

Abstract
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterized by oligodendrocytic cytoplasmic inclusions containing abnormally aggregated α-synuclein. This aggregation has been linked to the neurodegeneration observed in MSA. Current MSA treatments are aimed at controlling symptoms rather than tackling the underlying cause of neurodegeneration. This study investigates the ability of the antibiotic rifampicin to reduce α-synuclein aggregation and the associated neurodegeneration in a transgenic mouse model of MSA. We report a reduction in monomeric and oligomeric α-synuclein and a reduction in phosphorylated α-synuclein (S129) upon rifampicin treatment. This reduction in α-synuclein aggregation was accompanied by reduced neurodegeneration. On the basis of its anti-aggregenic properties, we conclude that rifampicin may have therapeutic potential for MSA.
Keywords: astrogliosis, dendrites, oligodendrocytes, protein aggregation, synucleopathy
Introduction
Multiple system atrophy (MSA) is a sporadic, progressive, neurodegenerative disease with an estimated 25 000–100 000 Americans currently diagnosed with the disorder, it is characterized by clinical symptoms that can subdivided into motor and autonomic categories. Motor abnormalities such as akinesia, ridigidy and postural instability, are classed as either Parkinsonian-type (MSA-P) or cerebellar (MSA-C) and reflect damage to the basal ganglia (striato-nigral degeneration) or cerebellum (olivo-pontocerebellar atrophy), respectively. About 80% of MSA cases are categorized as MSA-P, whereas the remaining 20% are classified as MSA-C. Autonomic symptoms of MSA include orthostatic hypotension, increased sweating and loss of bowel and/or bladder control and reflect damage to the autonomic nervous system. In addition to motor and autonomic symptoms, MSA patients develop behavioral alterations such as attention deficits that suggest frontal lobe impairment in MSA [1,2].
Neuropathologically, MSA is characterized by glial cytoplasmic inclusions of abnormally aggregated α-synuclein (α-syn) [2]. Immunohistochemical analysis of the inclusions shows that they stain with antibodies to a variety of proteins in addition to α-syn including ubiquitin, tubulin, synphilin-1, and DJ-1 [2]. Although glial cytoplasmic inclusions are the primary neuropathological hallmark of MSA, neuronal cytoplasmic and nuclear inclusions of α-syn have also been reported. Neuronal loss occurs in the striatum, cerebellum, brainstem and cortex, and is accompanied by astrogliosis, microgliosis and myelin loss [2,3]. Current treatments for MSA patients are aimed at controlling their symptoms, as yet there are no treatments that halt or reverse the disease progression itself.
Many lines of evidence highlight the pathological importance of α-syn aggregation, both in vitro [4] and in Parkinson's disease [5]. Although the mechanisms that lead to this aggregation remain to be determined, factors including mitochondrial dysfunction [6] and oxidative stress [7] have been implicated. Disease progression is thought to be causally linked to accumulation and aggregation of α-syn and much effort has gone into devising strategies to combat this accumulation and aggregation [8]. The majority of work has been conducted in relation to the neuronal α-syn aggregation characteristic of Parkinson's disease, whereas little has focused on mechanisms underlying the oligodendroglial aggregation seen in MSA.
Recent studies with rifampicin, a common antibiotic treatment for tuberculosis and leprosy, have highlighted its ability to inhibit α-syn aggregation and disaggregate preformed fibrils in vitro [9,10]. In this context, this study sought to examine the possible beneficial effects of rifampicin in transgenic (tg) mice expressing human α-syn under the myelin basic protein (MBP) promoter. These MBP-α-syn tg mice have been shown to exhibit the oligodendroglial aggregates of α-syn and motor deficits characteristic of MSA [11].
Materials and methods
Generation of myelin basic protein-α-syn tg mice
Mice expressing human α-syn under the control of the MBP promoter were generated as described [11]. Genomic DNA was extracted from tail biopsies and analyzed by PCR amplification as described [12]. These mice have previously been shown to develop α-syn inclusions in oligodendrocytes from 3 months of age and to display neuropathology including myelin loss and astrogliosis and are proposed to be a model for MSA [11]. Mice used for this study were an average of 12 months old upon onset of rifampicin (or saline) treatment.
