

Abstract
Long-lived mutants provide unique insights into the genetic factors that limit lifespan in wild-type animals. Most mutants and RNA-interference targets found to extend life, typically by 1.5- to 2.5-fold, were discovered in C. elegans. Several longevity-assurance pathways are conserved across widely divergent taxa, indicating that mechanisms of lifespan regulation evolved several hundred million years ago. Strong mutations to the C. elegans gene encoding AGE-1/PI3KCS achieve unprecedented longevity by orchestrating the modulation (predominantly silencing) of multiple signaling pathways. This is evident in a profound attenuation of total kinase activity, leading to reduced phosphoprotein content. Mutations to the gene encoding the catalytic subunit of PI3K (phosphatidylinositol 3-kinase) have the potential to modulate all enzymes that depend on its product, PIP3, for membrane tethering or activation by other kinases. Remarkably, strong mutants inactivating PI3K also silence multiple signaling pathways at the transcript level, partially but not entirely mediated by the DAF-16/FOXO transcription factor. Mammals have a relatively large proportion of somatic cells, and survival depends on their replication, whereas somatic cell divisions in nematodes are limited to development and reproductive tissues. Thus, translation of longevity gains from nematodes to mammals requires disentangling the downstream consequences of signaling mutations, to avoid their deleterious consequences.
Keywords: Lifespan, Longevity, Caenorhabditis elegans, Insulin, IGF-1 (insulin-like growth factor 1)
Introduction
Genetic mutations capable of extending metazoan lifespan were first discovered in the nematode C. elegans [1-8]. Mutations with substantial benefits to lifespan were later discovered in Drosophila, mice and rats, either by targeted quests for mutations in the corresponding pathway of these species [9-12] or by independent discovery [13-17]. Conserved mutations have also been found to increase “lifespan” of yeast, by several operational definitions [18-21].
Gene mutations, if random, are expected to impair function far more often than improve it [22]. Thus, finding that mutations frequently augment lifespan (e.g., see www.wormbase.org) implies that the mutated genes have primary functions other than ensuring survival. Genes that instead reduce longevity when they are mutated or knocked down have been termed “longevity assurance genes” (see [23,24]), on the assumption that their normal function serves to extend life. In this case, a possible trivial explanation is that they may affect essential functions or pathways, so that their impairment reduces physiological fitness. The appropriate test of a gene's role in longevity attainment is to assess whether life is extended through its over-expression. By studying single-gene mutations, RNA interference, and (more rarely) transgene overexpression, several signal-transduction pathways have been shown to modulate longevity [25-29]. Accumulating evidence indicates that these pathways intersect one another, to form a network of protective and regulatory genes that influence survival in diverse circumstances [28,30-39]. Environmental conditions, such as nutrients, stresses and pathogens, also impact lifespan. Although these “external” influences could perturb survival directly or by altering tissue metabolites, in several instances they have been shown to exert at least part of their effect through the same signal-transduction pathways that were demonstrated genetically to influence longevity [11,20,28,31,33,35,40-43].
A single-gene mutation in the nematode C. elegans was recently shown to increase adult survival by tenfold, exceeding the previous record by a factor of at least three [44]. Median lifespan was boosted from ∼16 days at 20°C, to over 5 months, with comparable effects on mean and maximal (90th percentile) lifespan. Four- to six-fold extensions of nematode lifespan had been achieved previously through a combination of two or three interventions [45-47]. Similarly, a ten-fold increase in yeast lifespan was recently attained by combining three interventions: two mutations plus severe caloric restriction [18]. These results suggest that in both taxa, several parallel mechanisms curtail normal lifespan, and that the benefits of subverting them are “additive” — meaning that their combined effect is roughly the sum of those seen for individual factors.
Nematode lifespan is limited by the normal operation of multiple genes
Reduction-of-function mutations that were discovered through mutagenesis screens, chiefly testing traits other than longevity, can individually enhance C. elegans lifespan by factors of 1.1- to nearly 3-fold. The first longevity-conferring mutation, an allele of age-1, came out of a sib-screen for long-lived mutants arising after chemical mutagenesis [1]. The temperature-sensitive (ts) age-1(hx546) mutant, when homozygous, extends life by 40-65% [8]. A very similar protocol, of EMS mutagenesis and sib screening for longevity, yielded only one gene, termed age-2 [48]; a third mutagenesis screen, in which four stress-response assays served as surrogates for lifespan, produced only additional alleles of age-1 [49]. These rather limited returns on sizeable investments of effort are all the more remarkable in view of the many longevity-enhancing mutations discovered by other means. The daf-2 mutation, initially discovered as a ts mutant featuring constitutive production of dauer larvae at ≥25°C [50], was much later tested for adult lifespan and found to extend its normal duration by almost two-fold [6]. When 15 independent daf-2 alleles were crossed into an isogenic background, they extended normal lifespan to quite variable degrees: from as little as 10%, up to 2.5-fold [51]. The protein encoded by daf-2 is a membrane receptor-kinase responsive to many insulinlike ligands (both agonists and antagonists), whereas the age-1 product is the p110 catalytic subunit of class-I PI3 kinase (PI3KCS), responsible for converting PIP2 to PIP3. These two kinases participate in the insulin/IGF-1 signaling (IIS) pathway of nematodes, which regulates dauer formation, fertility, stress response, and lifespan. Further downstream, and excluded from the nucleus by phosphorylation via the DAF-2/AGE-1/PDK-1/AKT kinase cascade, lies DAF-16/FOXO — a Forkhead or winged-helix transcription factor encoded by the daf-16 gene. When IIS is blocked, DAF-16 enters the nucleus to regulate transcription of several hundred genes, many of which are involved in survival and stress responses. All known phenotypes of either age-1 or daf-2 mutations can be largely or entirely reversed by a second mutation inactivating daf-16 [30,44,45,52,53]. Rescue experiments, restoring wild-type daf-2 or daf-16 to individual cell types in mutant worms, demonstrate that IIS effects on lifespan are tissue specific [54,55], and imply critical roles of neurons and gut in the regulation of nematode longevity.
Of all the life-prolonging mutations discovered in the nematode C. elegans, until recently none had surpassed the 1.5- to 3-fold increases reported over a decade ago for disruptions of the insulin/IGF-1 signaling (IIS) pathway [5,45,51,52]. Checking for convergence with the IIS pathway has thus become a “default test” for all new longevity mutations, to determine whether they are dependent or independent of DAF-16. Once IIS and other pathways were shown to extend lifespan in the nematode when disrupted, they were cross-checked in short order for corresponding effects in other species. In general, similar mutations have produced more modest life extensions in other taxa, and in many cases no effects were seen. In Drosophila, very few single-gene mutations increase longevity by more than 2-fold [10,56], and none have exceeded 1.5-fold in mice [9,10,12,13,16,57,58].
