

Abstract
Investigations with astroglial cells carry more prominence in drug abuse studies. However, due to earlier perception that astroglial cells were only passive bystanders in neural signal transmission, not many investigations were conducted on the toxicity of various abused drugs, like cocaine. The present study was aimed to discern the effect of cocaine on rat astroglioma cells and analyzed qualitatively for morphological features as well as vacuolation by phase contrast microscope, quantitatively for cytotoxicity, mitochondrial membrane potential by rhodamine- 123 fluorometric assay, and cell cycle analysis by flow cytometry. Based on population cell doubling time studies, glial cells were grown in 10% FBS in RPMI 1640 medium and treated with cocaine for 24 or 48 h. Microscopic assessments clearly demonstrated massive vacuolation and significant disruption at general architecture of glial cell morphology with cocaine. Chronic cocaine treatment (24 or 48 h) caused significant loss of cell viability. The sublethal dose of cocaine was found to be 4.307 and 3.794 mM at 24 and 48 h, respectively. Cocaine reduced the mitochondrial membrane potential in a dose dependent manner with ED50 of 4 mM after 24 h. Cell cycle analysis suggested dual inhibition at G0/G1 and G2/M phases after 24 and 48 h, respectively. In summary, our findings suggest that cocaine toxicity was due to loss of mitochondrial membrane potential, vacuolation, and dual inhibition of cell cycle phases. These results may shed light in understanding the onset of some early key events in cocaine-induced toxicity in glial cells.
Keywords: Glioma cells, Mitochondrial membrane potential, Population cell doubling time, Vacuolation
Introduction
Cocaine is a widely abused and addictive psychostimulant drug that acts on the central nervous system (CNS). Currently, about 3.6 million Americans use this drug on a regular basis [1]. The toxic effects of cocaine on several cell lines outside of CNS have been demonstrated earlier [2-5], but only a few studies have identified cocaine’s toxic or inhibitory role on cell cycle phases in different CNS cells [6]. Attention has focused primarily on the deleterious effects of cocaine on neurons. Studies have demonstrated that cocaine inhibits reuptake of neuronal dopamine, resulting in its prolonged presence at the synaptic cleft. Auto-oxidation of dopamine in the synaptic cleft was shown to cause neuronal death. Little, however, is known about toxic or inhibitory role of cocaine on glial cells.
Glial cells may be the first to experience the toxic effects of cocaine in the mammalian brain, since glial cells outnumber neurons by 10–1 [7]. Glial cells are critical for neuron survival and maintenance of fundamental patterns of circuits under normal circumstances in vivo. Malfunction of or damage to glial cells due to cocaine exposure may contribute to neuronal dysfunction, which may increase the risk for neurodegenerative disorders, such as Parkinson’s disease [8], schizophrenia, Alzheimer’s disease, and other late-onset neurologic disorders. Understanding the role of cocaine on the inhibition of glial cell proliferation and toxicity may lead to the development of new therapeutic approaches to neuronal disorders.
Previous in vitro studies of the effects of abused drugs, like cocaine and morphine, on CNS cells were performed in culture media either in the absence of serum proteins [9] or with reduced serum proteins [10], based on the assumption that brain cells do not have direct contact with serum proteins due to the blood brain barrier [9]. But it has been shown that exclusion of serum proteins by the blood brain barrier is not absolute [11], indicating the possibility of interaction between serum proteins and brain cells under normal circumstances. In addition, this barrier is more permeable to serum proteins in drug abusers [12], in people with compromised health [13], and in developing fetuses, enabling serum proteins direct access to intraneural domains. We believe that the results of serum-deprived studies provide an incomplete picture of the effect of cocaine on CNS cells and contribute little to the understanding of the correct cocaine dose for future acute toxicity studies in preclinical trials. Another important aspect, which has not received attention in most previous studies with cocaine on various cell lines was that no emphasis was given to quantify a sublethal toxic dose for cocaine [2, 3, 14], nor to follow any defined period of its treatment [2, 3, 15].
Our study had two objectives. To fill the gap left by serum deprived studies, the primary objective was to examine cocaine toxicity to glial cells in the presence of serum proteins (10% FBS) in the culture media and to quantify sublethal doses at different time points based on cell doubling studies. The secondary objective was to determine the effects of cocaine on mitochondrial membrane potential and cell cycle phases. Mitochondrial membrane potential is an important function for electron transport and oxidative phosphorylation in cells. Findings related to the interaction of cocaine with mitochondria may aid in understanding energy metabolism in these cells. Similarly, cocaine interaction at different glial cell cycle phases may help determine the early onset of some key events in these cells, which proliferate even in adult hood [16].
