

Abstract
Respiratory tract dendritic cells (DCs) are juxtaposed to directly sample inhaled environmental particles. Processing and presentation of these airborne Ags could result in either the development of immunity or tolerance. The purpose of this study was to determine the consequences of cigarette smoke exposure on DC function in mice. We demonstrate that while cigarette smoke exposure decreased the number of DCs in the lungs, Ag-induced DC migration to the regional thoracic lymph nodes was unaffected. However, cigarette smoking suppressed DC maturation within the lymph nodes as demonstrated by reduced cell surface expression of MHC class II and the costimulatory molecules CD80 and CD86. Consequently, DCs from cigarette smoke-exposed animals had a diminished capacity to induce IL-2 production by T cells that was associated with diminished Ag-specific T cell proliferation in vivo. Smoke-induced defects in DC function leading to impaired CD4+ T cell function could inhibit tumor surveillance and predispose patients with chronic obstructive pulmonary disease to infections and exacerbations.
The purpose of this study was to evaluate the effect of cigarette smoke exposure on lung dendritic cell (DC)4 function in vivo. DCs are bone marrow-derived APCs that populate lymphoid as well as nonlymphoid tissues such as the respiratory tract, a major portal of entry for Ags and microbes. Immature DCs are strategically located in the airway mucosa and lung parenchyma where they continuously sample their environment, taking up foreign airborne Ags, forming MHC-peptide complexes, and up-regulating costimulatory molecules (1). DCs then rapidly migrate to the regional thoracic lymph nodes (TLNs) where they stimulate the proliferation and differentiation of naive T cells (2–4).
Over 20% of adults in the U.S. regularly smoke cigarettes with an even greater prevalence of smoking worldwide. Cigarette smoking results in a variety of common and life-threatening diseases related to inflammation and immunity including chronic obstructive pulmonary disease (COPD) and lung cancer. COPD comprises emphysema, a result of inflammatory cell-mediated proteolytic destruction of lung tissue, as well as airway inflammation that is in part related to repeated airway infection. Lung cancer arises from cigarette-related mutations combined with defects in immune surveillance. DCs play a key role in lung immunity and hence potentially these cigarette-related disease processes.
Cigarette smoke has been shown to alter both innate and adaptive immune responses (5, 6) and some cigarette smoke constituents, such as oxygen radicals and endotoxin, can activate and exert a direct immunomodulatory effect on respiratory DCs. In vitro studies using bone marrow and monocyte-derived DCs exposed to varying doses of nicotine (7, 8) and cigarette smoke extract (9) have yielded contrasting results with respect to their effect on DC function.
Because of limitations in interpreting the biological relevance of exposure of individual cigarette smoke components as well as whole cigarette smoke extract on cells in culture, we assessed the effect of mainstream cigarette smoke exposure in vivo on lung DC number, migration to lymph nodes, and Ag presentation within the nodes. Despite the limitations of the classical maturation paradigm linked to the complexity and heterogeneity of the DC network (1, 10), we believe this approach provides a useful model to dissect the effects of cigarette smoke on lung DCs. DCs have a central role in controlling immune responses during infection, allergy, and cancer, therefore knowledge of how smoking affects this system of cells is likely key in elucidating the mechanisms by which smoking affects the immune system during disease processes.
Materials and Methods
Mice
C57BL/6 and BALB/c female mice 8 wk of age were obtained from Charles River Laboratories. Same age and gender nonsmoking control mice were housed in the same conditions as the smoke-exposed animals for the duration of the study. OVA-TCR-transgenic mice (DO11.10) on a BALB/c background were obtained from The Jackson Laboratory. All mice were housed in sterile microisolator cages. The Harvard Standing Committee for Animal Research at Harvard University School of Public Health approved all animal protocols.
Cigarette smoke exposure
Mice were exposed to cigarette smoke from four unfiltered cigarettes (two administered in the morning and two in the afternoon; University of Kentucky) 5 days/week for 1 mo using a smoking apparatus with the chamber adapted for mice, as previously described (11).
Instillation of macromolecule solutions into the trachea
Mice were anesthetized by i.p. injection of 2.5% avertin and then intubated as described (12) and 600 μg of FITC-conjugated OVA (OVA-FITC, screened for low endotoxin content; Molecular Probes) was administered intratracheally (i.t.) in a total volume of 60 μl of sterile PBS (Invitrogen Life Technologies) to both smoke-exposed and control mice. In some experiments, 600 μg of OVA (chromatographically purified, screened for low endotoxin content; Worthington Biochemical) was administered in a volume of 60 μl of sterile PBS to both smoke-exposed and control mice.