Rifampicin treatment
Thirty-four mice were used for this study and received daily intraperitoneal injections with either 25 mg/kg rifampicin (MBP-α-syn tg mice, n=10 and non-tg mice n=8) or saline (MBP-α-syn tg mice, n=8 and non-tg mice n=8) daily for 3 months. The rifampicin dose used in this study was based on published literature examining brain bioavailability after peripheral administration of rifampicin [13–15].
Tissue processing
Following NIH guidelines for the humane treatment of animals, under anesthesia mice were sacrificed and brains removed. The right hemibrain was immersion-fixed in 4% paraformaldehyde in pH 7.4 phosphate-buffered saline and serially sectioned at 40 μm with a Vibratome (Leica, Deerfield, Illinois, USA). The left hemibrain was kept at −80°C for biochemical analysis.
Immunohistochemistry
Forty micromolar vibratome sections were immunolabeled overnight with antibodies against α-syn using monoclonal (1 : 1000, BD Biosciences, San Jose, California, USA) or polyclonal (1 : 1000, Chemicon, Temecula, California, USA) antibodies, phosphorylated α-syn (1 : 1000, Millipore), nitrosylated α-syn (1 : 1000, Millipore), the dendritic marker, microtubule-associated protein-2 (MAP2; 1 : 1000, mouse monoclonal; Chemicon), the neuronal marker NeuN (1 : 1000, Chemicon) and the astroglial marker glial fibrillary acidic protein (GFAP), followed by incubation with species-appropriate secondary antibodies (1 : 2000, Vector Laboratories). Sections were transferred to SuperFrost slides (Fisher Scientific, California, USA) and mounted with anti-fading media (Vector Laboratories). The immunolabeled blind-coded sections were analyzed with the laser scanning confocal microscope (MRC1024, BioRad) to evaluate the area of the neuropil covered by MAP2 immunoreactive dendrites and the number of α-syn immunoreactive oligodendrocytes. Stereological analysis was also conducted, as previously described [16] to examine the neuronal density as evidenced by NeuN immunoreactivity and astrogliosis as determined by GFAP immunoreactivity.
Western blot analysis
Protein levels of total α-syn, phosphorylated α-syn (S129), nitrosylated α-syn and β-syn were determined by immunoblot analysis. Briefly, tissue was processed to obtain the detergent –insoluble fraction as previously described [11], 20 μg of total protein per mouse were loaded onto 10% Bis-Tris (Invitrogen) SDS-PAGE gels, transferred onto Immobilon membranes, incubated with antibodies against α-syn (1 : 1000, Chemicon), phosphorylated α-syn (S129) (1 : 1000, Millipore), nitrosylated α-syn (1 : 1000, Millipore) and β-syn (1 : 1000, Chemicon). After overnight incubation with primary antibodies, membranes were incubated in appropriate secondary antibodies, reacted with ECL, and developed on a VersaDoc gel-imaging machine (Bio-Rad, Hercules, CA). Anti-β-actin (1 : 1000, Sigma) antibody was used to confirm equal loading.
Statistical methods
Differences between groups were tested using one and two factor analysis of variance (ANOVA) with Fisher PLSD post-hoc tests. All the results are expressed as mean±SEM.
Results
Rifampicin reduced α-synuclein accumulation in myelin basic protein-α-syn tg mice
Consistent with previous studies, vehicle-treated MBP-α-syn tg mice displayed abundant inclusions immunoreactive for α-syn (Fig. 1a), phosphorylated α-syn (Fig. 1d) and nitrosylated α-syn (Fig. 1g). Rifampicin-treated MBP-α-syn tg mice displayed significantly reduced α-syn, phosphorylated α-syn and nitrosylated α-syn immunoreactivity in comparison to vehicle-treated MBP-α-syn tg mice (Fig. 1b, e and h-analyzed in c, f, i and l, respectively).
Fig. 1.