We employed rat C6 astroglioma cells in our research. Unlike other cell lines of the CNS, the rat glioma C6 cell line has the greatest number of genes whose expressions are similar to human brain tumors [17], as well as possessing enzymes found in normal glial cells [18]. Thus, the rat C6 astroglioma cell line is a widely accepted model cell line, which may represent a better in vitro model system for investigations of abused drugs, like cocaine.
Experimental Procedure
Materials
RPMI 1640 (modified), fetal bovine serum (FBS), penicillin/streptomycin sulfate, amphotericin B, phosphate-buffered saline (PBS) and l-glutamine were purchased from Media Tech (Herndon, VA, US). Crystal violet, rhodamine- 123, l-glutaraldehyde, trypan blue, cocaine–HCl, and EDTA (ethylene diamine tetraacetic acid) were supplied by Sigma Chemical Company (St. Louis, MO, US). All other routine chemicals were of analytical grade.
Preparation of Sample
The stock (1 M) as well as working stocks (80–180 mM) of cocaine hydrochloride in all our studies were prepared fresh in PBS just prior to assays and applied at different concentrations to glial cultures in minimum volume (5 μl/well) to prevent pH alterations of the medium. After the additions, the final pH in all culture wells was found to be around 7.4.
Cell Culture and Microscopy
The central nervous system derived rat C6 glial cell line (CCL-107) was purchased from American Type Culture Collection (Rockville, MD, US) and maintained as an adherent monolayer culture in complete RPMI-1640 medium, 2 mM l-glutamine, 10% FBS (v/v), 100 U/ml penicillin, 100 μg/ml streptomycin sulfate and 0.25 μg/ml amphotericin B. Cells were grown in a humidified atmosphere containing 5% CO2 in air at 37°C in an incubator, and sub-cultured twice a week. For cytotoxic studies, the culture was harvested by treating with 0.05% EDTA in PBS for 2 min or less, resulting in a single cell suspension. Cell counts and cell viability were assessed immediately by using 0.4% trypan blue dye exclusion assay on hemocytometer under a light microscope. Cell viability always exceeded 90%. Cells were diluted in complete RPMI medium, and then seeded in culture plates or dishes for the experiments. Cells were observed under inverted phase contrast IX-70 Olympus microscope (Olympus, Tokyo, Japan). Photomicrographs were taken by an ocular video-camera system (MD35 Electronic eyepiece, Zhejiang JinCheng Scientific & Technology Co., Ltd, HangZhou, China) using C-Imaging System Software (Compix Inc. Cranberry Township, PA, USA).
Population Cell Doubling Time
In order to analyze population cell doubling time (PCDT), the rat glial cells were plated in polystyrene, flat bottom 96-well (0.32 cm2) microtiter plates (BD Labware, US). The cells were seeded at a starting density of 5 × 103 cells per well in RPMI 1640 medium supplemented with 10% FBS. The cells were then allowed to adhere to wells in the incubator for 18–24 h. The number of cells at various time points, namely 0, 12, 24, 48 and 72 h, was evaluated by a dye uptake assay using crystal violet as described previously [19]. As per this method, intensity of violet color was proportional to number of live cells. The culture plates were read at 540 nm in a plate reader (Bio-Tek Instruments Inc, Wincoski, VT). The PCDT in hours was determined from average absorbance values (n = 32) of cells growing in exponential phase using the following equation [20]:
Where, A0 and A = absorbance at time zero and final, respectively, t = time difference between A0 and A.
Treatments with Cocaine
The cytotoxic studies were performed in 96-well microtiter plates. The cells were seeded at a starting density of 2 × 104 cells per well in a total volume of 195 μl growth medium supplemented with 10% FBS. The cells were then allowed to adhere to wells in the incubator for 18–24 h before drug exposure. Cells which were typically about 60–70% confluent were treated at six different concentrations (2–7 mM) of cocaine in a final volume of 5 μl. In all these studies, cells with medium alone or PBS in medium served as controls. Controls and the treated samples were always present in different wells of the same 96-well microtiter culture plates. These plates were incubated for 24 or 48 h continuously without further renewal of growth media in a 5% CO2 at 37°C with the plates capped in normal fashion. All studies were repeated at least twice (n = 12–36).
