

Abstract
The oxidative addition of PhX (X = I, Br, Cl) to the complexes Pd(PtBu3)2 (1), Pd(1-AdPtBu2)2 (2), Pd(CyPtBu2)2 (3), and Pd(PCy3)2 (4) were studied to determine the effect of steric properties on the coordination number of the species that undergoes oxidative addition and to determine if the type of halide affects the identity of this species. The kinetic data imply that the number of phosphines coordinated to the complex that reacts in the irreversible step of the oxidative addition processes for complexes 1-4 depends on the halide more than the steric properties of the ligands. The rate-limiting step of the oxidative addition of PhI occurred with L2Pd(0) in all cases, as determined by the lack of dependence of kobs on [PtBu3], [1-AdPtBu2], and [CyPtBu2] and the inverse dependence of the rate constant on [PCy3] when the reaction is initiated with Pd(PCy3)3. The irreversible step of the oxidative addition of PhCl occurred with a monophosphine species in each case, as signaled by an inverse dependence of the rate constant on the concentration of ligand. The irreversible step of the oxidative addition of PhBr occurred with a bisphosphine species, as signaled by the zeroth-order or small dependence of the rate constant on the concentration of phosphine. Thus, the additions of the less reactive chloroarenes occur through lower-coordinate intermediates than additions of the more reactive haloarenes.
INTRODUCTION
The oxidative addition of haloarenes to palladium(0) complexes is a fundamental organometallic reaction.1 It constitutes the first step in palladium-catalyzed reactions of haloarenes, such as aromatic amination,2-4 Heck,5,6 Suzuki,7-10 and Stille11,12 couplings. Many of these oxidative additions occur to phosphine-ligated Pd(0) species. Some of the most active catalysts for these reactions involve hindered alkylphosphine ligands that form bis-phosphine complexes of Pd(0).13-17 Because of the high activity of these catalysts, the mechanism of the oxidative addition to these bisphosphine complexes is important to determine. Moreover, it would be valuable to reveal the relationships between the reactivity of the isolated L2Pd(0) species in which L is a hindered trialkylphosphine and the Pd(0) reactive intermediates in which L is PPh3.18,19
The oxidative addition of haloarenes to L2Pd(0) complexes in which L is a trialkylphosphine could occur directly to the bisphosphine starting complex to form a four-coordinate product, or it could occur to a monophosphine intermediate20,21 that would form a three-coordinate arylpalladium halide complex22,23 as the immediate product. Previous studies have shown that the coordination number of the palladium species that undergoes oxidative addition and the structure of the complexes produced by oxidative addition are different for reactions of complexes containing various ligands.18,19,24-32,33,34-36
A majority of these studies have been conducted on complexes containing monophosphine ligands.18,19,24-29 The mechanism appears to depend on the steric and electronic properties of the ligand. For example, classic studies on the addition of ArI to Pd(PPh3)4 showed that this reaction occurs through the 14-electron intermediate Pd(PPh3)2 to produce a four-coordinate arylpalladium halide complex.18,19 In contrast, more recent studies on the oxidative addition of PhBr to Pd(P(o-Tol)3)2 implied that this addition occurred to a monophosphine intermediate to form a dimeric arylpalladium bromide complex containing a single phosphine per metal center.20 Addition of ArI to a eries of trialkylphosphine palladium complexes of the general formula Pd(CynPtBu3-n)2 (n = 0–3) has also been conducted. These authors concluded that complexes containing the bulkier phosphines (n = 0, 1) underwent addition of ArI after dissociation of ligand to form a monophosphine intermediate and that complexes containing the smaller phosphines (n = 2, 3) reacted through an associative pathway.27 The results of studies on reactions of PPh3 complexes in the presence of anions have also been published. For example, the 16-electron anionic [Pd(PPh3)2(OAc)]− generated in situ from Pd(OAc)2 and PPh3 is proposed to be the species that adds haloarenes when the reaction is conducted in the presence of acetate.25,26
Studies on oxidative addition to complexes of bidentate ligands have also been conducted. The oxidative addition of aryl bromides to [Pd(bisphosphine)2] (bisphosphine = 1,1’-bis(diphenylphosphino)ferrocene (DPPF), 2,2"-bis(diphenylphosphino-1,1’-binaphthyl) (BINAP)) occurred predominantly to Pd(bisphosphine), with a second pathway appearing to involve addition to [Pd(κ2-bidentate)(κ1-bidentate)].31,32 Prior studies on the oxidative addition of ArCl to Pd(dippp)2 (dippp=bis(diisopropylphosphino)propane) showed that this reaction occurred to the 14-electron Pd(dippp) intermediate to form cis-(dippp)Pd(Ph)(Cl) as the main product. In the presence of free phosphine, this complex equilibrates with trans-(κ1-dippp)2Pd(Ph)(Cl).28
The identity of the halide in the haloarene could affect the mechanism of oxidative addition for a given ligand. Recently, we reported in communication form that the oxidative addition of iodo-, bromo-, and chlorobenzene to the Pd(0) complexes of Q-phos (Q-phos = pentaphenylferrocenyl di-tert-butylphosphine) occurs with three different kinetic behaviors.21 Addition of PhI occurred by irreversible associative displacement of a phosphine; addition of PhBr occurred by rate-limiting dissociation of phosphine; and addition of PhCl occurred by reversible dissociation of phosphine, followed by rate-limiting oxidative addition.21
The diversity of these results from reactions of several complexes with various haloarenes under different reaction conditions illustrates the need for a systematic study of the factors that control the coordination number of the Pd(0) species that adds the haloarene. Thus, we have conducted such a study of the oxidative addition of iodo-, bromo-, and chlorobenzene to complexes of alkyl phosphines of varied size. This study has produced data that begin to clarify the effects of the halide in the haloarene and the effects of the size of the alkyl phosphine on the reaction mechanism. In brief, the number of phosphines coordinated to the complex that reacts in the irreversible step is more dependent on the identity of the halide than on the size of the cyclohexyl, tert-butyl, and 1-adamantyl phosphine ligands in this study.
RESULTS
1. Oxidative Additions Included in this Study and Characterization of Reaction Products
The oxidative addition of PhX (X = I, Br, or Cl) to PdL2 complexes 1-4 containing the trialkylphosphines PtBu3, 1-AdPtBu2, CyPtBu2, and PCy3 were studied. Scheme 1 summarizes the Pd(0) complexes 1–4 and their reactions to form the arylpalladium halide products 5–19. Although kinetic studies on the oxidative addition reactions show that the mechanism depends predominantly on the identity of the halide, the identity of the reaction products depended most strongly on the steric properties of the ligand. Thus, this section that describes the reactions and characterization of the products is organized by type of ligand.
Scheme 1.
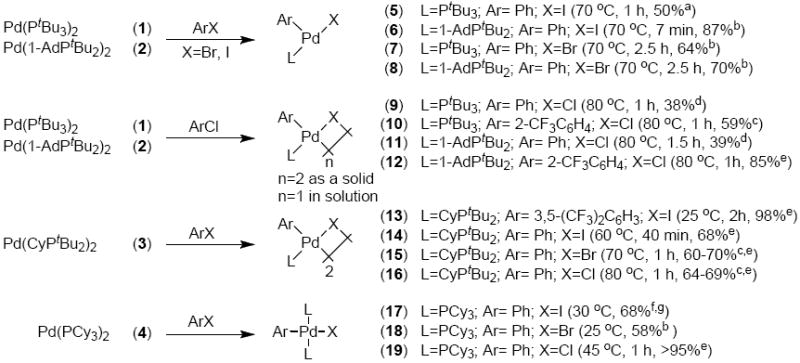
Complexes formed by oxidative addition of different ArX to L2Pd(0).
a see Ref. 22. b see Ref. 23. c see Supporting Information. d yield of product formed in situ, as determined by 31P NMR spectroscopy, at 50% conversion. The yield decreased at longer reaction times. e yield of product formed in situ, as determined by 31P NMR spectroscopy. f see Ref. 27. g see Ref. 39.
1.1. Additions of ArX to complexes of PtBu3 and 1-AdPtBu2
The reactions of PhI and PhBr with the Pd(0) complexes Pd(PtBu3)2 (1) and Pd(1-AdPtBu2)2 (2) has been shown to produce the known three-coordinate complexes 5–8 (Scheme 1) in 50 to 87% yield.22,23 These complexes have been shown previously to be stabilized by a weak agostic interaction of the metal with a phosphine C–H bond positioned at the open coordination site.
In the current work, the reaction of 1 with 1-chloro-2-trifluoromethylbenzene occurred at 80 °C in 1 h to produce the stable [(PtBu3)Pd(2-CF3C6H4)(Cl)]2 (10) in 59% isolated yield (Scheme 1). Reaction of 2 with 1-chloro-2-trifluoromethylbenzene occurred at 80 °C in 1 h to produce [(1-AdPtBu2)Pd(2-CF3C6H4)(Cl)]2 (12) in 85% yield as determined by 31P NMR spectroscopy. Complex 12 was isolated in 39% yield after reaction for 20 min at 100 °C and subsequent recrystallization. The reaction of 1 and 2 in neat PhCl at 80 °C formed [LPd(Ph)(Cl)]2 (9, L = PtBu3 and 11, L = 1-AdPtBu2), but the yield of the oxidative addition product at full conversion was low due to decomposition of the oxidative addition product. The yield of the oxidative addition products at 50% conversion was high (about 80% with respect to the amount of reacted Pd(0) species), but at higher conversions the arylpalladium halide products decayed.
Thus, phenylpalladium chloride complexes 9 and 11 were characterized after preparing them independently (Scheme 2). PtBu3-ligated complex 9 was isolated in 42% yield from the reaction of (Py)2Pd(Ph)(Cl) (20) with PtBu3 in toluene solvent under dynamic vacuum to evaporate the liberated pyridine. 1-AdPtBu2-ligated 11 was isolated in 86% yield from the reaction of N(octyl)4Cl with the known (1-AdPtBu2)Pd(Ph)(CF3SO3),37 which was obtained by the previously reported reaction of (1-AdPtBu2)Pd(Ph)(Br) (8) with AgOTf.23
Scheme 2.