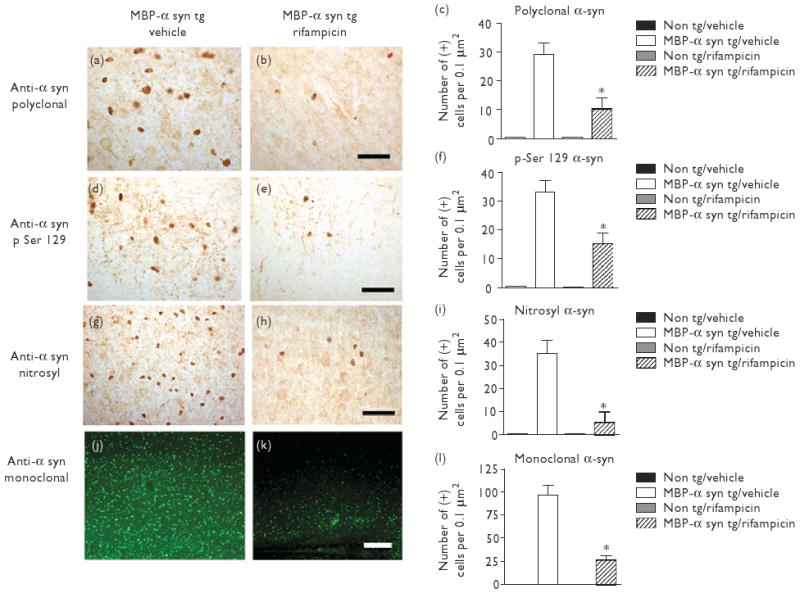
Rifampicin reduced α-syn accumulation in MBP-α-syn tg mice. Brightfield microscope images of α-syn immunoreactivity in vehicle-treated MBP-α-syn tg mice (a), rifampicin-treated MBP-α-syn tg mice (b), phosphorylated α-syn (S129) in vehicle-treated MBP-α-syn tg mice (d), rifampicin-treated MBP-α-syn tg mice (e), nitrosylated α-syn immunoreactivity in vehicle-treated MBP-α-syn tg mice (g), and rifampicin-treated MBP-α-syn tg mice (h). Confocal microscope images of α-syn immunoreactivity in vehicle-treated MBP-α-syn tg mice (j) and rifampicin-treated MBP-α-syn tg mice (k). Scale bars represent 50 μM (b, e, and h) and 100 μM (k). c, f, i, and l represent stereological analyses of the number of α-syn-immunoreactive neurons (polyclonal antibody), phosphorylated α-syn (S129), nitrosylated α-syn and monoclonal α-syn, respectively.*Significant difference between rifampicin-treated MBP-α-syn tg mice (n=10) in comparison with vehicle-treated MBP-α-syn mice (n=8) (P<0.05, one-way ANOVA and post-hoc Fisher). Alpha-syn, α-synuclein; ANOVA, analysis of variance; MBP, myelin basic protein.
In line with immunohistochemical results, western blot analysis revealed a significant increase in both monomeric and oligomeric forms of α-syn, phosphorylated α-syn (S129) and nitrosylated α-syn in vehicle-treated MBP-α-syn tg mice in comparison to non-tg controls (Fig. 2a). Rifampicin-treated MBP-α-syn tg mice displayed significantly lower levels of monomeric and oligomeric forms of α-syn, phosphorylated α-syn (S129) and nitrosylated α-syn immunoreactivity in comparison to vehicle-treated MBP-α-syn tg mice (Fig. 2a, analyzed in b, c, d and e respectively). No differences in β-syn immunoreactivity were evident (Fig. 2a).
Fig. 2.
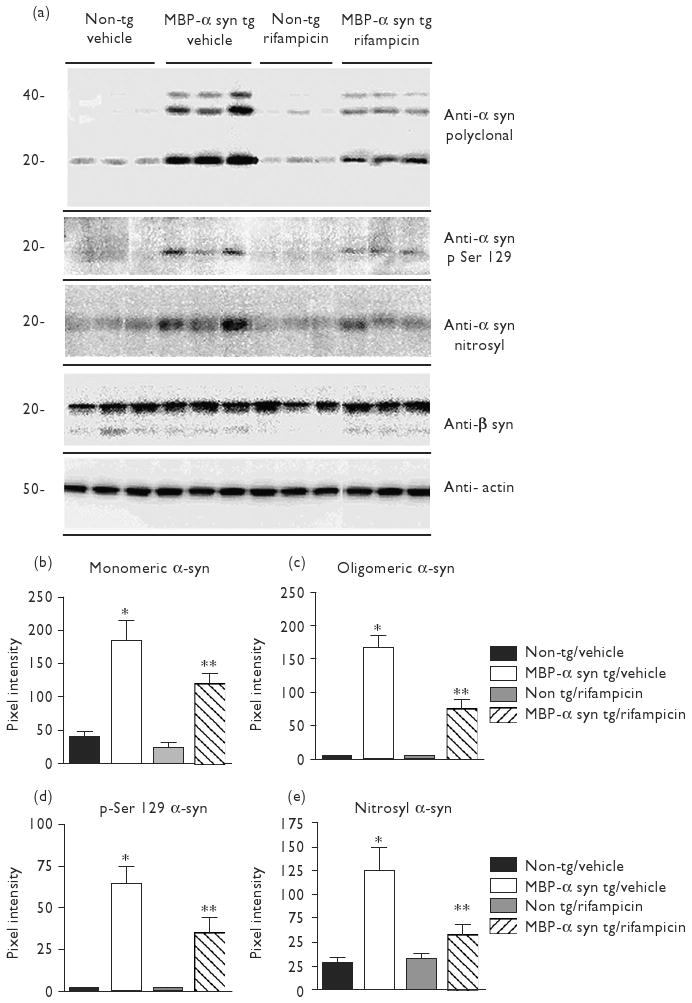
Rifampicin reduced α-syn accumulation in MBP-α-syn tg mice. Western blot analysis of α-syn, phosphorylated α-syn (S129), nitrosylated α-syn, and β-syn. Actin was used as a loading control (a). All blots were loaded with protein from the detergent-insoluble fraction. (b–e) Analyses of immunoreactivity for monomeric α-syn, oligomeric α-syn, phosphorylated α-syn (S129), and nitrosylated α-syn, respectively. Immunoreactivity signal was normalized over Actin. *Significant difference between vehicle-treated MBP-α-syn tg mice (n=8) in comparison with vehicle-treated non-tg controls (n=8) (P<0.05, one-way ANOVA and post-hoc Fisher). **Signifcant difference between rifampicin-treated MBP-α-syn tg mice (n=10) in comparison with vehicle-treated MBP-α-syn tg mice (n=8) (P<0.05, one-way ANOVA and post-hoc Fisher). Alpha-syn, α-synuclein; ANOVA, analysis of variance; MBP, myelin basic protein.