Preparation of lung single-cell suspensions
Mice were euthanized by CO2 narcosis. A thoracotomy was performed, followed by right heart catheterization with a 21-G (3/4) siliconized needle and the pulmonary circulation was perfused with 20 ml of sterile PBS to remove the intravascular pool of cells. A total of 2 ml of digestion medium (RPMI 1640 (obtained from Invitrogen Life Technologies), supplemented with 1 mg/ml collagenase type IV and 0.5 mg/ml DNase from bovine pancreas (both obtained from Sigma-Aldrich), was then injected i.t. using a 22-G catheter and the trachea was quickly sealed with a silk suture. The trachea and lungs were then removed. Lungs were carefully separated from the heart, thymus, and trachea and incubated at 37°C in an additional 3 ml of digestion medium for 30 min. Incubation was then prolonged for additional 30 min, with vigorous pipetting of the samples at 10-min intervals.
Subsequently, samples were passed over a 70-μm nylon cell strainer and RBC were lysed. The cell suspension was then incubated in calcium- and magnesium-free PBS containing 10 mM EDTA for 5 min at room temperature on a shaker, resuspended in staining buffer (PBS without Ca2+ or Mg2+, 5% FBS, 0.1% sodium azide, and 5 mM EDTA) and kept on ice until immunofluorescent labeling. Sodium azide was not added to the staining buffer when the cells were going to be used in functional studies.
Preparation of lymph node single-cell suspensions
For studies involving migration and costimulatory molecules, mice were euthanized by CO2 narcosis 24 h after i.t. injection of OVA-FITC or OVA, respectively. For T cell proliferation studies, mice were euthanized by CO2 narcosis 96 h after injection of OVA. Following thoracotomy, paratracheal and parathymic intrathoracic lymph nodes were removed under a stereomicroscope (Olympus SZ 60) and incubated at 37°C in 3 ml of lymph node digestion medium (1× HBSS (Cellgro Mediatech), with 2% 10 mM EDTA-treated FBS (HyClone), supplemented with 2.5 mg/ml collagenase type IV). After a 10-min incubation, lymph nodes were minced with 20-G 1(1/2) and 25-G (5/8) needles and incubated for an additional 10 min. Subsequently, samples were passed over a 70-μm nylon cell strainer, incubated in calcium- and magnesium-free PBS containing 10 mM EDTA for 5 min at room temperature on a shaker, resuspended in staining buffer, and kept on ice until immunofluorescent labeling. Due to the photosensitivity of the FITC material, lymph nodes and cell suspensions from OVAFITC-injected animals were protected from direct light.
Labeling of single-cell suspensions for flow cytometry
All staining reactions were performed at 4°C. All Abs were obtained from BD Pharmingen unless otherwise noted. Cells were preincubated for 20 min with Ig constant fragment FcR-blocking Ab to reduce nonspecific binding (anti-CD16/CD32, clone 2.4G2). For lung studies, cells were subsequently stained with PE-Cy5.5 hamster anti-mouse CD11c mAb (clone N418; Caltag Laboratories) and data acquisition was performed using the FL1/FL3 template, to allow assessment of the distribution of CD11cbright cells with regard to autofluorescence (AF), as previously described (13). PE-Cy 5.5 hamster IgG isotype control was used to determine background staining (Caltag Laboratories).
For migration studies, cells were stained with PE-Cy5.5 hamster anti-mouse CD11c mAb (Caltag Laboratories). For CCR7 expression studies, cells were stained with FITC anti-mouse I-Ek (14-4-4S) mAb, PE anti-mouse CCR7 (4B12; eBioscience), and PE-Cy5.5 hamster anti-mouse CD11c mAb (Caltag Laboratories). FITC mouse IgG2a, PE rat IgG2a (Caltag Laboratories), and PE-Cy 5.5 hamster IgG (Caltag Laboratories) isotype controls were used to determine background staining.
For costimulatory molecule expression studies, cells were stained with PE-Cy5.5 hamster anti-mouse CD11c mAb (Caltag Laboratories) and one of the following Ab: FITC anti-mouse CD80 (B7-1) (16-10A1), FITC anti-mouse CD86 (B7-2) (GL1), FITC anti-mouse CD40 (3/23), and FITC anti-mouse I-Ab (Aβb) (AF6-120.1). PE-Cy 5.5 hamster IgG (Caltag Laboratories), FITC hamster IgG2,k isotype control (anti-KLH), FITC hamster IgG2a, FITC mouse IgG2a (G155-178) isotype controls were used to determine background staining.
For T cell proliferation studies, cells were stained with PE mouse anti-mouse DO11.10 TCR mAb (clone KJ1-26; Caltag Laboratories). PE-conjugated mouse IgG2a isotype control was used to determine background staining (Caltag Laboratories).
Flow cytometry data acquisition was performed on a FACScan running CellQuest software (BD Biosciences). FlowJo software (Tree Star) was used for data analysis. For lung and migration studies, 50,000 total events were acquired for each sample. For CCR7 expression and T cell proliferation studies, 500,000 total events were acquired for each sample. Dead cells were gated out based on light scatter properties.
Data acquisition for experiments assessing costimulatory molecule expression in lymph nodes was performed on a DakoCytomation High Speed MoFlo Sorter running Summit 3.1 software, acquiring 500,000 events for each sample.