Greater extensions of lifespan can be attained by combining several interventions. In particular, specific pairings of mutated alleles for daf-2 and daf-12 (encoding a nuclear hormone receptor) can increase nematode longevity by 3.5-fold [45] or 4.4-fold [51]. Multiple interventions, such as a daf-2 mutation combined with germ-cell ablation or caloric restriction, can increase C. elegans lifespan by 4- to 6-fold [46,47]. In mice, a mutation impairing pituitary development extends lifespan by 35–40%; when combined with caloric restriction, it yielded a record-setting 1.8-fold extension [16]. Thus, there exist in each species multiple pathways for life extension, effects of which are largely additive. This is often assumed to imply their independence, although “additivity” can also arise in a single pathway if each of the constituent mutations disrupts it only partially.
Several clear patterns have emerged. As longer-lived species are examined, the gains to be reaped from interventions are diminished. This is precisely what would be expected if longevity is subject to natural selection like other life-history traits: longer-lived species presumably have already been subjected to selective pressure to establish life-extending mutations as the norm. Another recurring observation is that long-lived mutations generally confer resistance to stresses [15,59,60]. However, several natural genetic variants for lifespan, quantitative trait alleles, vary widely in resistance to specific stresses [61], so it may be unwise to rely on any single stress-resistance test as a short-term surrogate for assessing longevity [49,60].
Null mutations in the age-1 gene extend C. elegans lifespan by ten-fold
C. elegans strains carrying a nonsense mutation of the age-1 gene (either the mg44 or the m333 allele) cannot be maintained as homozygous-mutant lines, but instead are propagated as heterozygotes, in which the mutated gene copy is offset by a wild-type allele on a “balancer” chromosome, which also carries visible mutations as markers. These worms self-fertilize (C. elegans reproduction being primarily hermaphroditic), to yield roughly one-quarter of total progeny homozygous for the age-1 mutation. These age-1 homozygotes may be loosely termed the “F1” generation, and their progeny would then be “F2” homozygotes. F2 mutants for these nonsense alleles, although genetically identical to their F1 parents, are clearly quite different in many respects. They develop very slowly at 15° or 20°C, and their development arrests completely at 25°C [44]. The adults are far more resistant to both electrophilic and oxidative stresses, and live 10-fold longer than normal, wild-type worms under benign conditions ([44] and Figure 1). Their lifespans are uniformly extended, with 25th, 50th (median), 75th and 95th percentiles all nearly 10 times those seen in near-isogenic worms bearing wild-type age-1 alleles.
Figure 1. Survivals of F2 age-1(mg44) homozygotes vs. N2drm controls.
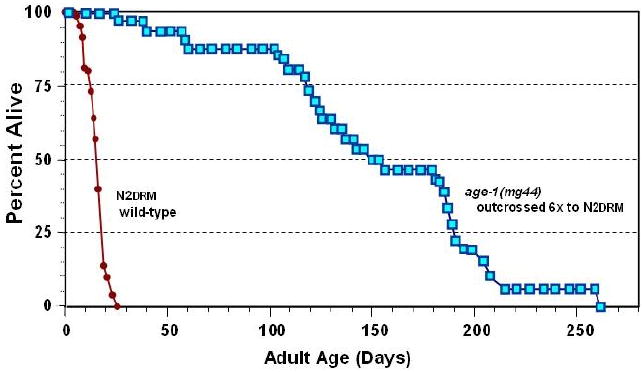
Survivals are plotted for age-1(mg44) F2 (squares) and N2drm (circles). (Redrawn from [44]).
In appearance and activity, they resemble normal worms at a tenth their age. The F1 homozygous generation, their parents, show intermediate stress resistance [44] and roughly a doubling of wild-type lifespan [62]. The dramatic difference between these two generations, despite identical genes, is almost certainly a result of normal components in the egg cytoplasm that gives rise to F1-homozygous worms. The parent that produced this cytoplasm was an age-1/+ heterozygote, which would have produced essentially normal age-1 mRNA, PI3K enzyme, and its PIP3 product.
The age-1 gene of C. elegans encodes the catalytic (p110) subunit of phosphatidylinositol 3-kinase. The PI3K enzyme family transfers a phosphate from ATP to the 3 position of the inositol ring at the head of phosphatidylinositides. Specific PI3K enzymes convert phosphatidylinositol (abbreviated PtdIns or PI) to the monophosphate, PtdIns(3)P; PtdIns(4)P to PtdIns(3,4)P2; and PtdIns(4,5)P2 (PIP2) to PtdIns(3,4,5)P3 (PIP3) (Figure 2). The mammalian tumor-suppressor gene Pten encodes the Pten phosphatase which removes the 3-phosphate from PIP3., thus opposing PI3K action. PTEN in nematodes, like Pten of mammals, converts PtdIns(3,4,5)P3 to PtdIns(4,5)P2, and PtdIns(3,4)P2 to PtdIns(4)P [63-65]. Mammals also have SHIP1 and SHIP2 phosphatidylinositol 5-phosphatases, which can deplete PIP3 by removal of the 5-phosphate to form PtdIns(3,4)P2. There are three classes of mammalian PI3K (I, II, and III), which differ in substrates, structure and activators. The C. elegans AGE-1 protein is a class-I PI3K catalytic subunit, based on its sequence and structure, which serves to convert PI(4,5)P2 to PI(3,4,5)P3.
Figure 2. Structure of PIP3.
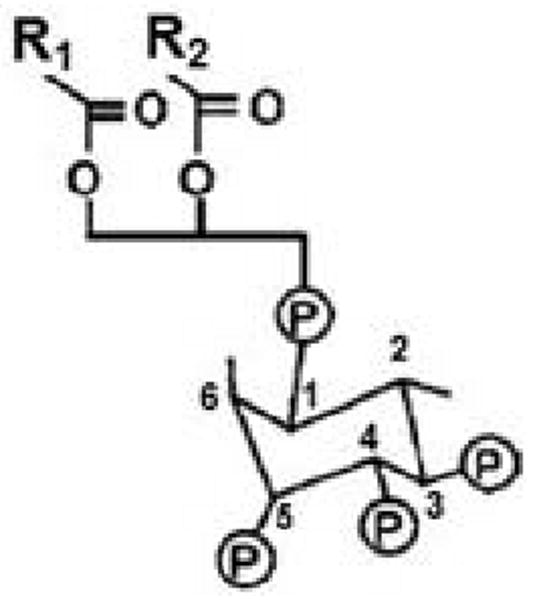
R1 and R2 are fatty-acid chains that vary among different molecules.