Rifampicin reduced neurodegeneration in myelin basic protein-α-syn tg mice
In order to evaluate whether the ability of rifampicin to reduce α-syn aggregation led to an amelioration of the neuropathological alterations previously reported in MBP-α-syn tg mice [11], dendritic complexity, evidenced by MAP2 immunoreactivity, neuronal number, determined by NeuN immunoreactivity and astrogliosis as determined by GFAP immunoreactivity were analyzed.
Consistent with earlier studies, analysis of neuropil area revealed a significant decrease in MAP2 immunoreactivity in vehicle-treated MBP-α-syn tg mice in comparison to vehicle-treated non-tg controls (Fig. 3a, b and d, e at higher magnification). Stereological analysis of neuronal number revealed a significant decrease in NeuN immunoreactivity in vehicle-treated MBP-α-syn tg mice in comparison to vehicle-treated non-tg controls (Fig. 3h and i). GFAP was significantly increased immunoreactivity in vehicle-treated MBP-α-syn tg mice in comparison with vehicle-treated non-tg controls Fig. 3l and m).
Fig. 3.
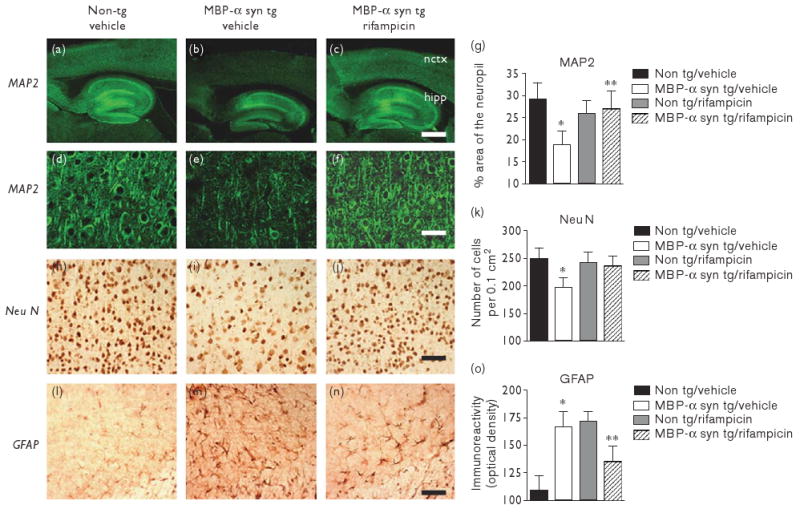
Rifampicin reduced neurodegeneration in MBP-α-syn tg mice. Low-power confocal microscopic images of MAP2 immunoreactivity in vehicle-treated non-tg mice (a), vehicle-treated MBP-α-syn tg mice (b), and rifampicin-treated MBP-α-syn tg mice (c). Higher power confocal microscopic images of MAP2 immunoreactivity in vehicle-treated non-tg mice (d), vehicle-treated MBP-α-syn tg mice (e), and rifampicin-treated MBP-α-syn tg mice (f). Neocortical analysis of the area of the neuropil covered by MAP2 immunoreactivity was performed (g). Bright-field images of NeuN immunoreactivity in vehicle-treated non-tg mice (h), vehicle-treated MBP-α-syn tg mice (i) and rifampicin-treated MBP-α-syn tg mice (j) and GFAP immunoreactivity in vehicle-treated non-tg mice (l), vehicle-treated MBP-α-syn tg mice (m) and rifampicin-treated MBP-α-syn tg mice (n). Stereological analysis of NeuN immunoreactive neurons (k) and analysis of GFAP immunoreactivity (o). Scale bars represent 200 μM (a–c) and 50 μM (d–n). *Significant difference between vehicle-treated MBP-α-syn tg mice (n=8) in comparison with vehicle-treated non-tg controls (n=8) (P<0.05, one-way ANOVA and post-hoc Fisher). **Significant difference between rifampicin-treated MBP-α-syn tg mice (n=10) in comparison with vehicle-treated MBP-α-syn tg mice (n=8) (P<0.05, one-way ANOVA and post-hoc Fisher). ANOVA, analysis of variance; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein.