In vivo T cell proliferation studies
CD4+ T cells were enriched from spleens of DO11.10 OVA-transgenic mice by magnetic bead separation under sterile conditions using a mixture of biotin-conjugated mAbs against CD8α (Ly-2) (rat IgG2a), CD11b (Mac-1) (rat IgG2b), CD45R (B220) (rat IgG2a), DX5 (rat IgM) and Ter-119 (rat IgG2b), followed by anti-biotin MicroBeads (colloidal superpara-magnetic MicroBeads conjugated to a monoclonal anti-biotin Ab, clone: Bio3-18E7.2; mouse IgG1) (Miltenyi Biotec). CD4+ DO11.10 T cells were subsequently labeled with 10 μM CFSE (Sigma-Aldrich) at 37°C for 10 min, as previously described (14) and then resuspended in sterile PBS. Mice received an i.v. injection of 10 × 106 CFSE-labeled DO11.10 T cells 24 h before i.t. injection of 600 μg of OVA in a volume of 60 μl of PBS. Four days later, T cell responses were analyzed in the draining TLNs by observing CFSE division profiles of live KJ1-26+CD4+ T cells.
In vitro T cell activation studies
To assess the ability of lung DCs to activate T cells, smoke-exposed mice and controls received an i.t. injection of 600 μg of OVA and 15 h later lungs were digested and the lung cell suspension was enriched for DC under sterile conditions by CD11c magnetic bead separation (clone N418; Miltenyi Biotec). The enriched cells were subsequently stained with PE hamster anti-mouse CD11c mAb (clone HL3; BD Pharmingen). Within the PE-CD11c+ population, lung DCs were identified as the low AF group and sorted by flow cytometry as described above (13). DCs were then cocultured with purified CD4+ DO11.10 TCR-transgenic T cells at a 1:100 ratio for 24 h at 37°C in tissue culture medium (RPMI 1640 supplemented with 10% FBS, 0.1% 2-ME (Invitrogen), 200 mM L-glutamine (Invitrogen), 1% penicillin/streptomycin (Invitrogen), and 1% HEPES (Sigma-Aldrich)). The induction of T cell activation by lung DCs derived from smoke-exposed mice and controls was determined by detection of IL-2 in the culture supernatants by ELISA as per manufacturer’s instructions (eBioscience).
For in vitro assessment of ability of lung-derived DCs to stimulate T cells, smoke-exposed mice and controls were euthanized by CO2 narcosis 24 h after i.t. injection of 600 μg of OVA-FITC and TLNs were extracted as described. Single-cell suspensions were obtained under sterile conditions and labeled with PE-Cy5.5 hamster anti-mouse CD11c mAb (Caltag Laboratories) as described. OVA-FITC+CD11c-PE-Cy5.5+ cells were sorted by flow cytometry and cocultured with purified CD4+ DO11.10 TCR-transgenic T cells at a 1:100 ratio for 24 h at 37°C. The induction of T cell activation by TLN-derived DCs obtained from smoke-exposed animals and controls was determined by detection of IL-2 in the coculture supernatants.
TLN cell culture
TLN cells were cultured in RPMI 1640 alone or with 40 μg of OVA/well at 8 × 105 cells/well in a flat-bottom, 96-well plate (BD Biosciences). After 4 days of culture, supernatants were harvested for cytokine measurement by commercial ELISA.
Data analysis
Data are expressed as means ± SEM. Statistical interpretation of the results is indicated in the figure legends. All statistical analysis was performed using SigmaStat statistical software (version 2.0; SPSS). Differences were considered statistically significant at p < 0.05.
Results
Cigarette smoke exposure decreases the number of lung DCs
We began by investigating the effect of cigarette smoke exposure on the number of lung DCs. Mice were exposed to cigarette smoke for 4 wk. Lung cells were isolated and flow cytometry was used to identify CD11c+ cells. Within the CD11c+ cell population, DCs were identified within the low AF gate as previously described (13) (Fig. 1A). Smoke-exposed mice showed a significant reduction in lung DCs compared with non-smoke-exposed controls. DCs represented 1.03% (±0.1) of the total lung cells in smoke-exposed mice compared with 1.57% (±0.1) in the controls ( p = 0.007), corresponding to a mean number of 568,889 (±68,124) lung DCs in smoke-exposed mice compared with 901,444 (±88,513) in the control group ( p = 0.01) (Fig. 1B). The number of total lung cells and lung macrophages (CD11c+/AFhigh) retrieved after lung digestion did not differ significantly between smoke-exposed and control mice (data not shown).
FIGURE 1.