All class-I PI3K enzymes are heterodimeric, consisting of a catalytic (p110) subunit, of which mammals have four varieties [65] but C. elegans has only one (AGE-1; see WormBase); and a regulatory subunit, of which mammals have seven known varieties [65], while worms are only known to have one, AAP-1 (WormBase). AGE-1 most closely resembles p110-α and -β catalytic subunits [62], narrowing its classification to class IA. Because nematodes have fewer PI3K varieties than mammals, AGE-1 may share some properties of mammalian class-IB PI3Ks. F39B1.1 encodes another C. elegans p110 protein, which is more closely akin to a mammalian class-II PI3K. The third nematode PI3K p110 gene, vps-34, is an ortholog of the yeast vps34 gene, encoding a class-III PI3KCS. Class-III PI3Ks are implicated in vesicular trafficking, autophagy, endocytosis and secretion [66]. In keeping with this, yeast VPS34 governs endocytosis, and C. elegans vps-34 is thought to be involved in endocytic trafficking between cell compartments, including that of low-density lipoprotein (LDL) receptors [67]. Class-II and -III PI3Ks phosphorylate the 3 position of PI(4,5)P2 with very low efficiency in vitro, where they show a marked preference for other PIP substrates [62]. If some substrate promiscuity also occurred in vivo, it would blunt the phenotype of age-1-null mutants.
Roles of phosphatidylinositol 3,4,5-phosphate (PIP3) in signal transduction
Under normal circumstances, PtdIns(3,4,5)P3 (PIP3) is roughly a thousand-fold less abundant than its precursor, PtdIns(4,5)P2, which is itself a quite scarce molecule. PIP3 levels increase up to 100-fold in activated cells [68], and are detectable in nematodes only after they are induced by starvation or oxidative stress [69]. The exceedingly low levels of this molecule, together with the fact that PTEN (which removes the 3-position phosphate of PIP3) acts as a tumor suppressor in mammals, imply that PIP3 is far from innocuous and must be stringently controlled to prevent undue cell proliferation. The role of PI3K in IIS is believed to proceed entirely through PIP3 generation [64,65,70], although the p110 catalytic subunit also has protein-kinase activity, as demonstrated by phosphorylation of its regulatory subunit [34,71]. PIP3 molecules carry two fatty-acid chains (R1 and R2 in Figure 2) that anchor it to the inner cell membrane. The hydrophilic phosphatidylinositol headgroup projects into the cytoplasm where it is avidly bound by proteins possessing a pleckstrin-homology (PH) domain. IIS proteins that dock to PIP3 include PDK-1 (phosphoinositide-dependent kinase 1), AKTs, and SGK-1 [65,72]. Once they are tethered to the membrane, the IIS components are effectively constrained to the same 2-dimensional surface, greatly promoting their interaction by elevating their effective local concentrations with respect to one another.
Tethered PDK-1 phosphorylates AKT-1 at a Thr residue, thereby activating it and allowing the AKT complex to phosphorylate the DAF-16/FOXO transcription factor. The latter is an inhibitory modification, restraining DAF-16 to the cytoplasm where it cannot act on its target genes [52,73]. PIP3 not only tethers the AKT complex to the membrane, but its binding to the AKT-1 pleckstrin-homology domain also allosterically exposes a phosphorylation site to PDK-1, which is thus able to activate AKT-1 [65,72]. Thus, in addition to a stoichiometric requirement for PIP3 to enable anchoring of AKT-1 (and many other signal-transduction molecules) to membranes, PIP3 also may play an essentially catalytic role to transiently alter the conformation of AKT-1 (and possibly other targets), thus permitting their site-specific phosphorylation. We hypothesize that PIP3 dissociates and is subsequently bound by other target molecules, in which case a single molecule of PIP3 may be sufficient to activate all of a cell's AKT-1, although more PIP3 would be needed for membrane-tethering of AKTs and other targets. Insofar as the anchoring role of PIP3 is a quantitative one (working, in effect, by mass action), whereas the allosteric requirement for activation is absolute, it is likely that removing the last traces of PIP3 from a cell would block the last vestiges of AKT-mediated signaling, and also any other pathways in which PIP3 plays a comparable allosteric role.
This may account for the markedly enhanced survival of F2 homozygous age-1(mg44) mutants relative to their F1 parents, in which there still retain traces of PIP3 carried over from their heterozygous parents. If so, then the secret of extreme longevity may be to remove any remaining traces of PIP3-dependent signaling — a state which only allows C. elegans to develop, quite slowly, at lower temperatures. In higher eukaryotes, with requirements for cell division not only during development but also in specific adult tissues, this will be far more challenging to achieve without sacrificing fitness under some circumstances.
Loss of PI3K disrupts multiple protein-kinase activities
We assessed kinase activity in normal and age-1-mutant worms using an in vitro phosphorylation assay. As illustrated in Figure 3, F2 age-1(mg44) lysates show remarkably low kinase activities toward endogenous substrates — less than 8% of levels seen in wild-type worms. Activity was also low in the F1 generation, although higher than in F2 worms (P<0.05; Figure 3c). Steady-state levels of phosphoproteins were accordingly depressed in F2 adults, by >40% relative to controls [30]. Kinase activity was also reduced ∼30% in age-1(hx5456), a widely-studied but weaker mutant allele (Fig. 3c). The decline in hx546 appears to be entirely mediated by DAF-16/FOXO or other transcription factors further downstream, since it is reversed in double mutants also lacking functional DAF-16/FOXO. However, only about half of the deficit is reversed in age-1(mg44) F2 adults, implying that this strong-mutant allele employs both DAF-16/FOXO-dependent and -independent routes (Figure 3, c and d). The F2 mutants are sterile, and consequently for comparison the other strains were all assessed when post-gravid. The presence of residual eggs in other strains could not account for the >12-fold decrease in kinase activity of F2 age-1(mg44) adults, since N2drm eggs actually contain less activity per μg of protein than do N2drm adults (Figure 3d).
Figure 3. Protein-kinase activity for endogenous substrates are reduced in age-1(mg44) F2 homozygotes.