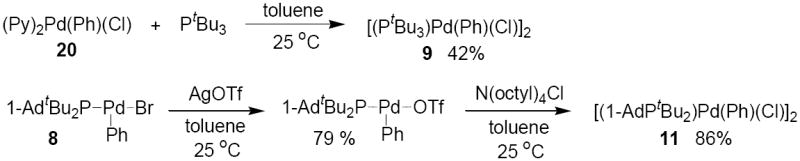
Routes for the synthesis of chloride complexes [(PtBu3)Pd(Ph)(Cl)]2 (9) and [(1-AdPtBu2)Pd(Ph)(Cl)]2 (11).
The nuclearity of the arylpalladium chloride complexes 10 and 11 depended on the phase. Our data indicate that both complexes are dimeric in the solid state and contain two bridging chlorides but are predominantly monomeric in solution. The solid-state structures of PtBu3-ligated, 2-CF3C6H4-complex 10 and 1-AdPtBu2-ligated, phenyl-complex 11 are shown in Figure 1. In this state, complex 10 contains two palladium atoms that are bridged nearly symmetrically by two μ2-chloride ligands. The two aryl groups are located anti to each other. The Pd–Cl bond distances range from 2.39 to 2.49 Å, and the two palladium atoms are separated by 3.61 Å. The Pd coordination planes intersect at an angle of 145.6°. The structure of 11 also contains two palladium atoms that are bridged by two μ2-chloride ligands, but the two aryl groups are syn to each other. In this case, the Pd–Cl(1) distances are shorter than the Pd–Cl(2) distances by about 0.1 Å, possibly caused by the steric congestion of the phosphines and the larger trans influence of the aryl groups located trans to Cl(1) than of the phosphines located trans to Cl(2). The two palladium atoms are separated by a longer distance of 3.78 Å, and the two coordination planes intersect at an angle of 166.3°.
Figure 1.

ORTEP diagrams of the complexes [(PtBu3)Pd(2-CF3C6H4)(Cl)]2 (10) and [(1-AdPtBu2)Pd(Ph)(Cl)]2 (11).
Solution molecular weight and NMR spectroscopic data indicate that the tri-tert-alkylphosphine-ligated arylpalladium chloride complexes 9-11 are monomeric in solution. The molecular weight measurements by the Signer method38 on the more stable complex 10, provided measurements of 530 g/mol in THF and 505 g/mol in benzene. These values are closer to the calculated molecular weight of the monomer (489.29 g/mol) than to that of the dimer (978.29 g/mol). The 31P NMR chemical shift of the arylpalladium chloride complexes 9-11 were similar, and ranged from 69–73 ppm. These chemical shifts are expected for a monomeric species, based on the chemical shifts of the monomeric arylpalladium bromide (60–65 ppm) and iodide (55–60 ppm) complexes. Thus, the spectroscopic and solution molecular weight data together provide evidence that 9 and 11 possess the same monomeric structure shown clearly for 10 in solution. The nuclearity of these species, however, do not affect our conclusions about the mechanism of oxidative addition because the steps that control a monomer-dimer equilibrium occur after the rate-limiting steps.
1.2. Additions of ArX to complexes of the less hindered CyPtBu2 and PCy3
The reactions of Pd(CyPtBu2)2 (3) with 3,5-(CF3)2C6H3I and PhI at 25 and 60 °C, respectively, produced the dimeric arylpalladium halide complexes [(CyPtBu2)Pd(Ar)(I)]2 (Ar = 3,5-(CF3)2C6H3, 13; Ar = Ph, 14)27 in 98% and 68% yields, as determined by 31P NMR spectroscopy. Complex 3 reacted with PhBr and PhCl over 1 h at 70 and 80 °C to produce the analogous bromide (15) and chloride (16) dimers in 70% and 64% yield. The complex Pd(PCy3)2 (4) reacted with PhI, PhBr, and PhCl at 25 °C to 45 °C to produce the known stable trans four-coordinate complexes 1727,39, 1823 and 19.40,41 These reactions occurred in high yield, as determined by 31P NMR spectroscopy; isolated yields depended on the solubility and crystallinity of the products.
The nuclearity of CyPtBu2-ligated 15 was determined by X-ray crystallography (Figure 2) and Signer solution molecular weight analysis. In the solid state, the complex contains two bridging μ2-bromide ligands and syn aryl groups. The core contains acute Br–Pd–Br angles of 82.79 and 82.53°, and the Pd coordination planes intersect at an angle of 20.7°. The Pd–Br(1) distances are longer than the Pd–Br(2) distances by about 0.1 Å, possibly because of the steric bulk the phosphines and the large trans influence of the aryl moiety.
Figure 2.
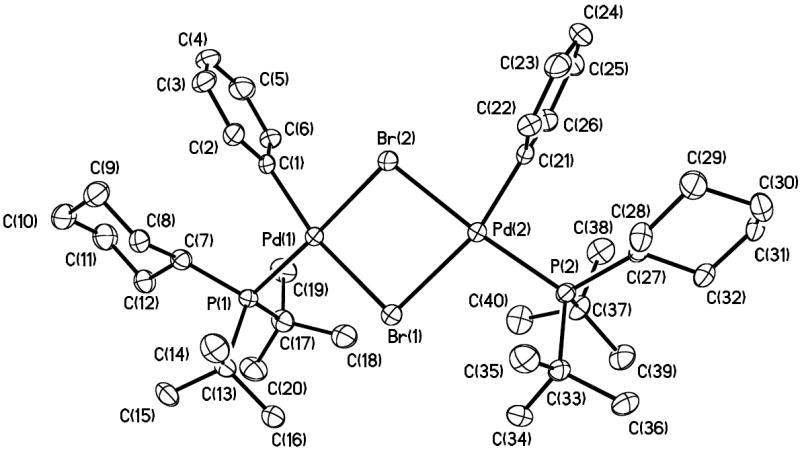
ORTEP of the complex [(CyPtBu2)Pd(Ph)(Br)]2 (15).
This complex 15 appears to dissociate to predominantly a monomeric species in solution. The molecular weight in THF as determined by the Signer method was 523 g/mol. This value is close to the 491.78 molecular weight of the monomer. Considering that the nuclearity of (1-AdPtBu2)-complex 10 was the same in THF and benzene by the Signer molecular weight measurement, that neither THF nor benzene coordinates to the metal center in either structure, and that the 31P NMR chemical shifts in these two solvents for this class of compound have generally been nearly identical, we assert that complex 15 is predominantly monomeric in solution.
2. Kinetic Studies
2.1. General considerations
The rates of oxidative addition of PhX (X = I, Br, Cl) to Pd(0)L2 complexes were measured for complexes containing the bulky trialkylphosphines PtBu3, 1-AdPtBu2, CyPtBu2, and PCy3. The mechanism of the oxidative addition depended largely on the identity of the halide. For this reason, the kinetic data are presented according to the type of haloarene undergoing reaction with the palladium(0) complexes. The data on the oxidative addition of iodoarenes and chloroarenes are simplest to interpret and are presented first. Our kinetic data on these reactions are plotted as the reciprocal of the rate constants to fit with the typical linear equations corresponding to reactions occurring through a preequilibrium, followed by an irreversible step. The data on the additions to bromoarenes are more complex. These data were treated in several ways and suggest that the additions occur in some cases by a combination of mechanisms.
The LPd(Ph)(X) products from oxidative addition of PhBr and PhCl to Pd(PtBu3)2 (1) and Pd(1-AdPtBu2)2 (2) were unstable at the temperatures of oxidative addition, leading to decomposition by cyclometallation at the phosphine to form L·HX as side product. This phosphonium salt has been shown to accelerate the rate of oxidative addition of aryl bromides.42 Because this autocatalysis has been shown to be suppressed by conducting reactions in the presence of the hindered phosphazene base tert-butylimino-trispyrrolidino phosphorane (BTPP),42 the rate constants for oxidative addition of PhBr and PhCl to 2 were obtained on reactions containing 30 to 60 mol% of phosphazene base. Because the addition of PhI to 1 and 2 occurred at lower temperatures than those that lead to the cyclometalation process, the oxidative addition of PhI occurs with an exponential decay of the starting Pd(0) complexes without autocatalysis in the presence or absence of phosphazene base.
Before presenting data on the kinetics of oxidative addition to PCy3 complex 4, comments on the coordination chemistry of this Pd(0) species are warranted. Spectroscopic studies show that the combination of PCy3 and 4 generates an equilibrium mixture of 4, free ligand, and the trisphosphine complex Pd(PCy3)3.43-45 The 31P NMR spectra of a solution consisting of 4 and 2 equiv. of PCy3 obtained between 20 and −40 °C contained two broad signals (δ 39 and 10 ppm) that corresponded to 4 and free PCy3. At lower temperatures (−60 °C to −80 °C), a sharp signal for Pd(PCy3)3 at 26 ppm and a sharp signal for the remaining PCy3 were observed. Thus, the Pd(PCy3)2 complex coordinates a third phosphine at low temperatures.
2.2. Kinetic studies of the oxidative addition of phenyl iodide
Kinetic studies were conducted on the oxidative addition of iodobenzene to Pd(0) complexes 1–4. In all cases, the rate constants were measured by 31P NMR spectroscopy. The rate constants for reactions of complexes 1–3 were obtained from the decay of the starting Pd(0) species. Because signals of PCy3-ligated 4 were broad in the presence of added PCy3, the rate constants for reaction of this complex were determined from the appearance of the oxidative addition product. The decay of complexes 1–3 and the appearance of product from reaction of 4 were clearly exponential with time, and the products were stable, except for the product ligated by PtBu3. Some of the arylpalladium iodide complex ligated by PtBu3 decomposed to form the iodo-bridged Pd(I) dimer [(PtBu3)Pd(μ-I)]2,27 but this subsequent process did not affect the decay of P(t-Bu)3 complex 1. Although autocatalysis was not observed for reactions of iodoarenes, the reaction of 1-AdPtBu2 complex 2 with PhI was carried out in the presence of 30 mol% phosphazene to be consistent with the reactions of 2 with PhBr and with ArCl.