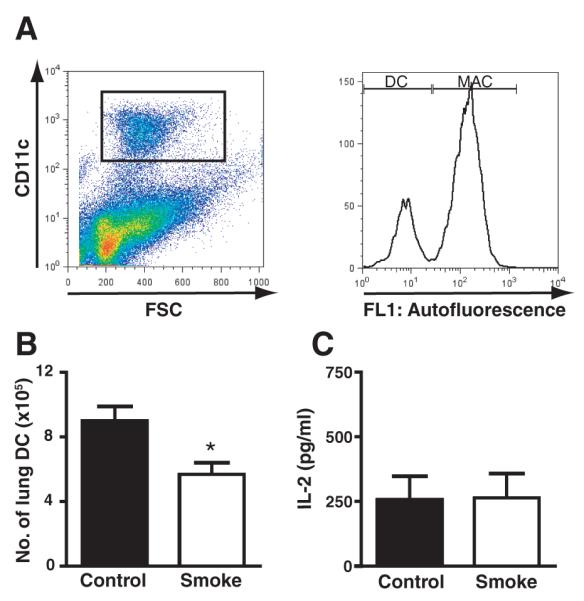
Cigarette smoke exposure decreases DC numbers in the lung. A, Lung cells were isolated from smoke-exposed and non-smoke-exposed control mice and flow cytometric analysis was used to identify DCs (CD11c+/AFlow). Macrophages (MAC) were identified as (CD11c+/AFhigh). B, Absolute numbers of lung DCs from A. Data represent means ± SEM; n = 8. Statistical analysis was performed using the Student t test; p < 0.05. C, CD11c+/AFlow lung DCs were isolated from smoke-exposed and control mice 15 h following OVA administration and cocultured with DO11.10 CD4+ T cells. IL-2 was measured in cell culture supernatants by ELISA.
To determine whether the reduction in DC number induced by smoke exposure had functional consequences, we tested the ability of residual lung DCs to act as stimulators of T cell proliferation in vitro. Lung DCs (CD11c+/AFlow) were sorted from smoke-exposed and control mice 15 h following i.t. injection of OVA and cocultured for 24 h with CD4+ DO11.10 cells (1:100). IL-2 levels were measured in cell culture supernatants by ELISA. On a per cell basis, IL-2 production by T cells did not differ significantly between smoke-exposed (264.7 ± 93.5 pg/ml) and control (257.1 ± 91.4 pg/ml) mice (Fig. 1C).
Cigarette smoke exposure does not inhibit migration of lung DCs to TLNs
We next determined whether cigarette smoke influences lung DC migration to the regional TLNs. TLN cells from smokers and controls were isolated 24 h after intratracheal injection of OVA-FITC and stained with an anti-CD11c mAb. Lung DCs were identified as described, and recently migrating, lung-derived DCs were defined as the FITC+ population (shown gated in Fig. 2A). Similar percentages of lung-derived nodal CD11c+ cells were observed in both smoke-exposed and control mice (16 ± 2% vs 14 ± 1.5%, respectively). Calculation of absolute numbers of recently lung-derived DCs in the TLNs demonstrated that the mean number of CD11c+FITC+ cells in the TLNs did not differ significantly between smokers and controls (14,541 ± 4,590 vs 10,535 ± 1,774, respectively) (Fig. 2B). Furthermore, the total number of TLN cells isolated following OVA-FITC administration was similar between the two groups (data not shown).
FIGURE 2.

Migration of lung DCs to the regional TLNs is unaffected by cigarette smoke. TLN cells were isolated from smoke-exposed and control mice 24 h following intratracheal administration of FITC-labeled OVA and flow cytometry was used to identify (A) recently migrated, lung-derived DC (CD11c+/FITC+). Shown are density plots from one representative mouse per group. B, Absolute numbers of CD11c+/FITC+ DCs from A. Data represent means ± SEM; n = 8. C, CCR-7 expression was identified within the CD11chigh/MHCIIhigh TLN DC population. A representative histogram from one of five mice is shown. The isotype control is denoted beneath the gray histogram.
The chemokine receptor CCR7 is thought to regulate migration of peripheral DCs to lymph nodes (15–17). Recently migrated (OVA-FITC+) DCs (CD11chighMHC class II (MHCII)high) express higher levels of CCR7 than the semimature/steady-state nodal DCs (OVA-FITC− and CD11chigh/intMHCint) as previously described (18). To assess CCR7 expression by migrated lung DCs in response to cigarette smoking, TLNs were extracted from smoke-exposed and control mice 24 h after i.t. injection of OVA, and the cells were stained with anti-MHCII (I-Ek), anti-CCR7, and anti-CD11c mAb. CCR7 expression in the CD11chighMHCIIhigh population did not differ between smoke-exposed and control cells (average mean fluorescence intensity 493 vs 512, respectively) (Fig. 2C).