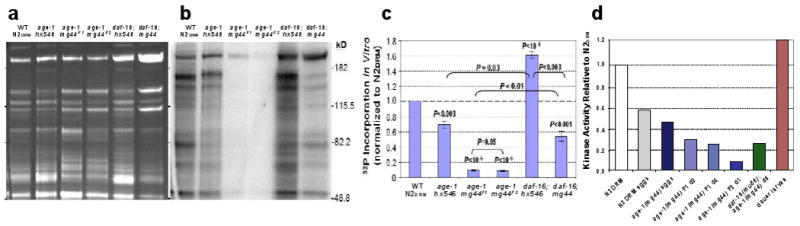
Day-6 adults were harvested, frozen in liquid nitrogen and ground over dry ice. Kinase activity of cleared, sonicated lysates was assessed by γ-32P-ATP incorporation per 20 μg protein sample, in 1 min at 30°C. Samples were electrophoresed on gels of 10% acrylamide/SDS. (a) Gel stained with SYPRO Ruby (invitrogen) for total protein. (b) 32P β-image (Molecular Dynamics Storm) from the gel in a, dried under vacuum. (c) Data summary from 2–3 independent expansions each, of strains N2drm, age-1(hx546), F1 age-1(mg44), F2 age-1(mg44) homozygotes, and daf-16 double mutants with each age-1 allele. (d) Additional controls show that in vitro kinase activity is lower in F2 homozygotes of age-1(mg44), even as day-1 adults, than in F1's at adult days 1 or 6; a second mutation, deleting most of the daf-16 gene, restores age-1(mg44) kinase activity to F1 levels; eggs laid by N2DRM or age-1(mg44) F1 adults are intermediate in kinase activity; and dauer larvae have even higher kinase activity than N2DRM adults. Adapted from [30].
Loss of PI3K leads to transcriptional inhibition of multiple signaling pathways
If kinase attenuation in age-1 mutants were only due to the absence of PIP3 needed to anchor or activate cellular kinases, it should be unaffected by loss of DAF-16/FOXO. As noted in the preceding section, this is clearly not the case. The deficit in age-1(hx546) kinase activity is fully reversed by a second mutation to the daf-16 gene (in fact it is “over-reverted”; see Figure 3c), while the same mutation restores just half of the kinase deficiency seen in F2 age-1(mg44) worms. To test for transcriptional regulation of IIS kinases, we used RT-PCR (real-time polymerase chain reaction) to quantify transcript levels of several IIS components. The data (Figure 4) provide compelling evidence for transcriptional silencing of IIS moieties in very long-lived F2 age-1 adults, while the weaker hx546 allele produced much less attenuation. Most of this inhibition, although not all, disappears in double mutants also defective for daf-16 — again implying that transcripts are attenuated by multiple routes.
Figure 4. Transcriptional suppression of IIS genes in age-1(mg44).
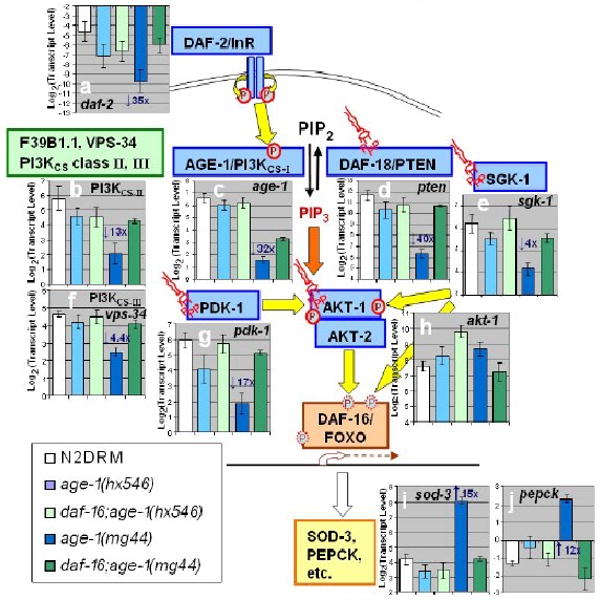
Transcript levels were assayed by real-time polymerase chain reaction (RT-PCR). Expression histograms are shown superimposed on a schematic diagram of IIS. Yellow arrows show protein phosphorylations (circled P's); orange arrows indicate binding of phosphatidylinositol 3,4,5-triphosphate (PIP3, “structural” symbols). Within each histogram, transcript mean ± SEM (steady-state) is shown on a log(2) scale, comparing wild-type to 4 age-1 mutant groups and to dauer larvae. For each group, fold changes are shown (e.g., “3×”), of age-1 (mg44)-F2 relative to N2drm. Post-gravid age-1(mg44) F1 homozygotes were at adult day 8–9; F2 homozygotes at day 10; N2drm, age1(hx546), and daf-16(mu86); age-1(mg44) double mutants, were all post-gravid adults at adult day 6; and N2drm dauer larvae 1 day after reaching 98% SDS-resistance. Transcript levels are means of 3 independent biological replicates, normalized to the mean of three control gene (β-actin, T08G5.3, and Y71D11.3) that did not differ significantly among strains.
Profound silencing is seen, in F2 age-1(mg44) adults, for genes encoding the insulin receptor, DAF-2 (a); all 3 PI3KCS classes: AGE-1, F39B1.1, and VPS-34 (b, c, f); SGK-1 (e) and PDK-1 (g) kinases that phosphorylate AKT-1 (although AKT-1 itself (h) is not transcriptionally modulated), and the DAF-18/PTEN phosphatase (d) that opposes AGE-1 kinase. As a result of disrupted IIS inhibition of DAF-16/FOXO, two of its positive target genes, sod-3 (i) and pepck (j), are strongly induced in age-1(mg44) F2 worms.
Intersections among pathways
Signal transduction pathways do not operate in isolation, but cross-talk with multiple other pathways [34,35,73-75]. Their interactions are complex, and are perhaps best viewed as a multidimensional fabric that can be “tugged” in many directions by various inputs [30]. Crosstalk is especially well documented between the IIS and the JNK and p38/MAPK stress- and cytokine-response pathways [35,74], invoking interactions with AMP-activated kinase (AMPK) and TOR complexes (see Figure 5). IIS is initiated by a membrane kinase-receptor, which responds to many insulinlike peptides, both agonists and antagonists [76]. In mammals, secretion of insulin is governed by Wnt/β-catenin signaling, and β-catenin coactivates FOXO [37]. The regulation of other insulin-like peptides, however, remains largely unexplored.
Figure 5. Interactions between IIS and other signal transduction pathways in C. elegans.