2.2.1. Kinetic studies of the oxidative addition of iodobenzene to PtBu3 complex 1
The rate constants for oxidative addition of PhI to PtBu3-ligated 1 to produce complex 5 were measured on reactions in toluene solvent at 70 °C. A plot of these 1/kobs vs 1/[PhI] for varied concentration of PhI is shown in Figure 3 (left) and indicates that the reaction is first-order in PhI. However, the observed rate constant did not change significantly as a function of [PtBu3] (Figure 3, right); the average observed rate constant over the concentration range of phosphine was kobs = 1.3 ± 0.2 × 10−3 s−1.
Figure 3.
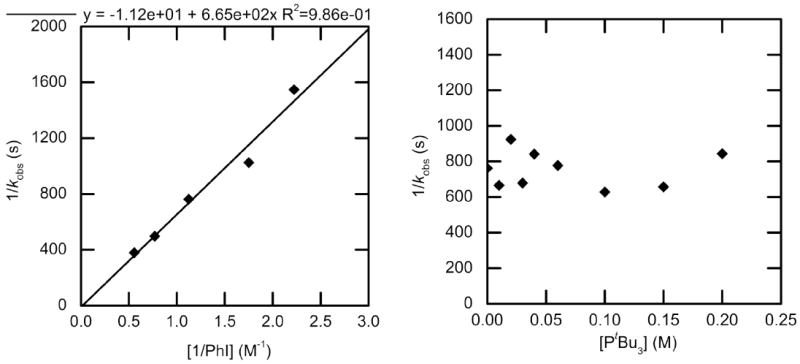
Dependence of the observed rate constant on the concentration of PhI (0.45–1.8 M) with no added PtBu3 (left) and on the concentration of PtBu3 (0-0.20 M) for the oxidative addition of PhI (0.90 M) (right) to Pd(PtBu3)2 (1) (0.040 M) in chlorobenzene at 70 °C.
2.2.2. Kinetic studies of the oxidative addition of iodobenzene to 1-AdPtBu2-ligated 2
The rate constants for the oxidative addition of PhI to 1-AdPtBu2-ligated 2 to form arylpalladium iodide complex 6 were measured in chlorobenzene at 50 °C. The orders for this reaction were similar to those for the oxidative addition of PhI to 1. The plot of 1/kobs vs 1/[PhI] with varied [PhI] shown in Figure 4 (left) indicates that the reaction is first order in PhI. Like the rate constants for the reaction of PhI with the PtBu3-ligated 1, the rate constants for addition to 2 did not change significantly when varying [1-AdPtBu2] (Figure 4, right); the average observed rate constant for reactions conducted with this range of phosphine concentration was 3.4 ± 0.4 × 10−4 s−1.
Figure 4.
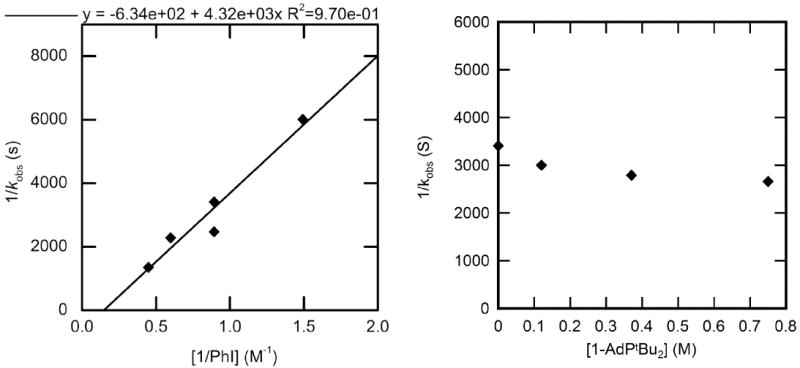
Dependence of the observed rate constant on the concentration of PhI (0.67–2.2 M) with no added 1-AdPtBu2 (left) and on the concentration of 1-AdPtBu2 (0-0.75 M) for the oxidative addition of PhI (1.1 M) (right) to Pd(1-AdPtBu2)2 (2) (0.025 M) in chlorobenzene at 50 °C.
2.2.3. Kinetic studies of oxidative addition of iodoarenes to CyPtBu2-ligated 3
The rate constants of the oxidative addition of PhI to CyPtBu2-ligated 3 to form phenylpalladium iodide complex 14 were measured in toluene at 50 °C. A plot of 1/kobs vs 1/[PhI] is provided in Figure 5 (left); these data show that the reaction is first order in PhI. The reaction rate did not depend significantly on the concentration of ligand, as shown in Figure 5 (right); the average observed rate constant for reactions conducted with this range of phosphine concentration was kobs = 1.1 ± 0.1 × 10−3 s−1. These data do not agree with previously published work,27 which stated that doubling the concentration of ligand decreased the rate of oxidative addition of 1-iodo-3,5-bistrifluoromethylbenzene by a factor of two. Thus, we investigated more closely the oxidative addition of haloarenes to CyPtBu2-ligated 3.
Figure 5.
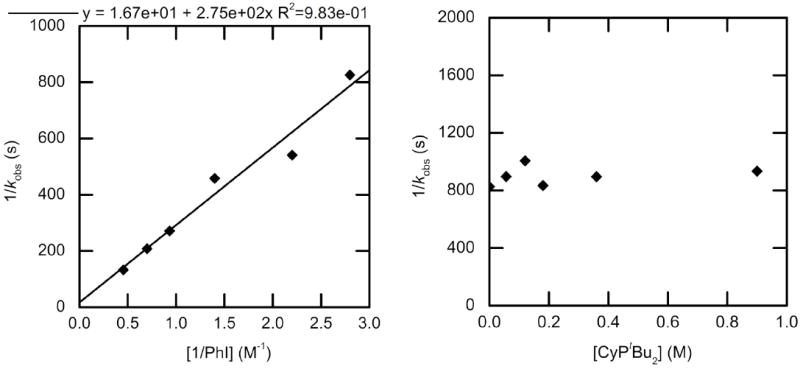
Dependence of the observed rate constant on the concentration of PhI (0.36–1.4 M) with no added CyPtBu2 in toluene at 50 °C (left) and on the concentration of CyPtBu2 (0-0.90 M) for the oxidative addition of PhI (0.36 M) in toluene at 50 °C to Pd(CyPtBu2)2 (3) (0.036 M).
The rate constants for the oxidative addition of 1-iodo-3,5-bistrifluoromethylbenzene to CyPtBu2-ligated 3 to form arylpalladium iodide complex 13 were measured at 25 °C in benzene with concentrations [3] = 0.036 M, [ArI] = 0.36 M, and concentrations of [CyPtBu2] ranging from 0–1.8 M. These substrates and conditions are identical to those reported previously.27 In contrast to the published data, the rate of the reaction was only slightly affected by the presence of excess ligand; the average value for kobs was 6.1 ± 0.6 × 10−4 s−1. We do not have an explanation for the difference between the previously published data and the data we report in this paper; however, the previous reactions were monitored to only two half-lives, and reactions at only two different concentrations of ligand were reported.
2.2.4. Kinetic studies of the oxidative addition of iodobenzene to PCy3-ligated 4
The reactions of PCy3-ligated 4 with PhI occurred to form phenylpalladium iodide complex 17 within minutes at room temperature. Thus, kinetic data on these oxidative additions were obtained on reactions conducted at −80 °C. Three series of reactions with iodobenzene were conducted, the first with varied [PhI], the second with [PCy3] ranging from 0.030–0.12, and the third with [PCy3] ranging from a lower 0.61–6.1 mM. The inverse of the rate constants for oxidative addition of PhI to the combination of 4 and PCy3 with varied [PhI] and [PCy3] are shown in Figure 6. These data show that the reaction is first order in PhI and inverse first order in PCy3 when concentration of ligand is high. As noted in detail in the introduction to this section, the trisphosphine-ligated Pd(0) is the major species in the presence of more than 0.030 M added PCy3 at −80 °C. Thus, this plot corresponds to data and rate expressions when the starting complex is the [Pd(PCy3)3] complex,46 and the inverse order in PCy3 indicates reversible dissociation of PCy3 from Pd(PCy3)3 prior to carbon-halogen bond cleavage. At low concentrations of added ligand, the major species observed in solution at −80 °C is Pd(PCy3)2 (4). Under these conditions, the rate of the reaction was zeroth order in added ligand; the average observed rate constant was 9.8 ± 0.1 × 10-4 s-1.
Figure 6.
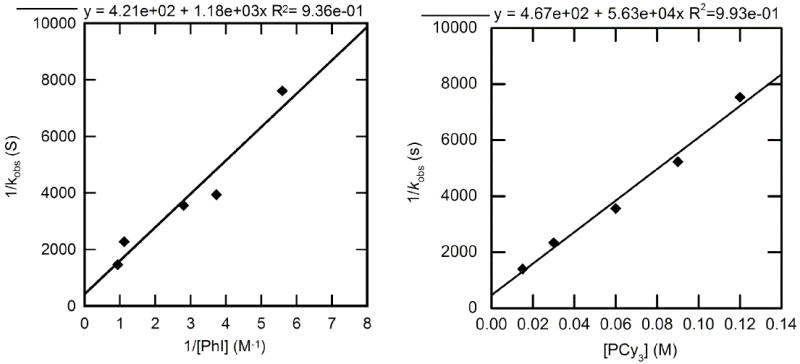
Dependence of the observed rate constant on the concentration of PhI (0.18–1.1 M) with [PCy3] = 0.056 M (left) and on the concentration of PCy3 (0.03-0.12 M) for the oxidative addition of PhI (0.36 M) (right) to Pd(PCy3)3 (4) (0.020 M) in toluene at −80 °C.
2.3. Kinetic studies of oxidative addition of chloroarenes
Kinetic studies on the oxidative addition of chloroarene to Pd(1-AdPtBu2)2 (2) were conducted with 2-CF3C6H4Cl because it formed a more stable arylpalladium chloride complex than did PhCl. These reactions were conducted in the presence of added phosphazene base to avoid potential autocatalysis by the side products from decomposition of the arylpalladium chloride complex. They were also conducted with higher concentrations of haloarene than the reactions of bromoarenes and iodoarenes to improve the reaction yields that are affected by product decomposition.47 The decay of 2 was measured by 31P NMR spectroscopy.