Cigarette smoke exposure impairs DC maturation
We next determined whether cigarette smoke exposure altered the maturation of TLN DCs following OVA exposure by assessing cell surface costimulatory molecule expression. TLNs from smoke-exposed and control mice were collected 24 h after i.t. injection of OVA. DCs were analyzed for costimulatory markers by two-color flow cytometry. This was done by pairing the labeling of CD80, CD86, MHCII, or CD40 with CD11c. A total of 5 × 105 total events were collected from the TLN cell suspension for each mouse. Cells were gated on CD11c+ and histograms for the second marker are shown in Fig. 3A. In the presence of smoke exposure, a lower percentage of CD11c+ cells expressed CD80, CD86, and MHCII hours after exposure to i.t. OVA than in control cells. The absolute number of CD11c+/CD80+ cells, CD11c+/CD86+ cells, and CD11c+/MHCIIhigh cells in the TLNs was also significantly reduced following smoke exposure (4,173 ± 885 CD11c+/ CD80+ cells in smokers vs 8,785 ± 1,747 in controls; 2,254 ± 656 CD11c+/CD86+ cells in smokers vs 6,097 ± 2,362 in controls; 1,878 ± 387 CD11c+/MHCIIhigh cells in smokers vs 3,359 ± 572 in controls; p < 0.05, six to eight mice per group for each costimulatory marker). No difference was observed for CD40 (5,730 ± 1,445 cells in smokers vs 6,228 ± 1,802 in controls) (Fig. 3B). The number of total TLN cells before and after OVA administration was not different between smoke-exposed and control mice (data not shown).
FIGURE 3.

Cigarette smoke exposure impairs DC maturation. A, TLN cells were isolated from smoke-exposed and control mice 24 h following intratracheal administration of FITC-labeled OVA and flow cytometry was used to identify costimulatory molecule expression on DCs. Cells were gated through CD11c and then evaluated for the distribution of CD80, CD86, CD40, and MHCII. Smokers are represented by gray histograms; black histograms show control mice. White histograms represent isotype staining controls. One representative histogram of eight is shown. B, Absolute numbers of CD11c+ cells expressing each costimulatory marker from A is shown. Data represent means ± SEM; n = 6 – 8. Statistical analysis was performed using the Student t test; p < 0.05.
Cigarette smoke exposure impairs T cell proliferation
To investigate the impact of cigarette smoke-induced impairment of DC maturation on T cell responses, we next assessed T cell-derived cytokine production by TLN cells in vitro. Mice were exposed to cigarette smoke for 4 wk. During the last week of exposure, mice received a single i.v. delivery of DO11.10 T cells, followed 24 h later by intratracheal administration of 600 μg of OVA. Four days later, TLN cells were isolated and placed in culture. OVA-specific production of IL-5, IL-10, IL-13, and IFN-γ was assessed in cell culture supernatants by ELISA. We observed similar levels of OVA-specific IL-5, IL-10, and IL-13 in both control and cigarette smoke-exposed mice (Table I). IFN-γ was not detected in any of the culture conditions (data not shown).
Table I.
In vitro OVA-specific cytokine productiona
| Control | Smoke | |
|---|---|---|
| IL-5 | ||
| Medium | 23.3 ± 17.5 | 39.7 ± 23.2 |
| OVA | 746.7 ± 69.8b | 549.1± 95.1b |
| IL-10 | ||
| Medium | 41.7 ± 16.0 | 51.0 ± 16.0 |
| OVA | 723.6 ± 96.9b | 643.9 ± 69.3b |
| IL-13 | ||
| Medium | 277.6 ± 147.4 | 392.7 ± 109.8 |
| OVA | 8880.7 ± 1058.5b | 9483.5 ± 1425.5b |
Smoke-exposed and control mice were injected i.v. with DO11.10T cells during the last week of exposure, followed 24 h later by intratracheal administration of 600μg of OVA. Four days later, TLN cells were isolated and placed in culture in the presence of either medium alone or OVA. Cytokine production was assessed in cell culture supernatants by ELISA. Data are expressed as means ± SEM (n = 9–11).Statistical analysis was performed using one-way ANOVA; p < 0.05.
Statistically significant compared to medium alone.
IL-2 is instrumental in promoting the clonal expansion of Agspecific T cells. To better understand the effect of smoking on DC-induced T cell proliferative responses, mice were exposed to cigarette smoke for 4 wk, followed by a single i.t. administration of FITC-labeled OVA. Twenty-four hours later, CD11c+/ OVA+ DCs were isolated from the TLNs and cocultured with CD4+ DO11.10 T cells. IL-2 production was measured in culture supernatants by ELISA. DCs from smoke-exposed mice induced significantly less IL-2 production from CD4 T cells than controls (434 ± 23 pg/ml vs 590 ± 19 pg/ml, respectively) (Fig. 4A).
FIGURE 4.

Cigarette smoke exposure inhibits lung-derived DC induced T cell proliferation in vitro and in vivo. A, Newly migrated CD11c+/OVA+ lung-derived DCs were isolated from the TLNs of smoke-exposed and control mice 24 h following intratracheal injection of FITC-labeled OVA and cocultured with DO11.10 CD4+ T cells. IL-2 levels were measured in culture supernatants by ELISA. Data represent means ± SEM of triplicate culture wells, each assayed in duplicate. Statistical analysis was performed using the Student t test; p < 0.05. B, Smoke-exposed and control mice were injected i.v. with CFSE-labeled DO11.10 T cells during the last week of exposure, followed 24 h later by intratracheal administration of 600 μg of OVA. Four days later, TLN cells were isolated and flow cytometry was used to assess the CFSE-staining profile of proliferating T cells. D denotes cell division cycle. C, The percentage of DO11.10 CD4 T cells in each cell division cycle was determined. Data represent means ± SEM; n = 6. One representative experiment of three is shown. Statistical analysis was performed using the Student t test; p < 0.05. D, Absolute numbers of DO11.10 CD4 T cells in the TLNs. Data represent means ± SEM; n = 9–11. Statistical analysis was performed using the Student t test; p < 0.05.