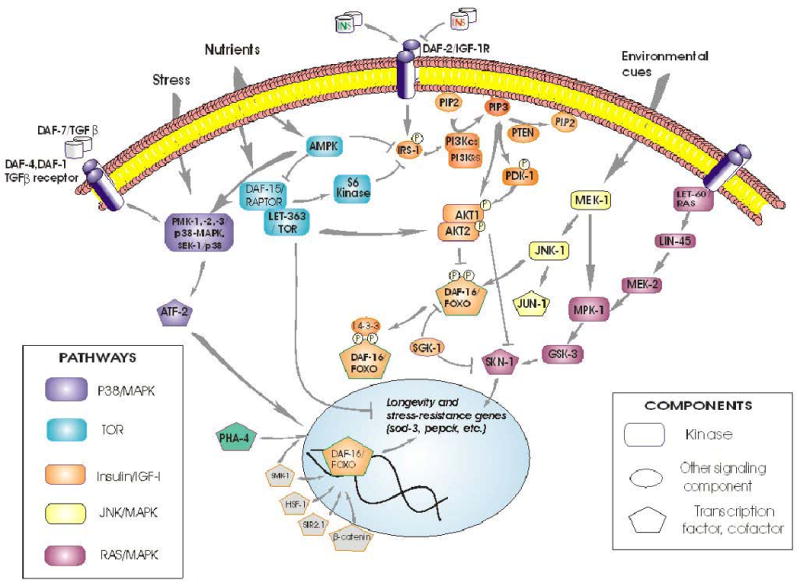
As an instructive example, AKTs can be activated by multiple inputs, including DNA-dependent protein kinase (DNA-PK) [77,78]. Multiple pathways converge through their common activation of AKTs, and AKT outputs are also diverse, utilizing many routes in addition to DAF-16/FOXO. In mammals (and presumably in other taxa), the critical PDK-1 phosphorylation of its target Thr in AKT-1 requires both a prior Ser phosphorylation, usually by TORC2 (target of rapamycin, complex 2), and also membrane- docking to PIP3 by the AKT-1 pleckstrin-homology domain [65]. AKTs directly activate at least 14 proteins, including IRS-1 (providing feedback reinforcement of its own activation), RAF-1, eNOS, NFkB, and several cell-cycling and anti-apoptotic genes. Indirectly, they promote synthesis of proteins (via TSCs and TORCs) as well as glycogen (via GSK-3). In addition, AKTs inhibit at least 11 mammalian targets, including FOXO, androgen receptors, and pro-apoptotic gene products [34,79,80].
The strong age-1(mg44) mutant allele shows transcriptional downregulation of genes encoding DAF-16-interacting factors SMK-1, PAR-5 and SIR-2.1; of TGF-β type I and II receptors DAF-1 and DAF-4, as well as the SMAD transcription factor DAF-3; of AMPK/TOR pathway components AAK-1/AMPK (but not AAK-2), LET-363/FRAP, and DAF-15/RAPTOR; and of ERK/MAPK components LET-60/RAS, LIN-45/ERK, MEK-1/ERK, MPK-1/MAPK, glycogen synthase kinase GSK-3, and the SKN-1 transcription factor. These inhibitions of gene expression are all reversed, at least in part, in daf-16; age-1 double mutants [30].
DAF-16/FOXO has been implicated as a central convergence point for diverse pathways, in several taxa separated by considerable evolutionary distances. Nuclear entry of DAF-16/FOXO is blocked by phosphorylation of at least 3 sites by the AKT complex and/or SGK-1. Stranded in the cytoplasm, this transcription factor is inactive and may be degraded. Nuclear exclusion is the principal, and perhaps sole, means of DAF-16/FOXO regulation through the IIS kinase relay. Several additional mechanisms are now known, by which non-IIS pathways can affect DAF-16/FOXO, thereby converging with IIS to modulate longevity and stress responses. At least 6 sites on DAF-16 are phosphorylated by AMP-activated protein kinase (AMPK), activating a specific subset of transcriptional targets that include a number of signal-transduction and oxidative-stress-resistance genes [42,81]. Crosstalk between IIS and nutrient sensing thus takes place within DAF-16 (or FOXO of mammals). Proteins that bind to DAF-16 can alter its nuclear entry, as do SIR-2 and 14-3-3 proteins [82,83], or serve as transcriptional coregulators such as SMK-1 [84] and β-catenin [28]; whereas HSF-1 is an independent transcription factor that nevertheless interacts with DAF-16 [85,86]. PHA-4 provides a particularly instructive example of a factor that acts in parallel to DAF-16, and yet interacts with it indirectly through competition for SMK-1, a shared coactivator [87]. Protein : protein interactions may be sensitive to site-specific modifications of DAF-16, which include phosphorylation [82,83], acetylation [31,88], as well as ubiquitinylation, which alters DAF-16 activity through proteasomal degradation [89]. Although distinct from IIS, all of these interactions obviously depend on the presence of DAF-16; nonetheless, there could be parallel effects on other transcription factors, which persist in its absence [28,82,85,87]. Thus, partial reversion of age-1(mg44) F2 traits by a second mutation to daf-16 (Figures 3 and 4) cannot be taken as evidence that AGE-1 acts only through the canonical IIS pathway.
The pervasiveness of crosstalk among pathways could be a consequence of the relatively limited repertoire of cellular responses to a much more varied range of stimuli. Cross-talk provides a concerted response to diverse signals arriving in an unpredictable sequence. The integration of signaling cascades could be compared to a neural-network algorithm for computer learning, in that the association between inputs and outputs cannot be defined by a set of rules.
The unprecedented improvement in longevity and stress resistance, seen in age-1(mg44) F2 adults bearing a single mutated gene, might be attributed to the profound realignment of signaling pathways when PIP3 is absent. The mechanisms affected clearly extend beyond those known to be directly entrained by IIS. Although IIS in C. elegans has often been portrayed as a linear pathway (supplemented by crosstalk to Sir2, MAPK, TOR, and AMPK), the central role of PIP3 depletion and the interplay with other pathways imply a distinctly nonlinear structure. Feedback loops, acting through both insulinlike peptides [90] and transcriptional inhibition of many kinases by DAF-16/FOXO [30], imply a more complex circuitry that cannot be adequately described as a collection of one-dimensional reaction chains. Most critically, the more elaborate the circuit, the greater the potential for the system to “hang” or become locked in a state of chronic repression and unresponsiveness to diverse signals (see Figure 6, adapted from [30]).
Figure 6. Three system states of a proposed positive-feedback loop.