Evaluation of Cytotoxicity of Cocaine
After 24 or 48 h of exposure, the cytotoxicity of cocaine was evaluated by dye uptake assay using crystal violet, which binds to nucleic acids [21], as described previously [19]. The average absorbance values of controls were taken as 100% cell viability. From the treated and control absorbance values, percent of cells killed were determined by the following equation: [1 − (T/C)] × 100, where T is average absorbance values of treated cells, and C is average absorbance values of control cells. If LC50 or ED50 values were not within the range of tested concentrations, then the concentrations were adjusted accordingly, and the experiments were repeated.
Evaluation of Glial Cell Vacuolation
Rat glial cells were seeded in complete medium in 96-well culture plates as described above. After treating with various concentrations of cocaine (2–6 mM) for 24 h, cells were fixed in 0.25% aqueous glutaraldehyde and stained with crystal violet as described earlier [19]. The presence of vacuoles in cells was photographed with inverted phase contrast IX-70 Olympus microscope under a 40X objective.
Estimation of Mitochondrial Membrane Potential
It was determined by employing laser dye fluorescent probe rhodamine- 123. Being a lipophilic cationic dye, this xanthene dye permeates easily and interacts with the negative charges on the inner membrane of mitochondria at a low concentration. Hence, its accumulation is proportional to mitochondrial membrane potential. Cocaine treatments were performed at six different concentrations (2–6 mM) for 24 h. At the end of incubation, monolayer cells were overlaid with 100 μl of 0.25% aqueous glutaraldehyde for fixation, containing rhodamine- 123 to yield a final concentration of 1 μM for 30 min at room temperature. The supernatant was discarded, and the plates were washed with tap water and air dried in the hood. Finally, 100 μl of 0.1% Triton X 100 in dPBS was added per well and incubated at 37°C for 1 h. The plates were read with the excitation filter set at 485 nm and the emission filter at 538 nm on a microplate Fluorometer model 7620, Version 5.02, Cambridge Technology, Inc., (Watertown, MA, US). These studies were repeated at least twice (n = 12).
Cell Cycle Progression by Flow Cytometry
The distribution of DNA in all cell cycle phases of cocaine treated glial cells was assayed by flow cytometer (BD Biosciences, San Jose, CA) as described elsewhere [22]. In keeping with the role of confluency on cell cycle stages, we seeded 1.3 × 106 cells for 24 h studies, and 0.7 × 106 cells for 48 h studies in RPMI 1640 complete medium in a final volume of 7 ml in 100 mm diameter plastic culture dishes and incubated over night. Cocaine hydrochloride was dissolved in PBS at 1 M and was used to achieve various concentrations (3–5 mM). It was added to dishes when cultures were sub-confluent (45–50%). Each concentration was performed in triplicate dishes. Incubations were done for 24 or 48 h in a 5% CO2 incubator at 37°C. Cells were harvested at indicated times after the addition of cocaine. The medium from each culture dish was aspirated and cells were dislodged by treating with 0.05% EDTA prepared in PBS for 1 min at room temperature. Then cells were collected in PBS (8 ml), transferred into falcon tubes and sedimented by centrifugation at 2,600 rpm for 7 min at room temperature. Each pellet was resuspended in 100 μl PBS and then gently vortexed. Cells were fixed in 5 ml of 100% ethanol (pre-cooled at −20°C), added drop-wise to each tube while vortexing, and incubated at 4°C for at least 24 h. The following day, the tubes were centrifuged at 2,600 rpm for 7 min and resuspended in 100 μl ethanol. After cells (75 μl/tube) were transferred into BD falcon tubes which were shielded from light, 1 ml of staining solution, containing final concentrations of 1.3 mg/ml ribonuclease A, 1 mg/ml d-glucose and 50 μg/ml propidium iodide, was added to each tube in the dark and incubated at room temperature for 1 h in the dark with occasional stirring. The proportion of cells in each stage was performed within 2 h by using a FACSCalibur flow cytometer. In each sample, a total of 10,000 individual events from the gated subpopulation were analyzed separately. CellQuest software was used for acquisition and analysis of the data, and the percentage of cells in each phase was determined by using ModFit LT 3.0 Software. These studies were repeated at least twice (n = 5–9) to confirm reliability of the results.