To assess the effect of smoking on T cell proliferation in vivo, mice were exposed to cigarette smoke for 4 wk. During the last week of exposure mice received a single i.v. delivery of CFSE-labeled DO11.10 T cells, followed 24 h later by intratracheal administration of 600 μg of OVA. Four days later, TLN cells were isolated and flow cytometry was used to assess the CFSE staining profile of proliferating T cells. Shown in Fig. 4B is a representative histogram (from one of three experiments, all with similar results, six mice per experimental group) illustrating CFSE division profiles of T cells from smoke-exposed and control animals. We demonstrate that compared with controls, cigarette smoke exposure attenuated proliferation of DO11.10 T cells (Fig. 4B). Specifically, smoking resulted in a significantly lower percentage of DO11.10 T cells undergoing six or more cycles of cell division compared with controls (Fig. 4C). To determine the consequence of impaired T cell proliferation, we quantified the absolute number of OVA-specific T cells in these animals and show that compared with controls, smoking was associated with the expansion of significantly fewer D011.10 T cells in the TLNs (Fig. 4D). The observed differences were not due to an effect of smoking on the trafficking of adoptively transferred cells to the TLNs because in the absence of OVA exposure, similar numbers of DO11.10 T cells migrated to the TLNs in both smokers and control animals (data not shown).
Discussion
The objective of this study was to determine the effects of cigarette smoke exposure on DC function in vivo. We demonstrate that while cigarette smoke exposure decreased the number of DCs in the lungs of mice, Ag-induced DC migration to the regional TLNs was unaffected. However, cigarette smoking suppressed DC maturation within the lymph nodes as demonstrated by reduced cell surface expression of MHC and the costimulatory molecules CD80 and CD86. Consequently, DCs from cigarette smoke-exposed animals had a diminished capacity to induce IL-2 production by T cells that was associated with diminished Ag-specific T cell proliferation in vivo.
Previous studies have shown that constituents within cigarette smoke impact DC function in vitro. Exposure of human monocyte-derived DCs and bone marrow-derived murine DCs to different doses of nicotine, for example, have led to conflicting reports; DC activation in one instance (8), but suppressed DC function in another (7). Other compounds within cigarette smoke may also have immunomodulatory potential. Indeed, cigarette smoke extract which, in addition to nicotine, contains many of the other substances that are found in cigarette smoke, inhibits DC function in a manner that is only partly dependent on nicotine (9). By exposing mice to mainstream tobacco smoke, we collectively assessed the impact of the >4000 constituents on the immune system. Furthermore, in vivo modeling of the DC response to OVA afforded the opportunity to recapitulate the complex cellular interactions between DCs and the surrounding tissue environment that accompanies the induction of immune responses.
We demonstrate that cigarette smoke exposure decreased the number of DCs in the lung. Our study is in agreement with a previous report demonstrating that 2–4 mo of smoke exposure significantly decreased the number of pulmonary CD11chigh MHCII+ DCs (19). In contrast, D’Hulst and colleagues (20) showed that cigarette smoke induced acute lung inflammation that was associated with increased numbers of DCs as early as 3 days following exposure that persisted for up to 6 mo. In the former study, a nose-only exposure system was used where the mainstream cigarette smoke from 2 cigarettes was delivered daily while in the latter mice were exposed whole body to the smoke from 20 cigarettes per day. Hence, differences between the two studies may be related to the amount of cigarette smoke delivered. Indeed, serum carboxyhemoglobin levels were ~3.6-fold increased in smokers over controls in the nose-only exposure system and ~8.3-fold increased in smokers in the whole body exposure system (21, 22). Furthermore, high-dose smoke exposure in the D’Hulst study (20) was associated with an inflammatory response suggestive of acute lung injury, a phenomenon less akin to the effects of chronic cigarette smoke exposure on humans.
To our knowledge, this is the first study to investigate the effect of cigarette smoke exposure on DC trafficking from the lung to the regional lymph nodes where they exert their action on T lymphocytes. We demonstrate that OVA-containing DCs from smoke-exposed and control mice were equally able to migrate from the lungs to the TLNs (Fig. 2). Moreover, smoking had no effect on DC cell surface expression of CCR7, an important chemokine receptor involved in the migration of DCs into lymphoid tissues (15–17). Thus, it is unlikely the decreased number of lung DCs associated with smoking is the result of enhanced migration to regional lymph nodes. More likely the half-life of lung DCs is diminished with smoke exposure, however, reduced numbers of monocyte precursors or impaired monocyte→DC differentiation may also account for the decreased number of lung DCs associated with smoking.