2.3.1. Kinetic studies of oxidative addition of 2-CF3C6H4Cl to 1-AdPtBu2-ligated 2
The rate constants for reactions of 2-CF3C6H4Cl with 1-AdPtBu2-ligated 2 to form arylpalladium chloride complex 12 were measured in toluene or neat ArCl at 100 °C in the presence of 0.015 M (60 mol %) phosphazene base (BTPP). The decay of 2 was exponential, showing that the additions of ArCl are first order in the palladium(0) complex. The plots of 1/kobs with varied [ArCl] and [1-AdPtBu2] are shown in Figure 7. The plot of 1/kobs vs 1/[ArCl] revealed a positive dependence of kobs on chloroarene. The plot of 1/kobs vs [1-AdPtBu2] showed an inverse dependence of kobs on [1-AdPtBu2], although this dependence was not simply inverse first order.
Figure 7.
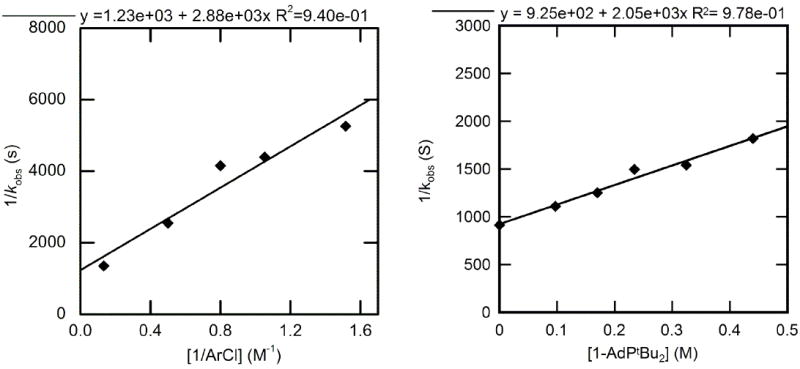
Dependence of the observed rate constant on the concentration of 2-CF3C6H4Cl (0.54–7.2 M) with [1-AdPtBu2] = 0.17 M (left) and on the concentration of 1-AdPtBu2 (0-0.44 M) for the oxidative addition of 2-CF3C6H4Cl (7.6 M) (right) to Pd(1-AdPtBu2)2 (2) (0.025 M) in toluene at 100 °C.
2.3.2. Kinetic Studies of oxidative addition of chlorobenzene to CyPtBu2-ligated 3
The rate constants for the oxidative addition of PhCl to CyPtBu2-ligated 3 to form phenylpalladium chloride complex 16 were measured in toluene at 100 °C in the absence of any added base. The plots of 1/kobs measured with varied [PhCl] and [CyPtBu2] are shown in Figure 8. The plot of 1/kobs vs 1/[PhCl] revealed a positive dependence of kobs on chlorobenzene. The plot of 1/kobs vs [CyPtBu2] revealed an inverse dependence of kobs on the concentration of ligand.
Figure 8.

Dependence of the observed rate constant on the concentration of chlorobenzene (1.0–7.9 M) with [CyPtBu2] = 0.32 M (left) and on the concentration of CyPtBu2 (0.32-0.94 M) for the oxidative addition of chlorobenzene (5.9 M) (right) to Pd(CyPtBu2)2 (3) (0.036 M) in toluene at 100 °C.
2.3.3. Kinetic studies of oxidative addition of chlorobenzene to PCy3-ligated 4
The rate constants for reactions of PCy3-ligated 4 with chlorobenzene to form complex 19 were measured in toluene at 70 °C. At the concentrations of [PCy3] investigated and the 70 °C temperature of the reaction, the starting Pd(0) species is the two-coordinate complex 4. The plots of 1/kobs obtained with varied [PhCl] and [PCy3] are shown in Figure 9 and are similar to the analogous plots for the additions of PhCl to 1-AdPtBu2-ligated 2 and CyPtBu2-ligated 3. The plot of 1/kobs vs 1/[PhCl] revealed a positive dependence of kobs on chloroarene; the plot of 1/kobs vs [PCy3] revealed an inverse dependence of kobs on the concentration of ligand.
Figure 9.
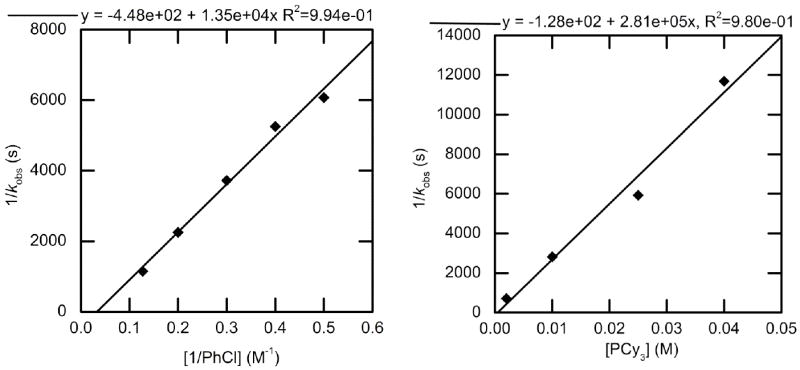
Dependence of the observed rate constant on the concentration of chlorobenzene (2.0–7.9 M) with [PCy3] = 0.009 M (left) and on the concentration of PCy3 (0.002-0.040 M) for the oxidative addition of chlorobenzene (3.9 M) (right) to Pd(PCy3)2 (4) (0.019 M) in toluene at 70 °C. Data for [PhCl] = 2.5, 3.3 M (left) are an average value from 2-3 runs.
2.4. Kinetic studies of oxidative addition of bromobenzene
As noted in section 2.1 and in published work,42 the arylpalladium bromide product from oxidative addition of bromobenzene to PtBu3-ligated 1 is unstable at elevated temperatures for extended times and undergoes cyclometalation to generate L•HBr. To avoid the problems associated with the generation of the phosphonium salt, the rate constants for oxidative addition of PhBr to 1 and 2 were measured in the presence of phosphazene base. Moreover, we focused on the oxidative addition of PhBr to 1-AdPtBu2-ligated 2 because the oxidative addition product (1-AdPtBu2)Pd(Ph)(Br) (8) was sufficiently stable to form in >90 % yield in the presence of the phosphazene base. In contrast, the reactions of CyPtBu2-ligated 3 and PCy3-ligated 4 occurred in high yield and with an exponential decay of the Pd(0) complex in the absence of any added base. Thus, the additions of PhBr to 3 and 4 were conducted in the absence of base.
2.4.1. Kinetic studies of oxidative addition of bromobenzene to 1-AdPtBu2-ligated 2
The reaction of PhBr with 1-AdPtBu2-ligated 2 to form phenylpalladium bromide complex 8 was studied at 90 °C. The reactions were conducted in the presence of 0.015 M (60 mol %) added phosphazene. A clear exponential decay of 2 was observed under these conditions. Thus, the additions of PhBr are first order in the Pd(0) complex. The kinetic data on the effect of the concentration of arene on the rate constant for oxidative addition of PhBr are plotted as kobs vs [PhBr]. This plot allows us to assess the potential that two mechanisms contribute to the observed rate constant. When such a plot is linear with a y-intercept, the double reciprocal plots shown for data on the oxidative addition of ArCl and ArI are nonlinear. Conversely, when the double reciprocal plot has a clear, non-zero y-intercept, the direct plot is curved.
The plot of kobs vs [PhBr] (Figure 10, left) was linear with a positive slope and nonzero intercept. Moreover, the rate constant for this oxidative addition to 1-AdPtBu2-ligated 2 did not depend on the concentration of ligand at high or low concentration of bromobenzene. The plot of the dependence of kobs on [1-AdPtBu2] is shown in Figure 10 (right) and shows the observed rate constant was independent of the concentration of added ligand (average kobs = 6.2 ± 1.2 × 10−4 s−1). The data at high concentration of PhBr contained some deviation, yet a change in [1-AdPtBu2] by a factor of four led to a variation in rate by less than a factor of two. Additionally, when [PhBr] = 2.5 M and [1-AdPtBu2] ranged from 0.10-0.50 M, the value of kobs varied by only 10-15% (2.5 ± 0.3 × 10−4 s−1). As described in the discussion section, these data suggest that two mechanisms for oxidative addition of bromoarenes occur simultaneously.
Figure 10.
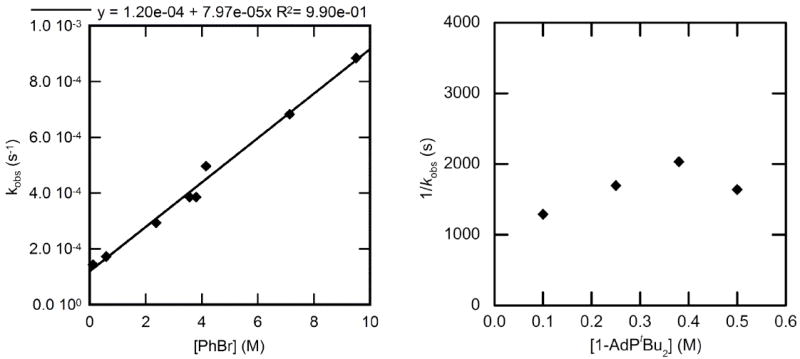
Dependence of the observed rate constant on the concentration of PhBr (0.12–9.5 M) with no added 1-AdPtBu2 (left) and on the concentration of 1-AdPtBu2 (0.10-0.50 M) for the oxidative addition of PhBr (8.5 M) (right) to Pd(1-AdPtBu2)2 (2) (0.025 M) in toluene at 90 °C.
2.4.2. Kinetic studies of oxidative addition of bromobenzene to CyPtBu2-ligated 3
The rate constants for the reaction of CyPtBu2-ligated 3 with PhBr to form phenylpalladium bromide complex 15 were measured in toluene at 70 °C. The plots of these data are shown in Figure 11. The observed rate constant depended positively on the concentration of bromoarene. Again, the plot of kobs vs [PhBr] contained a non-zero y-intercept. In contrast, the observed rate constant was independent of the concentration of added ligand; the average observed rate constant was 7.1 ± 0.7 × 10−4 s−1.
Figure 11.