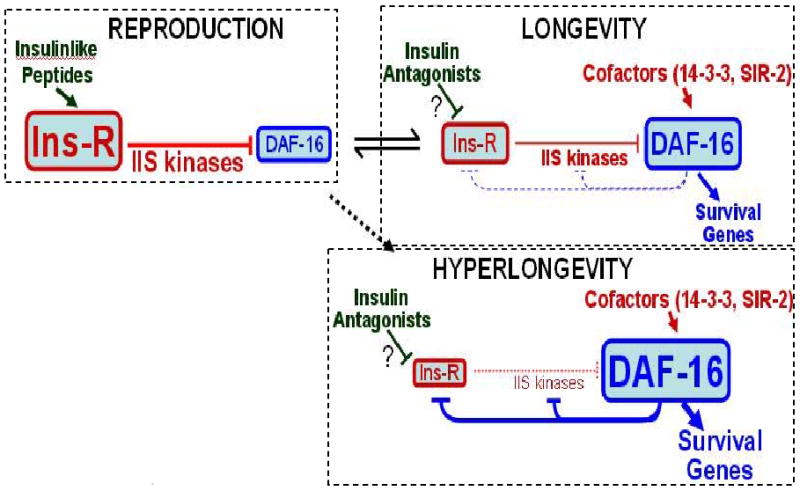
IIS leads through a series of kinase reactions to phosphorylation of DAF-16, sequestering it in the cytoplasm (reproduction mode). If insulinlike antagonists impede IIS, and/or coactivators reinforce DAF-16, the system switches to longevity mode where DAF-16 prevails and transcriptionally represses its own upstream regulatory kinases – promoting dauer formation in development, or life-extension in the adult. This “flip-flop” circuit, with opposing kinase and transcriptional signals, forms a positive-feedback loop. To recover from the dauer state, reproductive kinases must retain partial function, so favorable signals can shift the balance in their direction. Strong age-1 mutations “fuse the switch” in longevity mode, while conferring a distinctive transcript profile and greatly enhanced survival. Adapted from [30].
Tissue-specific determinants of metabolism and lifespan
D. melanogaster lifespan is increased when Cu/Zn-SOD transgenes are targeted to motor neurons, but not to other tissues or expressed globally [91]. This was interpreted to mean that lifespan is limited by oxidative damage to the nervous system, but that SOD overexpression in other tissues is not beneficial to survival, or may even oppose it. IIS modulation of C. elegans longevity also hinges on neurons. Mutations impairing daf-2 (which encodes the insulin/IGF-1 receptor) or age-1 (encoding PI3K) increase lifespan [6,8,92-95], whereas this effect is reversed when wild-type age-1 or daf-2 transgenes are targeted just to neurons (but not when they are expressed in muscle or the digestive tract) [54]. This implies that worm longevity is limited by metabolic activity or signaling of neurons. Effects of age-1 and daf-2 mutants on lipid metabolism, however, are most potently reversed when the corresponding wild-type genes are expressed in muscle cells [54]. Nerve and intestine cells are responsible for making most of the worm's INS-7, the one insulin-like peptide that has been demonstrated to limit lifespan [90]. Longevity enhancement by mutations that disrupt sensory-neuron function [4] also indicates that neurons play a key role in setting C. elegans lifespan,
Mouse lifespan increases when Igf-1R signaling is suppressed globally [12], implying a similar role for Igf-1 signaling in mammals. Mice in which one copy of the Irs2 gene has been knocked out, whether globally or only in the brain, live 17–18% longer than controls [96]. Although this establishes that IIS attenuation in the brain is sufficient to extend life, it is not known whether the signaling involves receptors for insulin or Igf-1, or hybrid receptors [97,98]. Irs-2 was long thought to respond exclusively to insulin receptor [96], but Igf-1R has been shown to phosphorylate both Irs-1 and Irs-2 [32,99]. This could be viewed either as Igf-1 signaling through Irs molecules [97,98], or as inter-pathway crosstalk, just as Jnk, Ampk, and S6k impinge on IIS through Irs phosphorylation [35,43,100]. PI3K p110γ can be deleted without extending life, possibly because this isoform is normally expressed only in macrophages and a few other tissues, where its absence interferes with innate immunity and inflammation [101].
If this were translated to mammals, could humans (like Methuselah) surpass 900 years?
Invertebrate models have led to the discovery of new genetic pathways, and also of unexpected consequences from altering known pathways and genes. Advantages of these model systems include the ease of conducting genetic studies, large-scale screens, and even selection experiments. Moreover, a relatively brief lifespan permits direct assessment of longevity where the equivalent study in any mammalian species would be far more costly and time consuming.
Invertebrate systems have been validated as useful models for human aging and age-associated debilities, by overwhelming evidence that at least some mechanisms determining lifespan are remarkably conserved from nematodes to mammals. Strong age-1 mutations, which increase worm lifespan ten-fold, are central to insulin/IGF-1 signaling. IIS is well established as a highly and broadly conserved pathway which extends life when abrogated in nematodes, insects and mice. Mechanisms may thus also be conserved, underlying the effects of extreme IIS disruption in the worm, but it will be neither simple nor straightforward to translate them for the benefit of mammals. That would only be possible if an intervention could be applied selectively to just nondividing cells so as to avoid lethality and cell-cycle arrest in essential mammalian cell compartments.
A two-fold or ten-fold increase in nematode lifespan may not, however, translate to a proportional enhancement of human survival. Extrapolation from worms to mammals is risky at best, and it cannot be assumed that interventions will result in comparable life extension factors. Longevity gains from dietary restriction, or from mutations studied previously, yield smaller benefits to Drosophila than to nematodes, and smaller still to mammals. This is not unexpected, since mammals have evolved to live many times the worm's lifespan, and humans live nearly twice as long as the next longest-lived primate. From an evolutionary perspective, mammals and their ancestors have already undergone several hundred million years of natural selection favoring traits that could directly or indirectly favor increased longevity, and may thus have already settled on gene sequences that promote lifespan. Moreover, the very notion of a “life-extension factor” that could apply across taxa presumes a linear response rarely seen in biology.
Even if tenfold life extension, realized in nematodes and yeast, proves to be unattainable in humans, there is still ample room for pharmacological extension of healthy human life beyond present levels, and even beyond what could be achieved by dietary restriction or more modest attenuation of IGF-1 signaling. This possibility, first hinted in the nematode, would perhaps never have been indicated by studies of mammals or any other metazoan dependent on continuing cell division, since AKT signaling is essential for cell replication. Substantial improvements in healthy lifespan may be achievable, but will require striking a delicate balance between two opposing cell states, replication and quiescence.