Although cigarette smoke did not impact DC trafficking from the lung to the lymph nodes, DC maturation was affected. Twenty-four hours following OVA administration, the number of CD11c+ cells expressing the maturation markers CD80 (B7-1), CD86 (B7-2), and MHCII were significantly reduced in the TLNs of smoke-exposed mice. Interestingly, CD40 expression was not modified by smoke exposure. The CD40-CD40L pathway has been implicated in the production of matrix metalloproteinases (23–25) therefore cigarette smoke could potentially impair DC maturation while promoting matrix metalloproteinase production and tissue destruction, important processes in the pathogenesis of COPD.
The diminished DC maturation status associated with smoking could be the result of increased numbers of T regulatory cells (Tregs) in the draining lymph nodes. Indeed, Tregs have been demonstrated to suppress the up-regulation of CD80 and CD86 on DCs in vitro (26). To address this, we assessed the number of Foxp3+ Tregs in the TLNs of smoke-exposed animals. We did not observe any difference in the number of OVA-specific T regulatory cells in the TLNs of smoke-exposed mice vs controls (data not shown). Therefore, it is unlikely that Tregs influenced the maturation status of DCs in our experimental system.
Cigarette smoke-induced inhibition of DC maturation should manifest itself in functional impairment of T cell activation. Within the TLNs, we observed a decreased ability of smoke-exposed DCs to induce Ag-specific T cell proliferation. Engagement of the TCR with the peptide/MHC complex and B7/CD28 costimulation is necessary to promote T cell survival, enhance production and stabilization of IL-2, and facilitate T cell-cycle progression (27). We compared the ability of TLN lung-derived DCs from smokers and controls to stimulate OVA-specific DO11.10 CD4+ T cell proliferation in vitro and in vivo. OVA-loaded DCs obtained from the lungs of both non-smoke-exposed and smokers displayed similarly modest ability to induce IL-2 production by DO11.10 CD4+ T cells. Upon transit to the regional lymph nodes, however, control DCs acquired a significantly greater capacity to stimulate proliferation of CD4+ T cells than smoke-exposed DCs.
We observed that on a per cell basis, smoking had no effect on the capacity of lung-derived TLN DCs to induce T cell cytokine production. D011.10 CD4+ T cells from control and cigarette smoke-exposed mice produced similar levels of OVA-specific IL-5, IL-10, and IL-13 following restimulation in vitro. Collectively, our findings suggest a specific effect for smoking on T cell responses; execution of the T cell effector program remains intact, but diminished clonal expansion in the TLNs means that fewer cells are available to perform these duties.
Diminished CD4+ T cell proliferation that results from impaired lung DC maturation caused by smoking may have important consequences for CD4+ T cell function with respect to immunity to microbes, allergens, and in tumor surveillance. CD4+ T cells control virtually all adaptive immune responses to protein Ags and only a few cell types that express MHCII molecules can function as APCs for CD4+ T cells, the most effective being DCs. Hence, the defects in DC function induced by smoking may impair CD4+ T cell-mediated adaptive immunity. These findings may also help explain the observation that mice exposed to cigarette smoke have impaired immune responsiveness to adenovirus (19).
In a broader sense, impaired DC function may contribute to the development of chronic bronchitis in smokers and exacerbations of COPD that are usually mediated by respiratory infections. It is also possible that impaired DC function induced by cigarette smoking may have implications for tumor surveillance. In fact, there is evidence to suggest that COPD is associated with an increased incidence of lung cancer (28). Although it is recognized that the main mechanism of tumor immunity is killing of tumor cells by CD8+ cytotoxic lymphocytes (CTL), the role of CD4+ cells in cancer immunity is less clear. However, CD4+ cells provide additional signals for the differentiation of naive CD8+ T cells into functional CTLs, thus impaired CD4+ T cell function induced by impaired DC function might interfere with CD8+ T cell function. Furthermore, DCs can be involved in cross-presentation of tumor Ags to CD8+ T cells.
In conclusion, we provide evidence that cigarette smoke exposure causes specific defects in DC maturation and suppresses the proliferation of CD4+ T cells in thoracic regional lymph nodes in mice. These studies link the effects of cigarette smoke on DC function to the known consequences of cigarette smoke on immunity and lung disease.
Acknowledgments
We thank Richard J. Riese and Sara Tomassetti for their involvement with the project and Laura Prickett for flow cytometry assistance.
Footnotes
This work was supported by National Institutes of Health Grants HL029594 and HL054853.
- DC
- dendritic cell
- TLN
- thoracic lymph node
- i.t.
- intratracheal
- AF
- autofluorescence
- MHCII
- MHC class II
- Treg
- T regulatory cell
Disclosures The authors have no financial conflict of interest.