Statistical Analysis
The experimental results were presented as mean ± standard error mean (SEM). The data were analyzed for significance by one-way ANOVA and then compared by Dunnett’s multiple comparison tests using GraphPad Prism Software, Version 3.00 (San Diego, CA, US). The test values of P < 0.05 and P < 0.01 were considered significant and highly significant, respectively. All bar graphs were plotted between the concentration of cocaine and the percentage of cell viability using the above software program. In case of line graphs, lethal dose values were obtained by plotting graphs between cocaine concentrations on the x-axis versus percent of total cell population (live and dead) on the y-axis. The lethal concentration (LC50) values, representing the millimolar concentration of cocaine needed to kill 50% of glial cells, was determined from the graphs where both curves crossed [23].
Results
Population Doubling Time of Glial Cells
Population cell doubling time (PCDT) studies were performed on glial cells maintained in 10% FBS. The initial lag phase under these conditions was found to be approximately 24 h, and thereafter the average PCDT of glial cells was 19.87 h (Fig. 1). Based on these observations, 24 and 48 h incubation periods were selected for further studies.
Fig. 1.
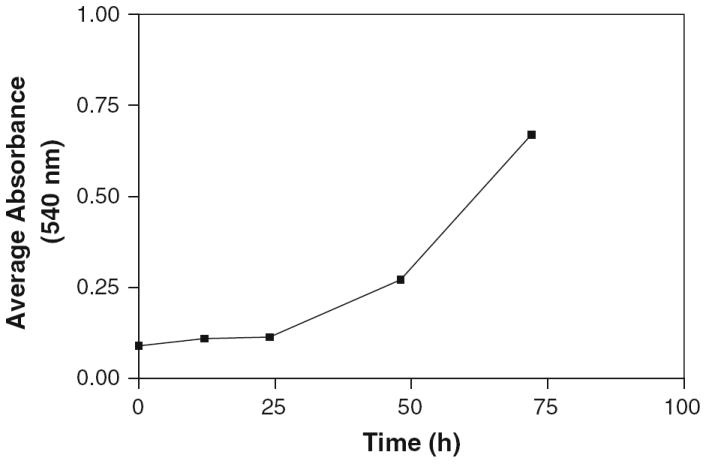
Population doubling period of rat glial cells. Cells with starting density of 5 × 103 per well in 96-well plates (n = 4, and each value is the average absorbance of eight wells, so final n at each time point = 32) with complete RPMI 1640 medium containing 10% FBS were incubated for designated time points. The cells were stained with crystal violet and the absorbance at 540 nm was read in a plate reader. The absorbance values at any time point were so close to each other that error bars were not seen on the plot
Effect of Cocaine on Glial Cell Morphology and Viability
Log phase growing glial cells were treated with varying concentrations of cocaine. Under our experimental conditions, preliminary studies with 1 mM or less cocaine showed no apparent effects on glial cell viability even after 48 h. Based on these results, the concentrations of cocaine were adjusted between 2 and 7 mM, and the experiments were repeated. The morphological features were assessed under a phase contrast microscope. Microscopic observations and photo documentation revealed significant morphological differences between untreated control and cocaine treated cells after 24 and 48 h. The cells not only became small and round at higher doses but also lost anchorage property (Fig. 2).
Fig. 2.