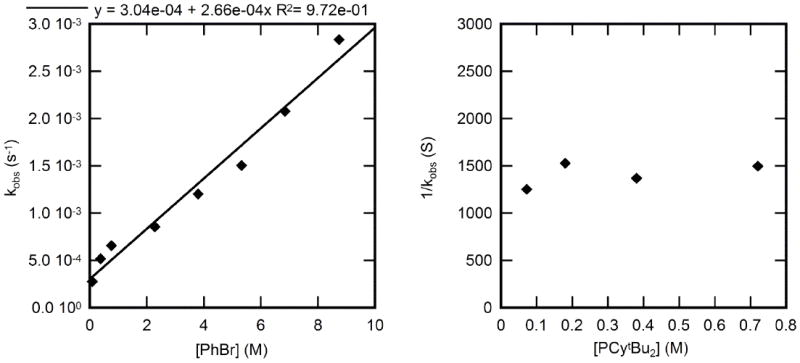
Dependence of the observed rate constant on the concentration of PhBr (0.09- 8.7 M) with [CyPtBu2] = 0.32 M (left) and on the concentration of CyPtBu2 (0.07-0.72 M) for the oxidative addition of PhBr (1.5 M) (right) to Pd(CyPtBu2)2 (3) (0.036 M) in toluene at 70 °C.
2.4.3. Kinetic studies of oxidative addition of bromobenzene to PCy3-ligated 4
The rate constants for reactions of PCy3-ligated 4 with bromobenzene to form arylpalladium bromide complex 18 were measured in toluene at 10 °C. The plots of these data are shown in Figure 12. These plots reveal a positive dependence of kobs on [ArBr] and a nearly zeroth-order dependence on [PCy3]. A change in the concentration of PCy3 by a factor of six led to a decrease in rate constant by less than a factor of two.
Figure 12.
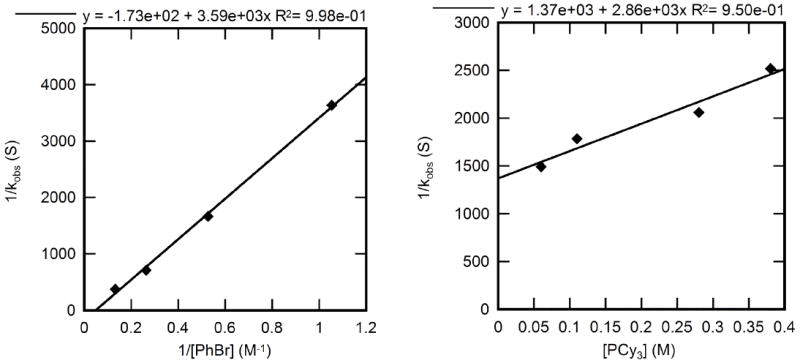
Dependence of the observed rate constant on the concentration of PhBr (0.95– 7.6 M) with [PCy3] = 0.19 M (left) and on the concentration of PCy3 (0.060-0.38 M) for the oxidative addition of PhBr (1.9 M) (right) to Pd(PCy3)3 (4) (0.019 M) in toluene at 10 °C.
DISCUSSION
1. Structures of the palladium and arylpalladium halide reactants and products
The most stable Pd(0) complexes of the bulky phosphines PtBu3,48 1-AdPtBu222 and CyPtBu227 are the bisphosphine complexes PdL2.49 In contrast, the most stable Pd(0) complex of PCy3 depended on the temperature and the concentration of added PCy3. At temperatures below −70 °C with [PCy3] < 0.03 M, the complexes Pd(PCy3)3 and bis ligated Pd(PCy3)2 were observed by 31P NMR spectroscopy.18 At the same temperature with [PCy3] > 0.03 M, the trisphosphine complex Pd(PCy3)3 was the only Pd(0) complex observed. At room temperature or above, the signals due to the free and coordinated ligand are broad due to exchange on the NMR time scale, but the bisphosphine complex Pd(PCy3)2 is the major Pd(0) species present.
The identities of some of the oxidative addition products have been published. The product from addition of PhX (X= Cl, Br, I) to the Pd(0) complex of PCy3 are bisphosphine, four-coordinate species,23,27,39-41 but the products from addition of ArX to complexes of the more hindered ligands contain a single ligand and are either dimeric with bridging halides27 or monomeric.22,23 Further, the structure of the arylpalladium halide complexes depended in some cases on the identity of the halide.21 For example, the arylpalladium bromide and iodide complexes containing the ligands PtBu3 and 1-AdPtBu2 are monomeric three-coordinate complexes, while the arylpalladium chloride complexes are mainly monomers in solution and dimers in the solid state. The kinetic data analyzed in the following sections show that the reactant and product structures do not correlate with either the coordination number of the species undergoing oxidative addition or the initial product formed by oxidative addition in many cases.
2. Potential mechanisms for oxidative addition
Three possible mechanisms were considered for the oxidative addition of ArX to the palladium complexes L2Pd(0). In the first mechanism, oxidative addition would take place by direct reaction of ArX with the starting bisphosphine complex L2Pd(0) to form a four-coordinate arylpalladium halide complex. This addition would be followed in some cases by dissociation of ligand and subsequent dimerization, depending on the phosphine or haloarene used. The rate of reaction by this pathway would be first order in the concentration of haloarene and independent of the concentration of ligand (Scheme 3).
Scheme 3.
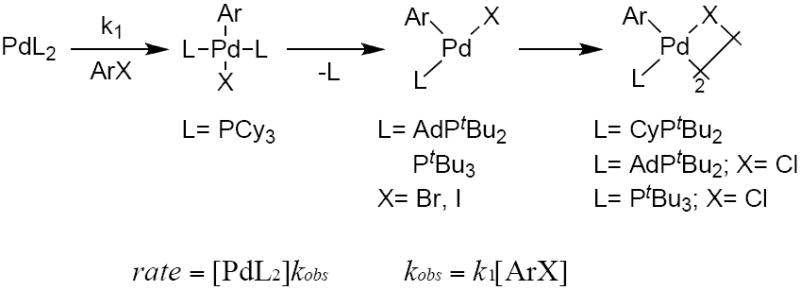
Oxidative addition of ArX by direct reaction with PdL2.50
Alternatively, the reaction could occur by reversible or irreversible associative displacement of the phosphine in PdL2 by the haloarene, as shown in Scheme 4. This first step would form a monophosphine intermediate coordinated by a haloarene that contains an intact C–X bond. Carbon–halogen bond cleavage would then form a three-coordinate arylpalladium halide complex. This three-coordinate complex could be the final product, or it could undergo dimerization or recoordination of ligand to form the final reaction product. If the initial associative displacement of phosphine were reversible, then the reaction rate would depend on the concentration of both the haloarene and the ligand. If the initial associative displacement of phosphine were irreversible (k3 ≫ k−2[L]), then the reaction rate would depend only on the concentration of haloarene. Thus, the orders of the reaction in haloarene and ligand alone cannot distinguish between irreversible, direct carbon-halogen bond cleavage by the L2Pd complex (Scheme 3) and irreversible displacement of ligand by the haloarene, followed by carbon-halogen bond cleavage by a monophosphine intermediate (Scheme 4). However, both mechanisms occur by reaction of the aryl halide with a bis-ligated Pd(0) species. In some cases, we were able to distinguish between these two mechanisms by the kinetic behavior of the reverse reaction.
Scheme 4.
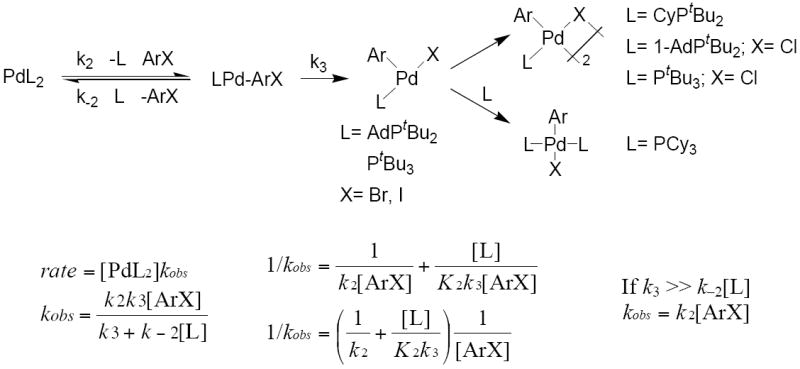
Oxidative addition of ArX after associative displacement of ligand from PdL2.50
Finally, the oxidative addition could occur by dissociation of a phosphine followed by oxidative addition of ArX to the resulting LPd intermediate to form a three-coordinate arylpalladium halide complex (Scheme 5). This dissociation of phosphine could be reversible or effectively irreversible (i.e. k5[ArX] ≫ k−4[L]) under the reaction conditions. If the ligand dissociation is fully reversible, then the reaction will depend on the concentration of both the haloarene and the ligand. If dissociation of ligand is effectively irreversible, then the rate of the reaction would be independent of the concentration of the haloarene and added ligand. In this case, the observed rate constant would be equal to k4. After the addition, dimerization of the three-coordinate complex or coordination of a second ligand to form a square planar complex could occur, depending on the identity of the phosphine or the halide (Scheme 5).
Scheme 5.
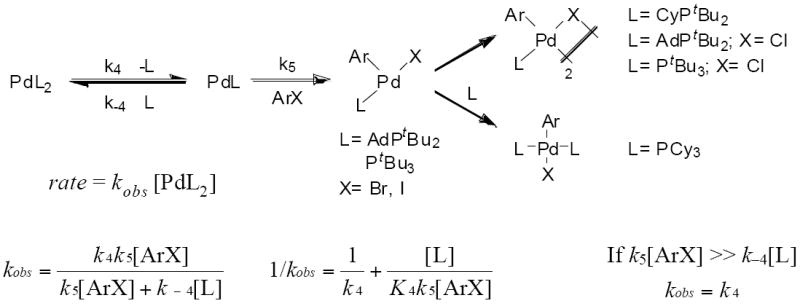
Oxidative addition of ArX after dissociation of ligand from PdL2.50
All of these mechanisms considered predict a positive dependence of kobs on [ArX], which is consistent with the observations. However, the predicted differences in the dependence on [L] can be used to differentiate the mechanisms. Direct oxidative addition to PdL2 (Scheme 3) is excluded if there is an inverse dependence of kobs on [L]. Oxidative addition to PdL (Scheme 5) is excluded if there is no dependence on [L] but a positive dependence on [ArX]. Associative displacement of L by ArX (Scheme 4) is excluded if the plot of 1/kobs vs 1/[ArX] has a significant y-intercept, but such analysis requires extremely accurate data, and we could not use this y-intercept to distinguish between the mechanisms in Schemes 4 and 5 in most cases. These predictions apply to systems in which the L2Pd(0) complex is the starting complex. The palladium complex of PCy3 adopts an L3Pd structure with added phosphine at low temperature. Thus, reactions under these conditions are analyzed separately. Most generally, the analysis of our data shows that the identity of the halide, rather than the identity of the ligands in this study has a larger effect on whether the irreversible step of the mechanism involves a bisphosphine or monophosphine complex.