Acknowledgments
Grant support was provided by the U.S. Dept. of Veteran Affairs (Research Career Scientist Award, RJSR) and the National Institute on Aging (NIH grant P01 AG20641, RJSR)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klass MR. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev. 1983;22:279. doi: 10.1016/0047-6374(83)90082-9. [DOI] [PubMed] [Google Scholar]
- 2.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1:119. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nature Genetics. 2001;28:139. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 4.Apfeld J, Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature. 1999;402:804. doi: 10.1038/45544. [DOI] [PubMed] [Google Scholar]
- 5.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 6.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 7.Cypser JR, Johnson TE. The spe-10 mutant has longer life and increased stress resistance. Neurobiol Aging. 1999;20:503. doi: 10.1016/s0197-4580(99)00085-8. [DOI] [PubMed] [Google Scholar]
- 8.Friedman DB, Johnson TE. Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol. 1988;43:B102–B109. doi: 10.1093/geronj/43.4.b102. [DOI] [PubMed] [Google Scholar]
- 9.Clancy DJ, Gems D, Harshman LG, et al. Extension of Life-Span by Loss of CHICO, a Drosophila Insulin Receptor Substrate Protein. Science. 2001;292:104. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 10.Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A Mutant Drosophila Insulin Receptor Homolog That Extends Life-Span and Impairs Neuroendocrine Function. Science. 2001;292:107. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 11.Bonkowski MS, Rocha JS, Masternak MM, Al Regaiey KA, Bartke A. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc Natl Acad Sci U S A. 2006;103:7901. doi: 10.1073/pnas.0600161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 13.Kurosu H, Yamamoto M, Clark JD. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marden JH, Rogina B, Montooth KL, Helfand SL. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc Natl Acad Sci U S A. 2003;100:3369. doi: 10.1073/pnas.0634985100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin YJ, Seroude L, Benzer S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science. 1998;282:943. doi: 10.1126/science.282.5390.943. [DOI] [PubMed] [Google Scholar]
- 16.Bartke AJ, Wright JC, Mattison JA, Ingram DK, Miller RA, Roth GS. Extending the lifespan of long-lived mice. Nature. 2001;414:412. doi: 10.1038/35106646. [DOI] [PubMed] [Google Scholar]
- 17.Migliaccio E, Giorgio M, Mele S, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 18.Wei M, Fabrizio P, Hu J, et al. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008;4:e13. doi: 10.1371/journal.pgen.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith ED, Kennedy BK, Kaeberlein M. Genome-wide identification of conserved longevity genes in yeast and worms. Mech Ageing Dev. 2007;128:106. doi: 10.1016/j.mad.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 20.Barbieri M, Bonafe M, Franceschi C, Paolisso G. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am J Physiol Endocrinol Metab. 2003;285:E1064–E1071. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- 21.Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- 22.Taddei F, Radman M, Maynard Smith J, Toupance B, Gouyon PH, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- 23.McElwee JJ, Schuster E, Blanc E, et al. Evolutionary conservation of regulated longevity assurance mechanisms. Genome Biol. 2007;8:R132. doi: 10.1186/gb-2007-8-7-r132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayyadevara S, Dandapat A, Singh SP, et al. Life span and stress resistance of Caenorhabditis elegans are differentially affected by glutathione transferases metabolizing 4-hydroxynon-2-enal. Mech Ageing Dev. 2007;128:196. doi: 10.1016/j.mad.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102:4494. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 29.Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell. 2003;2:131. doi: 10.1046/j.1474-9728.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 30.Tazearslan C, Ayyadevara S, Bharill P, Shmookler Reis RJ. Positive feedback between transcriptional and kinase suppression in nematodes with extraordinary longevity and stress resistance. PLoS Genet. 2009 doi: 10.1371/journal.pgen.1000452. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 32.Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br J Cancer. 2006;95:1220. doi: 10.1038/sj.bjc.6603354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- 34.Gami MS, Iser WB, Hanselman KB, Wolkow CA. Activated AKT/PKB signaling in C. elegans uncouples temporally distinct outputs of DAF-2/insulin-like signaling. BMC Dev Biol. 2006;6:45. doi: 10.1186/1471-213X-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo M, Yanase S, Ishii T, Hartman PS, Matsumoto K, Ishii N. The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech Ageing Dev. 2005;126:642. doi: 10.1016/j.mad.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 36.McColl G, Vantipalli MC, Lithgow GJ. The C. elegans ortholog of mammalian Ku70, interacts with insulin-like signaling to modulate stress resistance and life span. FASEB J. 2005;19:1716. doi: 10.1096/fj.04-2447fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rulifson IC, Karnik SK, Heiser PW, et al. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw WM, Luo S, Landis J, Ashraf J, Murphy CT. The C. elegans TGF-beta Dauer pathway regulates longevity via insulin signaling. Curr Biol. 2007;17:1635. doi: 10.1016/j.cub.2007.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundaram MV. RTK/Ras/MAPK signaling. WormBook. 2006:1. [Google Scholar]
- 40.Gems D, McElwee JJ. Broad spectrum detoxification: the major longevity assurance process regulated by insulin/IGF-1 signaling? Mech Ageing Dev. 2005;126:381. doi: 10.1016/j.mad.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 42.Greer EL, Dowlatshahi D, Banko MR, et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 44.Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008;7:13. doi: 10.1111/j.1474-9726.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 45.Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arantes-Oliveira N, Berman JR, Kenyon C. Healthy animals with extreme longevity. Science. 2003;302:611. doi: 10.1126/science.1089169. [DOI] [PubMed] [Google Scholar]
- 47.Houthoofd K, Braeckman BP, Johnson TE, Vanfleteren JR. Life extension via dietary restriction is independent of the Ins/IGF-1 signalling pathway in Caenorhabditis elegans. Exp Gerontol. 2003;38:947. doi: 10.1016/s0531-5565(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 48.Yang Y, Wilson DL. Characterization of a life-extending mutation in age-2, a new aging gene in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 1999;54:B137–B142. doi: 10.1093/gerona/54.4.b137. [DOI] [PubMed] [Google Scholar]
- 49.Duhon SA, Murakami S, Johnson TE. Direct isolation of longevity mutants in the nematode Caenorhabditis elegans. Dev Genet. 1996;18:144. doi: 10.1002/(SICI)1520-6408(1996)18:2<144::AID-DVG7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Riddle DL, Swanson MM, Albert PS. Interacting genes in nematode dauer larva formation. Nature. 1981;290:668. doi: 10.1038/290668a0. [DOI] [PubMed] [Google Scholar]