Rifampicin ameliorated the loss of dendritic complexity, with the area of the MAP2-immunoreactive neuropil being comparable between rifampicin-treated MBP-α-syn tg mice and vehicle-treated non-tg controls (Fig. 3c, f and analyzed in g). Rifampicin also ameliorated neuronal loss with increased NeuN immunoreactivity levels being evident in rifampicin-treated MBP-α-syn tg mice in comparison to vehicle-treated MBP-α-syn tg mice, levels of NeuN immunoreactivity in rifampicin-treated MBP-α-syn tg mice were comparable with those seen in non-tg controls (Fig. 3j and analyzed in k). Rifampicin of MBP-α-syn tg mice also resulted in significantly lower levels of GFAP immunoreactivity in comparison with vehicle-treated MBP-α-syn tg mice (Fig. 3n and analyzed in o).
Discussion
This study sought to investigate possible beneficial effects of the antibiotic rifampicin in the MBP-α-syn tg model of MSA. We demonstrated that rifampicin was able to reduce α-syn aggregation in MBP-α-syn tg mice, this was accompanied by reduced neurodegeneration, reflected by a restoration of dendritic complexity as evidenced by MAP2 immunoreactivity, neuronal number as evidenced by NeuN immunoreactivity and a reduction in astrogliosis as evidenced by the astrocytic marker GFAP. In addition to displaying reduced levels of total α-syn, rifampicin-treated MBP-α-syn tg mice also displayed reduced phosphorylated α-syn (S129) immunoreactivity in comparison with vehicle-treated MBP-α-syn tg mice. The serine-129 phosphoepitope has been shown to be pathogenically important [17] and reduction in the immunoreactivity for phosphorylated α-syn in the rifampicin-treated mice may suggest a possible mechanism by which rifampicin exerts its neuroprotective effect.
The mechanisms by which rifampicin reduces α-syn aggregation are under investigation, rifampicin is one of a growing list of compounds including curcumin and current treatments for Parkinson's disease such as selegiline, dopamine, and bromocriptine, which are reported to inhibit the formation of α-syn fibrils and destabilize preformed fibrils [9,10,18]. The exact mechanisms by which aggregation of α-syn leads to neurodegeneration remain unclear, however, it is becoming increasing apparent that agents that reduce or reverse the aggregation process are neuroprotective [8,10,18]. We propose that the antifibrillogenic and fibril-destabilizing actions of rifampicin play a key role in its ability to reduce neurodegeneration in the MBP-α-syn tg mice. Alpha-syn aggregation has been linked to oxidative stress with antioxidant compounds having been shown to exert beneficial effects in vitro [10]. In this context, it is interesting to note that treatment with rifampicin, which has itself been reported to have antioxidant properties, led to decreased nitrosylated α-syn immunoreactivity, in comparison with vehicle-treated MBP-α-syn tg mice.