3. Mechanism of oxidative addition of iodoarenes
3.1. Overview
The kinetic data for addition of iodobenzene to all four Pd(0) complexes 1–4 imply that the irreversible (“rate determining”) step involves reaction of the iodoarene with a palladium complex ligated by two phosphines. The reactions of iodobenzene with complexes 1–3 were zeroth order in ligand and first order in iodobenzene. The reactions of iodobenzene with complex 4 depended inversely on the concentration of added ligand when the concentration of the added ligand was high, but this kinetic behavior was observed because the starting complex 4 was converted to the trisphosphine complex in the presence of this amount of added ligand.
3.2. Addition of PhI to L2Pd Complexes 1–3 ligated by PtBu2, 1-AdPtBu2 and CyPtBu2
Our kinetic data indicate that the “rate-determining” or first irreversible step of the reaction of iodobenzene with bisphosphine complexes 1 –3 ligated by PtBu3, 1-AdPtBu2 and CyPtBu2 involves the starting bisphosphine species. This step can occur either by direct oxidative addition to the starting Pd(0) species shown in Scheme 3 or by irreversible displacement of the coordinated phosphine by iodoarene shown in Scheme 4 (k3 ≫ k−2[L]). Both rate equations are zeroth order in the concentration of added ligand and first order in the concentration of iodoarene. Although the two pathways cannot be distinguished by the reaction orders of the forward process, we were able to distinguish between these two mechanisms by the kinetic behavior of the reverse reaction we studied previously, reductive elimination of iodoarene from the arylpalladium halide product.21
If the carbon–halogen bond cleavage during the oxidative addition process occurs directly by the PdL2 complex (Scheme 3), then the principle of microscopic reversibility dictates that the elimination of ArI must also occur from a bisphosphine palladium complex. In this case, reductive elimination from the LPd(Ar)(I) complex would be first order in the concentration of ligand. In contrast, if carbon–halogen bond cleavage during the oxidative addition process occurs by a monophosphine species generated by associative displacement of the ligand by the iodoarene (Scheme 4), then microscopic reversibility would dictate that the elimination of ArI must occur from a monophosphine palladium complex, and reductive elimination from the LPd(Ar)(I) complex would be zeroth order in added ligand.
We previously showed that the rate of reductive elimination of 2-iodotoluene from (PtBu3)Pd(o-tol)(I) was independent of the concentration of added ligand (Scheme 6).21 Thus, we conclude that the oxidative addition of PhI to 1 and 2 occurs by irreversible, associative displacement of a phosphine, followed by cleavage of the C–X bond by the resulting haloarene complex (Scheme 4). This mechanism is the same as that proposed for oxidative addition of iodobenzene to the Pd(0) complex ligated by the hindered ferrocenylphosphine Q-phos.21
Scheme 6.
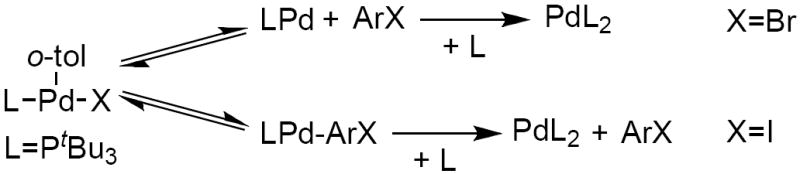
Mechanisms for the reductive elimination of ArX from 3-coordinate aryl palladium(II) halide complexes.
The mechanism of reductive elimination of haloarene did not provide clear information on the mechanism of the oxidative addition of iodoarenes to CyPtBu2-ligated 3 because reductive elimination of ArI from complexes ligated by CyPtBu2 occurred in low yield.51 The smaller size of the CyPtBu2 ligand in complex 3 provides the potential that the oxidative addition of iodobenzene occurs directly to 3, but the presence of a single phosphine in the product argues against this pathway. Thus, we favor a pathway involving irreversible displacement of the phosphine by the iodoarene. However, in either case, the kinetic data show that the rate-determining step during the reaction of PhI with CyPtBu2-ligated 3 involves a bisphosphine complex. This conclusion contrasts with the dissociative mechanism deduced previously by others for reactions of CyPtBu2-ligated 3 with iodoarenes.27
3.3. Mechanism of the oxidative addition of PhI to Pd(PCy3)3
Because the trisphosphine complex Pd(PCy3)3 is the major Pd(0) complex at the −80 °C temperature of the oxidative addition process, interpretation of the kinetic data for oxidative addition of iodobenzene to the PCy3-ligated Pd(0) complex is slightly different from interpretation of the data for oxidative addition of iodobenzene to the other two complexes. Scheme 7 shows the possible mechanisms for oxidative addition of PhI to the trisphosphine species Pd(PCy3)3.
Scheme 7.
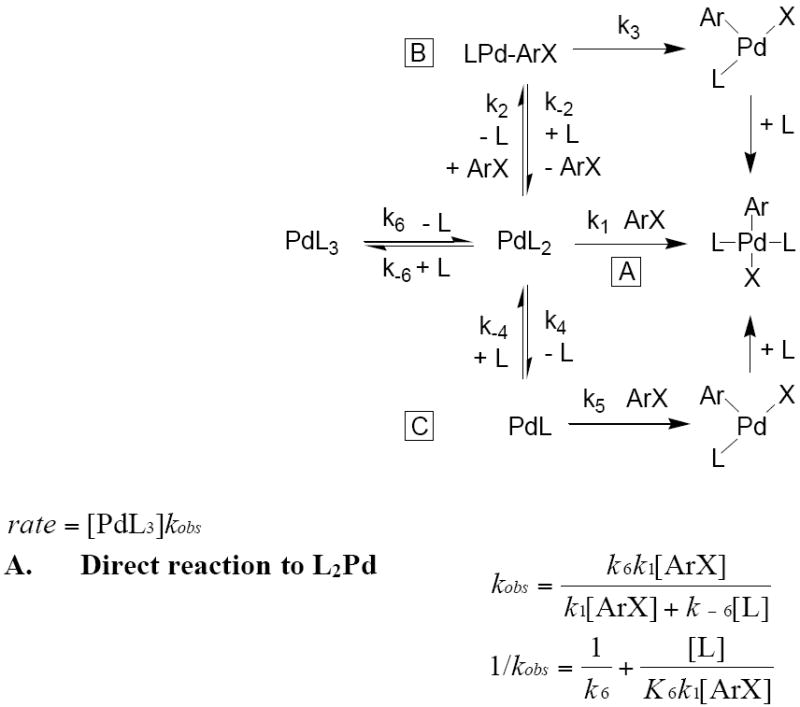
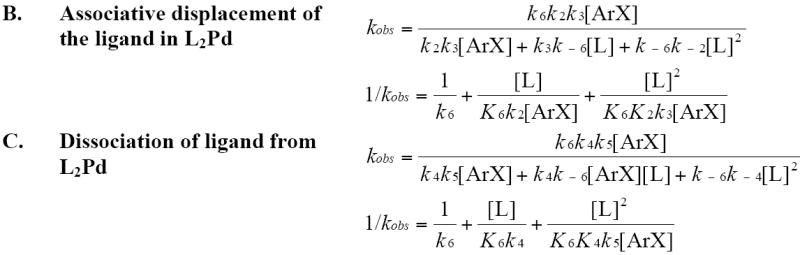
Possible mechanisms for the oxidative addition of ArI to PdL3 L = PCy3.50
The observed inverse dependence on added ligand shows that the oxidative addition process occurs after loss of one (Path A) or two (Path B) ligands to generate Pd(PCy3)2 or Pd(PCy3), respectively. The data could not be obtained over a wide enough range of concentrations with sufficient accuracy to distinguish between an inverse first-order and an inverse second-order dependence on the concentration of added ligand that would correspond to reaction of the iodoarene with Pd(PCy3)2 and Pd(PCy3), respectively. However, we were able to distinguish between addition to Pd(PCy3)2 and addition to Pd(PCy3) by conducting the oxidative addition with low enough concentrations of added ligand at −80 °C that PdL2 was the major species in solution. Under these conditions, the reaction was independent of the concentration of added ligand. Therefore, we conclude that the dependence on the concentration of ligand observed at high concentrations of added phosphine resulted from the reversible dissociation of one phosphine from Pd(PCy3)3 to form Pd(PCy3)2. We then conclude that Pd(PCy3)2 reacts with PhI by irreversible reaction between PhI and the bisphosphine complex. This irreversible reaction could occur by Path A in Scheme 7 involving irreversible oxidative addition to Pd(PCy3)2 or by a version of Path B in which the ArI irreversibly displaces a phosphine from Pd(PCy3)2.
4. Mechanism of oxidative addition of chloroarenes
The data on the oxidative addition of chloroarenes to Pd(0)L2 complexes 2, 3 and 4 containing the ligands 1-AdPtBu2, CyPtBu2, and PCy3 imply that the irreversible step in these processes involves a monophosphine palladium(0) complex. Each of the reactions depended positively on the concentration of chlorobenzene and inversely on the concentration of added ligand. Thus, the number of phosphines in the complex involved in the rate-determining step for addition of chloroarenes is the same for complexes containing all the ligands studied and is different from the number of phosphines contained in the complex involved in the rate-determining step for addition of iodoarenes.
