microvolume bioassays. Cytometry A. 2003;53:55–65. doi: 10.1002/cyto.a.10035. [DOI] [PubMed] [Google Scholar]
- 20.Young SM, Bologa CM, Fara D, Bryant BK, Strouse JJ, Arterburn JB, Ye RD, Oprea TI, Prossnitz ER, Sklar LA, Edwards BS. Duplex high-throughput flow cytometry screen identifies two novel formylpeptide receptor family probes. Cytometry A. 2009;75:253–63. doi: 10.1002/cyto.a.20645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids. 2008;73:870–3. doi: 10.1016/j.steroids.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 22.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 23.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- 24.Watson CS, Alyea RA, Jeng YJ, Kochukov MY. Nongenomic actions of low concentration estrogens and xenoestrogens on multiple tissues. Mol Cell Endocrinol. 2007;274:1–7. doi: 10.1016/j.mce.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas P, Dressing G, Pang Y, Berg H, Tubbs C, Benninghoff A, Doughty K. Progestin, estrogen and androgen G-protein coupled receptors in fish gonads. Steroids. 2006;71:310–6. doi: 10.1016/j.steroids.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–7. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 28.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 29.Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res. 2006;12:6359–66. doi: 10.1158/1078-0432.CCR-06-0860. [DOI] [PubMed] [Google Scholar]
- 30.Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, Prossnitz ER. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol. 2007;196(386):e1–11. doi: 10.1016/j.ajog.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, Verschraegen CF, Hathaway HJ, Joste NE, Prossnitz ER. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol. 2009 Jun 5; doi: 10.1016/j.ygyno.2009.05.015. [Epub ahead of print] PMID: 19501895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prossnitz ER, Sklar LA, Oprea TI, Arterburn JB. GPR30: a novel therapeutic target in estrogen-related disease. Trends Pharmacol Sci. 2008;29:116–23. doi: 10.1016/j.tips.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Boivin B, Vaniotis G, Allen BG, Hébert TE. G protein-coupled receptors in and on the cell nucleus: a new signaling paradigm? J Recept Signal Transduct Res. 2008;28:15–28. doi: 10.1080/10799890801941889. [DOI] [PubMed] [Google Scholar]
- 34.Goetzl EJ. Diverse pathways for nuclear signaling by G protein-coupled receptors and their ligands. FASEB J. 2007;21:638–42. doi: 10.1096/fj.06-6624hyp. [DOI] [PubMed] [Google Scholar]
- 35.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–68. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 36.Gobeil F, Fortier A, Zhu T, Bossolasco M, Leduc M, Grandbois M, Heveker N, Bkaily G, Chemtob S, Barbaz D. G-protein-coupled receptors signalling at the cell nucleus: an emerging paradigm. Can J Physiol Pharmacol. 2006;84:287–97. doi: 10.1139/y05-127. [DOI] [PubMed] [Google Scholar]
- 37.Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, Bossolasco M, Bkaily G, Peri K, Varma DR, Orvoine R, Chemtob S. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can J Physiol Pharmacol. 2006;84:377–91. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]
- 38.Marrache AM, Gobeil F, Zhu T, Chemtob S. Intracellular signaling of lipid mediators via cognate nuclear G protein-coupled receptors. Endothelium. 2005;12:63–72. doi: 10.1080/10623320590933815. [DOI] [PubMed] [Google Scholar]
- 39.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–8. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, Sklar LA, Arterburn JB, Prossnitz ER. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. 2007;2:536–44. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 41.Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. 1999;140:5455–8. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- 42.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, Katzenellenbogen JA. Hormone-PAMAM dendrimer conjugates: polymer dynamics and tether structure affect ligand access to receptors. Angew Chem Int Ed Engl. 2006;45:7243–8. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- 44.Savchuk NP, Tkachenko SE, Balakin KV. Rational Design of GPCR-Specific Combinational Libraries Based on the Concept of Privileged Substructures. In: Oprea TI, editor. Chemoinformatics in Drug Discovery. Wiley VCH; Weinheim: 2005. pp. 287–313. [Google Scholar]
- 45.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 46.Noel SD, Keen KL, Baumann DI, Filardo EJ, Terasawa E. Involvement of G-Protein Coupled Receptor 30 (GPR30) in Rapid Action of Estrogen in Primate LHRH Neurons. Mol Endocrinol. 2009;23:349–59. doi: 10.1210/me.2008-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- 48.Dun SL, Brailoiu GC, Gao X, Brailoiu E, Arterburn JB, Prossnitz ER, Oprea TI, Dun NJ. Expression of estrogen receptor GPR30 in the rat spinal cord and in autonomic and sensory ganglia. J Neurosci Res. 2009;87:1610–9. doi: 10.1002/jnr.21980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn J, Dina OA, Goswami C, Suckow V, Levine JD, Hucho T. GPR30 estrogen receptor agonists induce mechanical hyperalgesia in the rat. Eur J Neurosci. 2008;27:1700–9. doi: 10.1111/j.1460-9568.2008.06131.x. [DOI] [PubMed] [Google Scholar]
- 50.Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Andò S, Maggiolini M. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- 51.Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009;28:523–32. doi: 10.1038/emboj.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teng J, Wang ZY, Prossnitz ER, Bjorling DE. The G protein-coupled receptor GPR30 inhibits human urothelial cell proliferation. Endocrinology. 2008;149:4024–4034. doi: 10.1210/en.2007-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pang Y, Dong J, Thomas P. Estrogen signaling characteristics of Atlantic croaker G protein-coupled receptor 30 (GPR30) and evidence it is involved in maintenance of oocyte meiotic arrest. Endocrinology. 2008;149:3410–3426. doi: 10.1210/en.2007-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Prossnitz ER, Roy SK. G protein-coupled receptor 30 expression is required for estrogen stimulation of primordial follicle formation in the hamster ovary. Endocrinology. 2008;149:4452–4461. doi: 10.1210/en.2008-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, Rosenbaum JS, Vandenbark AA, Offner H. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol. 2009;182:3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M. Regulatory Role of G Protein-Coupled Estrogen Receptor for Vascular Function and Obesity. Circ Res. 2009;104:288–91. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–56. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- 59.Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421–7. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oprea TI, Bologa CG, Boyer S, Curpan RF, Glen RC, Hopkins AL, Lipinski CA, Marshall GR, Martin YC, Ostopovici-Halip L, Rishton G, Shoichet BK, Ursu O, Vaz RJ, Waller CL, Waldmann H, Sklar LA. A crowdsourcing evaluation of the NIH chemical probes. Nature Chem Biol. 2009;5:441–447. doi: 10.1038/nchembio0709-441. [DOI] [PMC free article] [PubMed] [Google Scholar]