Rifampicin has long been a component of the treatment for leprosy and as such there is a wealth of evidence regarding its therapeutic tolerance. Studies have also shown that rifampicin can cross the blood–brain barrier, though CSF levels are much lower than those found in the plasma [13–15]. In this study we administered rifampicin intraperitoneally and demonstrate an effect in the brain, indicating that regardless of issues with bioavailability, sufficient levels of rifampicin can cross the blood–brain barrier to have a beneficial effect.
Conclusion
This study presents the first in-vivo evidence that rifampicin is capable of reducing monomeric, oligomeric, and phosphorylated α-syn accumulation and of ameliorating the neuronal loss, reduction of dendritic complexity and increased astrogliosis associated with increased expression of α-syn. We demonstrate the beneficial effects of rifampicin on the neuropathology associated with α-syn aggregation, it would be interesting to expand this and assess its effects on clinical symptoms. Results from this study indicate that rifampicin may have potential be used to treat disorders which are characterized by α-syn accumulation, such as MSA, and unlike current treatments, may go someway toward treating the underlying cause.
Acknowledgments
This study was funded by NIH Grants NS044233, AG18440 and AG022074.
References
- 1.Beyer K, Ariza A. Protein aggregation mechanisms in synucleinopathies: commonalities and differences. J Neuropathol Exp Neurol. 2007;66:965–974. doi: 10.1097/nen.0b013e3181587d64. [DOI] [PubMed] [Google Scholar]
- 2.Wakabayashi K, Takahashi H. Cellular pathology in multiple system atrophy. Neuropathology. 2006;26:338–345. doi: 10.1111/j.1440-1789.2006.00713.x. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida M. Multiple system atrophy: alpha-synuclein and neuronal degeneration. Neuropathology. 2007;27:484–493. doi: 10.1111/j.1440-1789.2007.00841.x. [DOI] [PubMed] [Google Scholar]
- 4.El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E, et al. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 1998;440:71–75. doi: 10.1016/s0014-5793(98)01418-5. [DOI] [PubMed] [Google Scholar]
- 5.Batelli S, Albani D, Rametta R, Polito L, Prato F, Pesaresi M, et al. DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson's disease and involvement of HSP70. PLoS ONE. 2008;3:e1884. doi: 10.1371/journal.pone.0001884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 7.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Windisch M, Wolf HJ, Hutter-Paier B, Wronski R. Is alpha-synuclein pathology a target for treatment of neurodegenerative disorders? Curr Alzheimer Res. 2007;4:556–561. doi: 10.2174/156720507783018343. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Zhu M, Rajamani S, Uversky VN, Fink AL. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol. 2004;11:1513–1521. doi: 10.1016/j.chembiol.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Ono K, Yamada M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 2006;97:105–115. doi: 10.1111/j.1471-4159.2006.03707.x. [DOI] [PubMed] [Google Scholar]
- 11.Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 13.Mindermann T, Landolt H, Zimmerli W, Rajacic Z, Gratzl O. Penetration of rifampicin into the brain tissue and cerebral extracellular space of rats. J Antimicrob Chemother. 1993;31:731–737. doi: 10.1093/jac/31.5.731. [DOI] [PubMed] [Google Scholar]
- 14.Mindermann T, Zimmerli W, Gratzl O. Rifampin concentrations in various compartments of the human brain: a novel method for determining drug levels in the cerebral extracellular space. Antimicrob Agents Chemother. 1998;42:2626–2629. doi: 10.1128/aac.42.10.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yulug B, Kilic U, Kilic E, Bahr M. Rifampicin attenuates brain damage in focal ischemia. Brain Res. 2004;996:76–80. doi: 10.1016/j.brainres.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Kuczenski R, Everall IP, Crews L, Adame A, Grant I, Masliah E. Escalating dose-multiple binge methamphetamine exposure results in degeneration of the neocortex and limbic system in the rat. Exp Neurol. 2007;207:42–51. doi: 10.1016/j.expneurol.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 18.Ono K, Hirohata M, Yamada M. Anti-fibrillogenic and fibril-destabilizing activities of anti-Parkinsonian agents for alpha-synuclein fibrils in vitro. J Neurosci Res. 2007;85:1547–1557. doi: 10.1002/jnr.21271. [DOI] [PubMed] [Google Scholar]