More specifically, the rate data for the addition of PhCl to the complexes 2–4 were consistent with the rate equations for the mechanisms in Schemes 4 and 5 involving reversible loss of a ligand. These rate data include linear plots of 1/kobs vs 1/[ArCl] and 1/kobs vs [L] for reactions of PhCl with all three Pd(0) complexes.
The pathway in Scheme 4 involves reversible associative displacement of ligand by the haloarene, and the pathway in Scheme 5 involves a reversible sequence of dissociation of ligand and association of haloarene. In principle, these two pathways can be distinguished by the y-intercepts of the plots of 1/kobs vs 1/[ArCl] and 1/kobs vs [L]. Reaction by either mechanism predicts that a plot of 1/kobs vs [L] will have a non-zero y-intercept. However, the mechanism in Scheme 4 predicts that a plot of 1/kobs vs 1/[ArCl] will lack a y-intercept, while the mechanism in Scheme 5 predicts that the same plot would have a non-zero y-intercept. Moreover, the mechanism in Scheme 5 predicts that the y-intercept of the plot of 1/kobs vs 1/[ArCl] would be identical to the y-intercept of the plot of 1/kobs vs [L]. For the reactions of 1-AdPtBu2 complex 2, the kinetic behavior predicted for reaction by initial dissociation of ligand was clearly observed. As shown in Figure 7, the plot of 1/kobs vs. [L] has a clear non-zero y-intercept. Moreover, the y-intercepts of the plots of 1/kobs vs. [L] and 1/kobs vs. 1/[ArCl] are within experimental error of each other (925 s and 1230 s). Finally, the values for K4k5 deduced from these plots (5.9 × 10-5 s-1 and 6.4 × 10-5 s-1) are within experimental error of each other. We could not distinguish between these two mechanisms as confidently for reactions of the CyPtBu2 and PCy3 complexes by this analysis because the y-intercepts of their plots of 1/kobs vs 1/[ArCl] were close to zero. However, the values of K4k5 calculated from Figures 8 and 9 were similar to each other in both cases. This equivalence, and the clear dissociative mechanism for reaction of 2, suggest that complexes 3 and 4 also react with aryl chlorides by a dissociative mechanism.
Most generally, our data show unambiguously that the irreversible step for addition of chloroarenes involves a monophosphine species and that the number of phosphine ligands in the Pd(0) species involved in the irreversible step of the oxidative addition of chloroarenes is identical for each of the Pd(0) complexes in this study. Moreover, these data show that the number of phosphines in the Pd(0) species in the irreversible step of the oxidative additions of chloroarenes is different than that in the Pd(0) species that reacts in the irreversible step of the oxidative additions of iodoarenes.
5. Mechanism of oxidative addition of bromoarenes
Like the rate data on the oxidative addition of PhCl and PhI, the rate data on the oxidative addition of PhBr to each of the L2Pd(0) complexes 2–4 were closely related to each other. The oxidative additions of bromoarenes to all three complexes depended positively on the concentration of bromoarene, and each reaction occurred with little (L=PCy3) or no (L=1-AdPtBu2, CyPtBu2) dependence on the concentration of ligand. This lack of dependence of kobs on the concentration of ligand is similar to that for oxidative addition of PhI and is distinct from the strong dependence of kobs on the concentration of ligand for oxidative addition of PhCl. As shown below, a detailed assessment of the dependence of the rate constant on the concentration of bromoarene implies that two pathways for the oxidative addition of bromoarenes occur concurrently. One pathway clearly occurs by irreversible dissociation of ligand, as was deduced for reaction of the chloroarene, and the second pathway could occur by irreversible associative displacement of ligand by the bromoarene, as was deduced for the reactions of iodobenzene.
The dependence of kobs on [L] was nearly zeroth-order for the addition of PhBr to all three complexes. Thus, the irreversible step of these oxidative addition processes involves a bisphosphine Pd(0) species. Again, the coordination number of the species involved in the irreversible step of the oxidative addition of bromoarenes was identical for all of the Pd(0) species in this study.
Although this conclusion is the most concrete and the major conclusion we wish to draw from our studies on the oxidative addition of bromoarenes is the coordination number of the species reacting in the rate-limiting step of the major pathway, we can draw some additional conclusions about the mechanism of the oxidative addition of bromoarenes. The presence of a y-intercept implies that the reactions likely occur by two competing mechanisms involving bisphosphine species. For reactions of complexes 2 and 3, plots of kobs vs [PhBr] were linear, but they also contained a non-zero y-intercept. The non-zero y-intercepts in these plots suggest two pathways involving bisphosphine complexes in the rate-determining step compete, one that is dependent on the concentration of bromoarene and one that is independent of the concentration of bromoarene. Of the mechanisms considered, only the mechanism involving initial, irreversible dissociation of ligand (Scheme 5) is independent of the concentration of ligand and haloarene (kobs = k4). Thus, we conclude that the value of the y-intercept roughly corresponds to the rate constant for reaction by this path.52
Two other paths predict rate constants that would be independent of the concentration of ligand and positively dependent on the concentration of haloarene: direct C–X bond cleavage to form a four-coordinate intermediate (Scheme 3) and irreversible associative displacement of ligand, followed by carbon–halogen bond cleavage (Scheme 4). We consider the pathway involving direct C–X bond cleavage to form a four-coordinate intermediate the least likely. Besides forming a crowded intermediate, microscopic reversibility implies that the elimination of ArBr from LPd(Ar)(Br) would occur through a four-coordinate intermediate. Previous studies on reductive elimination of ArBr from (PtBu3)Pd(o-tol)Br implied that the elimination involves a three-coordinate intermediate (Scheme 6).53 Although reductive elimination of ArBr from aryl palladium(II) halide complexes of AdPtBu2 and CyPtBu2 have not been reported, the steric properties of these phosphines make the formation of a bisphosphine intermediate L2Pd(Ar)(X) and a change in mechanism from that of the reactions of PtBu3 complex 1 unlikely.
Therefore, we conclude, based on the presence of the y-intercepts, that the addition of PhBr to complexes 2 and 3 most likely occurs by two competitive paths involving rate-determining reactions of bisphosphine complexes to give rise to mono-phosphine intermediates. One pathway could occur by irreversible dissociation of ligand. The second by irreversible associative displacement of ligand prior to the carbon-halogen bond cleavage step (Scheme 8).
Scheme 8.
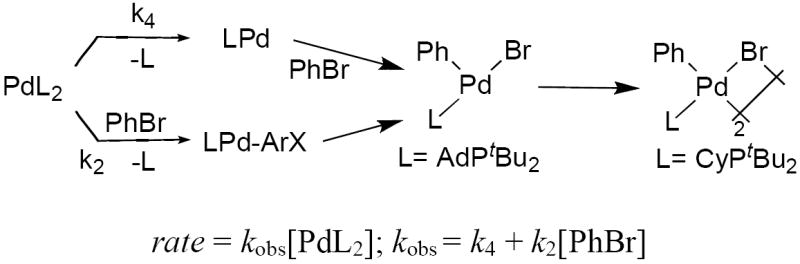
Two concurrent mechanisms for the oxidative addition of PhBr to 2 and 3.
In contrast, the oxidative addition of PhBr to PCy3-ligated 4 appears to occur by a single mechanism involving associative displacement of the ligand or oxidative addition to the two-coordinate species. The rate of the oxidative addition of PhBr to PCy3-ligated 4 was measured at a temperature in which the major Pd(0) species was the L2Pd complex. The data in Figure 12 showed that this oxidative addition occurred with a positive dependence on the concentration of bromobenzene and only a weak dependence on the concentration of added ligand. The small slope and large y-intercept of the plot of 1/kobs vs [L] is consistent with the rate equations corresponding to nearly irreversible associative substitution of PCy3 by PhBr (Scheme 4) or nearly irreversible oxidative addition to the two-coordinate species to generate a four-coordinate product (Scheme 3). The small slope implies that the product of the equilibrium and rate constants in the second term of the equation (K2k3) are large, relative to the rate constant (k2) in the first term. Because both processes would be favorable with the smaller PCy3 ligand, we cannot distinguish between these two mechanisms. However, we can conclude in general that a major pathway for reactions of bromoarenes with the L2Pd(0) complexes 2-4 occurs by irreversible reaction of the bromoarene with the bisphosphine complex. This irreversible reaction could occur by associative displacement of ligand followed by oxidative addition or by direct oxidative addition.
CONCLUSIONS
The observed kinetic data are summarized in Table 1, and this table shows that the dependence of the rate constants depends on the identity of the halide more than the steric bulk of the ligand. The reactions of iodobenzene were all zeroth order in added ligand, the reactions of bromobenzene were all zeroth, or nearly zeroth order, in added ligand, and the reactions of chlorobenzene were all inverse first order in added ligand. Moreover, all reactions depended positively on the concentration of haloarene. Thus, all the reactions of the iodoarenes occur by rate-determining reaction with a bisphosphine complex, while all of the reactions of the chloroarene occur by rate-determining reaction with a monophosphine complex. The reactions of the bromoarenes occur by rate-determining reaction of a bisphosphine complex. Our data imply that the reactions of chloroarenes occur by reversible formation of a monophosphine intermediate, while the reactions of iodobenzene occur by an irreversible reaction of the starting bisphosphine species. The oxidative additions of bromoarenes appear to occur by a combination of irreversible reaction of the bromoarene with the two-coordinate Pd(0) species and dissociation of phosphine prior to reaction with the bromoarene. In particular, the reactions of bromoarenes with Pd(1-AdPtBu2)2 (2) appeared to occur by a combination of both associative and dissociative substitution of the ligand by haloarene, and this result reveals the fine balance between these two mechanisms for generation of the monophosphine intermediates.
Table 1.