- 51.Gems D, Sutton AJ, Sundermeyer ML, et al. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics. 1998;150:129. doi: 10.1093/genetics/150.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tissenbaum HA, Ruvkun G. An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics. 1998;148:703. doi: 10.1093/genetics/148.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- 54.Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science. 2000;290:147. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- 55.Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- 56.Rogina B, Reenan RA, Nilsen SP, Helfand SL. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science. 2000;290:2137. doi: 10.1126/science.290.5499.2137. [DOI] [PubMed] [Google Scholar]
- 57.Tirosh O, Schwartz B, Zusman I, Kossoy G, Yahav S, Miskin R. Long-lived alpha MUPA transgenic mice exhibit increased mitochondrion-mediated apoptotic capacity. Ann N Y Acad Sci. 2004;1019:439. doi: 10.1196/annals.1297.080. [DOI] [PubMed] [Google Scholar]
- 58.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 59.Murakami S. Stress resistance in long-lived mouse models. Exp Gerontol. 2006;41:1014. doi: 10.1016/j.exger.2006.06.061. [DOI] [PubMed] [Google Scholar]
- 60.Lithgow GJ, Walker GA. Stress resistance as a determinate of C. elegans lifespan. Mech Ageing Dev. 2002;123:765. doi: 10.1016/s0047-6374(01)00422-5. [DOI] [PubMed] [Google Scholar]
- 61.Shmookler Reis RJ, Kang P, Ayyadevara S. Quantitative trait loci define genes and pathways underlying genetic variation in longevity. Exp Gerontol. 2007;41:1046. doi: 10.1016/j.exger.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 62.Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 63.Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- 64.Vanhaesebroeck B, Leevers SJ, Ahmadi K, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 65.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 66.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 67.Roggo L, Bernard V, Kovacs AL, et al. Membrane transport in Caenorhabditis elegans: an essential role for VPS34 at the nuclear membrane. EMBO J. 2002;21:1673. doi: 10.1093/emboj/21.7.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pettitt TR, Dove SK, Lubben A, Calaminus SD, Wakelam MJ. Analysis of intact phosphoinositides in biological samples. J Lipid Res. 2006;47:1588. doi: 10.1194/jlr.D600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Weinkove D, Halstead JR, Gems D, Divecha N. Long-term starvation and ageing induce AGE-1/PI 3-kinase-dependent translocation of DAF-16/FOXO to the cytoplasm. BMC Biol. 2006;4:1. doi: 10.1186/1741-7007-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vanhaesebroeck B. Charging the batteries to heal wounds through PI3K. Nat Chem Biol. 2006;2:453. doi: 10.1038/nchembio0906-453. [DOI] [PubMed] [Google Scholar]
- 71.Remenyi A, Good MC, Lim WA. Docking interactions in protein kinase and phosphatase networks. Curr Opin Struct Biol. 2006;16:676. doi: 10.1016/j.sbi.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 72.Stokoe D, Stephens LR, Copeland T, Gaffney PRJ, Reese CB, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of Protein Kinase B. Science. 1997;277:567. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 73.Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 74.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2006;2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pierce SB, Costa M, Wisotzkey R, et al. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 2001;15:672. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dragoi AM, Fu X, Ivanov S, et al. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J. 2005;24:779. doi: 10.1038/sj.emboj.7600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sester DP, Brion K, Trieu A, et al. CpG DNA activates survival in murine macrophages through TLR9 and the phosphatidylinositol 3-kinase-Akt pathway. J Immunol. 2006;177:4473. doi: 10.4049/jimmunol.177.7.4473. [DOI] [PubMed] [Google Scholar]
- 79.Shtilbans V, Wu M, Burstein DE. Current overview of the role of Akt in cancer studies via applied immunohistochemistry. Ann Diagn Pathol. 2008;12:153. doi: 10.1016/j.anndiagpath.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 80.Gills JJ, Castillo SS, Zhang C, et al. Phosphatidylinositol ether lipid analogues that inhibit AKT also independently activate the stress kinase, p38alpha, through MKK3/6-independent and -dependent mechanisms. J Biol Chem. 2007;282:27020. doi: 10.1074/jbc.M701108200. [DOI] [PubMed] [Google Scholar]
- 81.Greer EL, Oskoui PR, Banko MR, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 82.Araiz M, Chateau MT, Galas S. 14-3-3 regulates life span by both DAF-16-dependent and - independent mechanisms in Caenorhabditis elegans. Exp Gerontol. 2008;43:505. doi: 10.1016/j.exger.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, Tissenbaum HA. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:741. doi: 10.1016/j.mad.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 84.Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006;124:1039. doi: 10.1016/j.cell.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 85.Greer EL, Brunet A. Different Dietary Restriction Regimens Extend Lifespan by both Independent and Overlapping Genetic Pathways in C. elegans. Aging Cell. 2009 doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 87.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 88.Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf) 2008;192:19. doi: 10.1111/j.1748-1716.2007.01780.x. [DOI] [PubMed] [Google Scholar]
- 89.Li W, Gao B, Lee SM, Bennett K, Fang D. RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination. Dev Cell. 2007;12:235. doi: 10.1016/j.devcel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 90.Murphy CT, Lee SJ, Kenyon C. Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:19046. doi: 10.1073/pnas.0709613104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Phillips JP, Parkes TL, Hilliker AJ. Targeted neuronal gene expression and longevity in Drosophila. Exp Gerontol. 2000;35:1157. doi: 10.1016/s0531-5565(00)00117-0. [DOI] [PubMed] [Google Scholar]
- 92.Johnson TE. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science. 1990;249:908. doi: 10.1126/science.2392681. [DOI] [PubMed] [Google Scholar]
- 93.Ayyadevara S, Dandapat A, Singh SP, Zimniak L, Shmookler Reis RJ, Zimniak P. Lifespan extension in hypomorphic daf-2 mutants of Caenorhabditis elegans is partially mediated by glutathione transferase CeGSTP2-2. Aging Cell. 2005;4:299. doi: 10.1111/j.1474-9726.2005.00172.x. [DOI] [PubMed] [Google Scholar]
- 94.Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 95.Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141:1399. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 97.Kulkarni RN. Receptors for insulin and insulin-like growth factor-1 and insulin receptor substrate-1 mediate pathways that regulate islet function. Biochem Soc Trans. 2002;30:317. doi: 10.1042/bst0300317. [DOI] [PubMed] [Google Scholar]
- 98.Blakesley VA, Scrimgeour A, Esposito D, Le RD. Signaling via the insulin-like growth factor-I receptor: does it differ from insulin receptor signaling? Cytokine Growth Factor Rev. 1996;7:153. doi: 10.1016/1359-6101(96)00015-9. [DOI] [PubMed] [Google Scholar]
- 99.Kim B, van Golen CM, Feldman EL. Insulin-like growth factor I induces preferential degradation of insulin receptor substrate-2 through the phosphatidylinositol 3-kinase pathway in human neuroblastoma cells. Endocrinology. 2005;146:5350. doi: 10.1210/en.2005-0356. [DOI] [PubMed] [Google Scholar]
- 100.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 101.Chang JD, Sukhova GK, Libby P, et al. Deletion of the phosphoinositide 3-kinase p110gamma gene attenuates murine atherosclerosis. Proc Natl Acad Sci U S A. 2007;104:8077. doi: 10.1073/pnas.0702663104. [DOI] [PMC free article] [PubMed] [Google Scholar]