References
- 1.Villadangos JA, Heath WR. Life cycle, migration and antigen presenting functions of spleen and lymph node dendritic cells: limitations of the Langerhans cells paradigm. Semin. Immunol. 2005;17:262–272. doi: 10.1016/j.smim.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Lipscomb MF, Masten BJ. Dendritic cells: immune regulators in health and disease. Physiol. Rev. 2002;82:97–130. doi: 10.1152/physrev.00023.2001. [DOI] [PubMed] [Google Scholar]
- 4.Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am. J. Respir. Crit. Care Med. 2005;172:530–551. doi: 10.1164/rccm.200410-1384SO. [DOI] [PubMed] [Google Scholar]
- 5.Holt PG, Keast D. Environmentally induced changes in immuno-logical function: acute and chronic effects of inhalation of tobacco smoke and other atmospheric contaminants in man and experimental animals. Bacteriol. Rev. 1977;41:205–216. doi: 10.1128/br.41.1.205-216.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sopori M. Effects of cigarette smoke on the immune system. Nat. Rev. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 7.Nouri-Shirazi M, Guinet E. Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology. 2003;109:365–373. doi: 10.1046/j.1365-2567.2003.01655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aicher A, Heeschen C, Mohaupt M, Cooke JP, Zeiher AM, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003;107:604–611. doi: 10.1161/01.cir.0000047279.42427.6d. [DOI] [PubMed] [Google Scholar]
- 9.Vassallo R, Tamada K, Lau JS, Kroening PR, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J. Immunol. 2005;175:2684–2691. doi: 10.4049/jimmunol.175.4.2684. [DOI] [PubMed] [Google Scholar]
- 10.Sousa C. Reis e. Dendritic cells in a mature age. Nat. Rev. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 11.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 12.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170–177. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 14.Lyons AB, Hasbold J, Hodgkin PD. Flow cytometric analysis of cell division history using dilution of carboxyfluorescein diacetate succinimidyl ester, a stably integrated fluorescent probe. Methods Cell Biol. 2001;63:375–398. doi: 10.1016/s0091-679x(01)63021-8. [DOI] [PubMed] [Google Scholar]
- 15.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 16.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- 17.Fainaru O, Shseyov D, Hantisteanu S, Groner Y. Accelerated chemokine receptor 7-mediated dendritic cell migration in Runx3 knockout mice and the spontaneous development of asthma-like disease. Proc. Natl. Acad. Sci. USA. 2005;102:10598–10603. doi: 10.1073/pnas.0504787102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsland BJ, Battig P, Bauer M, Ruedl C, Lassing U, Beerli RR, Dietmeier K, Ivanova L, Pfister T, Vogt L, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. 2005;22:493–505. doi: 10.1016/j.immuni.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Robbins CS, Dawe DE, Goncharova SI, Pouladi MA, Drannik AG, Swirski FK, Cox G, Stampfli MR. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. Am. J. Respir. Cell Mol. Biol. 2004;30:202–211. doi: 10.1165/rcmb.2003-0259OC. [DOI] [PubMed] [Google Scholar]
- 20.D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur. Respir. J. 2005;26:204–213. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- 21.Gaschler GJ, Zavitz CC, Bauer CM, Skrtic M, Lindahl M, Robbins CS, Chen B, Stampfli MR. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2008;38:218–226. doi: 10.1165/rcmb.2007-0053OC. [DOI] [PubMed] [Google Scholar]
- 22.Bracke KR, D’Hulst I, Maes AT, Demedts IK, Moerloose KB, Kuziel WA, Joos GF, Brusselle GG. Cigarette smoke-induced pulmonary inflammation, but not airway remodelling, is attenuated in chemokine receptor 5-deficient mice. Clin. Exp. Allergy. 2007;37:1467–1479. doi: 10.1111/j.1365-2222.2007.02808.x. [DOI] [PubMed] [Google Scholar]
- 23.Gotoh H, Kawaguchi Y, Harigai M, Hara M, Saito S, Yamaguchi T, Shimada K, Kawamoto M, Tomatsu T, Kamatani N. Increased CD40 expression on articular chondrocytes from patients with rheumatoid arthritis: contribution to production of cytokines and matrix metalloproteinases. J. Rheumatol. 2004;31:1506–1512. [PubMed] [Google Scholar]
- 24.Nagashima H, Aoka Y, Sakomura Y, Uto K, Sakuta A, Aomi S, Kurosawa H, Hagiwara N, Kawana M, Kasanuki H. Matrix metal-loproteinase 2 is suppressed by trapidil, a CD40-CD40 ligand pathway inhibitor, in human abdominal aortic aneurysm wall. J. Vasc. Surg. 2004;39:447–453. doi: 10.1016/j.jvs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Malik N, Greenfield BW, Wahl AF, Kiener PA. Activation of human monocytes through CD40 induces matrix metalloproteinases. J. Immunol. 1996;156:3952–3960. [PubMed] [Google Scholar]
- 26.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Keir ME, Sharpe AH. The B7/CD28 costimulatory family in autoimmunity. Immunol. Rev. 2005;204:128–143. doi: 10.1111/j.0105-2896.2005.00242.x. [DOI] [PubMed] [Google Scholar]
- 28.Skillrud DM, Offord KP, Miller RD. Higher risk of lung cancer in chronic obstructive pulmonary disease: a prospective, matched, controlled study. Ann. Intern. Med. 1986;105:503–507. doi: 10.7326/0003-4819-105-4-503. [DOI] [PubMed] [Google Scholar]