Summary of the experimental observations and suggested mechanisms for the oxidative addition of ArX to complexes 2–4.
| Pd(1-AdPtBu2)2 | Pd(CyPtBu2)2 | Pd(PCy3)n n = 2,3 | General Conclusion | ||
|---|---|---|---|---|---|
| ArI | Order in ArI | 1 | 1 | 1 | The ArI reacts with the PdL2 species |
| Order in L | Zeroth | Zeroth | -1 for n = 3, 0 for n = 2 | ||
| Conclusion | Irreversible associative displacement of L by ArI | Reaction of ArI with L2Pd | |||
| ArCl | Order ArCl | 1 | 1 | 1 | The C–X bond cleavage step occurs after reversible dissociation or displacement of one phosphine from PdL2 to form PdL or LPd–ArX |
| Order in L | -1 | -1 | -1 | ||
| Conclusion | Reversible exchange of L with PhCl prior to C–X bond cleavage. | ||||
| ArBr | Order ArBr | 1 | 1 | 1 | The C–X bond cleavage step appears to occur by two pathways of similar energy. One major pathway clearly occurs by reaction of ArBr with the L2Pd(0) species. This step could occur by irreversible displacement of one L to form LPd–ArX or by irreversible direct C-X bond cleavage. The second, less defined pathway is zeroth order in ArBr, and appears to occur by irreversible dissociation of L to form LPd. |
| y-intercept of kobs vs [ArBr] | non-zero | non-zero | zero | ||
| Order in L | Zeroth | Zeroth | Nearly zeroth | ||
| Conclusion | One path by reaction of the ArBr with L2Pd, most likely by associative displacement of L to form LPd–ArX. A second competing pathway appears to occur that is zeroth order in ArBr, most likely by initial dissociation of L, followed by rapid reaction with ArBr. | Reaction of the ArBr with the bisphosphine complex L2Pd as the major pathway (by nearly irreversible associative displacement of L by PhBr prior to C–X bond cleavage or nearly irreversible direct oxidative addition) | |||
We propose that the iodoarenes react with the bisphosphine species in an irreversible process because they are softer and more reactive than the other haloarenes, while addition of chloroarenes requires generation of the more reactive monophosphine species because they are poorer ligands and require a more reactive intermediate to cleave the less reactive C–Cl bond. These conclusions are supported by the low barriers for oxidative addition of chloroarenes to monophosphine palladium(0) species and high barriers for oxidative addition of chloroarenes to bisphosphine palladium(0) species calculated recently by DFT methods.34,36
Supplementary Material
Acknowledgments
We thank the NIH RM-58108 for support of this work. We also thank Johnson-Matthey for a gift of palladium salts.
Footnotes
Supporting Information Available: Crystallographic data for 10, 11, and 15, experimental procedures, representative decays, and characterization of reactants. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Crabtree RH. The Organometallic Chemistry of the Transition Metals. 4. John Wiley & Sons, Inc; Hoboken, NJ: 2005. [Google Scholar]
- 2.Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i., editor. Vol. 1. John Wiley & Sons, Inc; Hoboken, N.J: 2002. p. 1051. [Google Scholar]
- 3.Hartwig JF. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; Weinheim, Germany: 2002. p. 107. [Google Scholar]
- 4.Yang BH, Buchwald SL. J Organomet Chem. 1999;576:125. [Google Scholar]
- 5.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 6.Shibasaki M, Vogl EM, Ohshima T. Adv Synth Catal. 2004;346:1533. [Google Scholar]
- 7.Suzuki A. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; Weinheim, Germany: 2002. p. 53. [Google Scholar]
- 8.Zapf A. In: Transition Metals for Organic Synthesis. 2. Beller M, Bolm C, editors. Vol. 1. Wiley-VCH; Weinheim, Germany: 2004. p. 211. [Google Scholar]
- 9.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419. [Google Scholar]
- 10.Herrmann WA. In: Applied Homogeneous Catalysis with Organometallic Compounds. 2. Cornils B, Herrmann WA, editors. Vol. 1. Wiley-VCH; Weinheim, Germany: 2002. p. 591. [Google Scholar]
- 11.Espinet P, Echavarren AM. Angew Chem Int Ed. 2004;43:4704. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]
- 12.Stille JK. Angew Chem Int Ed Engl. 1986;25:508. [Google Scholar]
- 13.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 14.Stambuli JP, Kuwano R, Hartwig JF. Angew Chem Int Ed Engl. 2002;41:4746. doi: 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]
- 15.Littke AF, Fu GC. Angew Chem Int Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Surry DS, Buchwald SL. Angew Chem Int Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunel JM. Mini-Reviews in Organic Chemistry. 2004;1:249. [Google Scholar]
- 18.Amatore C, Pfluger F. Organometallics. 1990;9:2276. [Google Scholar]
- 19.Fauvarque J-F, Pflüger F. J Organomet Chem. 1981;208:419. [Google Scholar]
- 20.Hartwig JF, Paul F. J Am Chem Soc. 1995;117:5373. [Google Scholar]
- 21.Barrios-Landeros F, Hartwig JF. J Am Chem Soc. 2005;127:6944. doi: 10.1021/ja042959i. [DOI] [PubMed] [Google Scholar]
- 22.Stambuli JP, Buhl M, Hartwig JF. J Am Chem Soc. 2002;124:9346. doi: 10.1021/ja0264394. [DOI] [PubMed] [Google Scholar]
- 23.Stambuli JP, Incarvito CD, Buehl M, Hartwig JF. J Am Chem Soc. 2004;126:1184. doi: 10.1021/ja037928m. [DOI] [PubMed] [Google Scholar]
- 24.Amatore C, Broeker G, Jutand A, Khalil F. J Am Chem Soc. 1997;119:5176. [Google Scholar]
- 25.Amatore C, Carre E, Jutand A, Mbarki MA. Organometallics. 1995;14:1818. [Google Scholar]
- 26.Amatore C, Jutand A. J Organomet Chem. 1999;576:254. [Google Scholar]
- 27.Galardon E, Ramdeehul S, Brown JM, Cowley A, Hii KK, Jutand A. Angew Chem Int Ed. 2002;41:1760. doi: 10.1002/1521-3773(20020517)41:10<1760::aid-anie1760>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 28.Portnoy M, Milstein D. Organometallics. 1993;12:1665. [Google Scholar]
- 29.Lewis AKdK, Caddick S, Cloke GN, Billingham NC, Hitchcock PB, Leonard J. J Am Chem Soc. 2003;125:10066. doi: 10.1021/ja035565k. [DOI] [PubMed] [Google Scholar]
- 30.Alcazar-Roman LM, Hartwig JF, Rheingold AL, Liable-Sands LM, Guzei IA. J Am Chem Soc. 2000;122:4618. [Google Scholar]
- 31.Alcazar-Roman LM, Hartwig JF. Organometallics. 2002;21:491. [Google Scholar]
- 32.Shekhar S, Ryberg P, Hartwig JF. Org Lett. 2006;8:851. doi: 10.1021/ol0528890. [DOI] [PubMed] [Google Scholar]
- 33.For computational studies of these effects, see the following three references.
- 34.Lam KC, Marder TB, Lin Z. Organometallics. 2007;26:758. [Google Scholar]
- 35.Ahlquist M, Fristrup P, Tanner D, Norrby P-O. Organometallics. 2006;25:2066. [Google Scholar]
- 36.Ahlquist M, Norrby P-O. Organometallics. 2007;26:550. [Google Scholar]
- 37.Stambuli JP. Ph.D. Dissertation, Yale University. 2003 [Google Scholar]
- 38.White LM, Morris RT. Anal Chem. 1952;24:1063. [Google Scholar]
- 39.Ozawa F, Kawasaki N, Okamoto H, Yamamoto T, Yamamoto A. Organometallics. 1987;6:1640. [Google Scholar]
- 40.Huser M, Youinou MT, Osborn JA. Angew Chem. 1989;101:1427. [Google Scholar]
- 41.Macgregor SA, Roe DC, Marshall WJ, Bloch KM, Bakhmutov VI, Grushin VV. J Am Chem Soc. 2005;127:15304. doi: 10.1021/ja054506z. [DOI] [PubMed] [Google Scholar]
- 42.Barrios-Landeros F, Carrow BP, Hartwig JF. J Am Chem Soc. 2008;130:5842. doi: 10.1021/ja711159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mann BE, Musco A. J Chem Soc Dalton Trans Inorg Chem. 1975:1673. [Google Scholar]
- 44.Musco A, Kuran W, Silvani A, Anker MW. J Chem Soc Chem Commun. 1973:938. [Google Scholar]
- 45.Mitchell EA, Baird MC. Organometallics. 2007;26:5230. [Google Scholar]
- 46.As we treated the data here, the linear curve fit at low [PCy3] corresponds to the hypothetical case of reaction of [Pd(PCy3)3] under these conditions. We did not fit the data to the full system that would include situations in which a significant concentration of both the [Pd(PCy3)3] and [Pd(PCy3)2] complexes are present.
- 47.Some reactions contained almost neat haloarene. However, the rates of these reactions are not strongly affected by solvent polarity (ref 18 and qualitative measurements with compound 2), and the dielectric constant of chlorobenzene is between that of benzene and THF. Thus, a high concentration of ArCl should not give rise to a large medium effect.
- 48.Yoshida T, Otsuka S. Inorg Synth. 1990;28:113. [Google Scholar]
- 49.Otsuka S, Yoshida T, Matsumoto M, Nakatsu K. J Am Chem Soc. 1976;98:5850. [Google Scholar]
- 50.The derivation of the rate expression is included in the Supporting Information.
- 51.Roy AH, Hartwig JF. Organometallics. 2004;23:1533. [Google Scholar]
- 52.Unimolecular decomposition of the Pd(0) complex can be excluded as a significant contributor to the value of the y-intercept because Pd(1-AdPtBu2)2 (2) was stable in toluene at 100 °C. Our values for the y-intercepts of the plots of kobs vs. [PhBr] in Figures 10 and 11 are similar. If these correspond to the rate constants for oxidative addition by an irreversible dissociation of ligand, one might expect the value of the y-intercept to be larger for reaction of the complex containing the more sterically bulky 1-AdPtBu2 compared to that containing CyPtBu2. However, comparison of these y-intercept values is complicated because they represent only an approximate value for the rate of ligand dissociation. A quantitative value for the rate constant for ligand dissociation k4 cannot be obtained from simple extrapolation of the plot of kobs vs [ArBr] over the linear range investigated because the plots become non-linear as [ArBr] approaches zero.
- 53.Roy AH, Hartwig JF. J Am Chem Soc. 2003;125:13944. doi: 10.1021/ja037959h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.