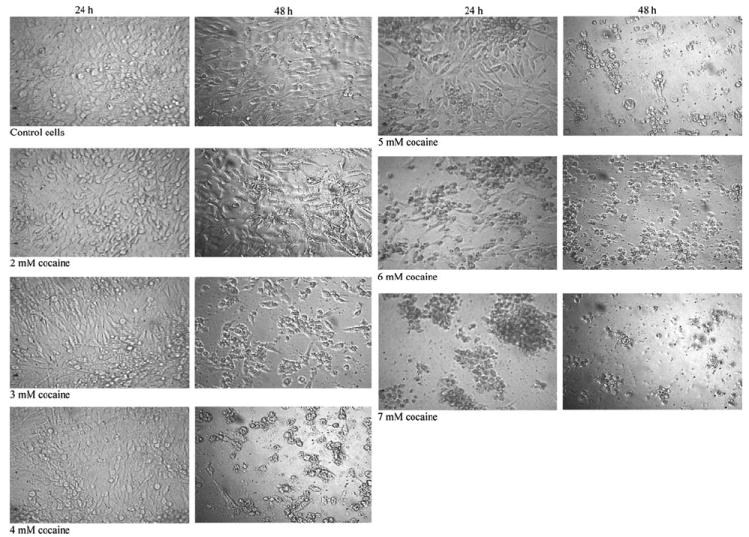
Comparison of cocaine induced rapid morphological alterations of glial cells. Cells in complete RPMI 1640 medium were treated continuously with 2–7 mM cocaine for 24 or 48 h. Unstained cells were photographed using an inverted phase contrast IX-70 Olympus microscope with 20X objective
As illustrated in Fig. 3, six different concentrations of cocaine were tested against glial cells with cytotoxic effect on the cells being analyzed after 24 and 48 h. It was observed that cocaine caused significant (P < 0.01) loss of cell viability at all concentrations compared to control cells. These results strongly indicated that toxicity of cocaine to glial cells was dose and time dependent. The LC50 at the 24 h time period was 4.307 mM cocaine, which was similar to an earlier report (4.30 mM cocaine, 24). After 48 h, the LC50 value further decreased to 3.794 mM. No significant intra-variability (<0.1%) was observed with different batches of serum in our studies. Consistent with earlier reports [25-27], cocaine at 2 mM or above induced vacuole formation immediately in glial cells (Fig. 4).
Fig. 3.
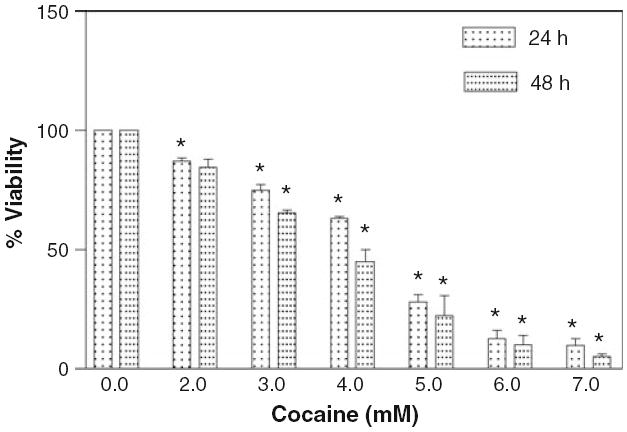
Effect of cocaine on glial cell viability. The cells (2 × 104 per well) were seeded in 96 well plates with complete RPMI 1640 medium containing 10% FBS and were treated with various concentrations of cocaine for 24 or 48 h. Data were represented as mean ± SEM (n = 36, *P < 0.01, highly significant in comparison to corresponding controls, One-way ANOVA, Dunnett’s multiple comparison test)
Fig. 4.
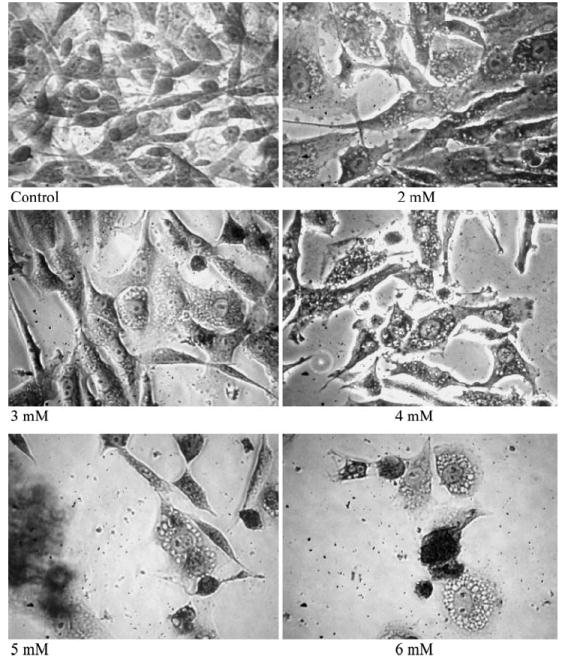
Induction of vacuoles in cocaine treated cells. Glial cells were treated with 2–6 mM cocaine continuously for 24 h in the incubator. Monolayer cells were fixed and stained with 0.1% Crystal violet dye, and photographed using inverted phase contrast IX-70 Olympus microscope with 40X objective
Decrease in Mitochondrial Membrane Potential
The retention of fluorescent probe rhodamine- 123 is proportional to mitochondrial membrane potential. In our study, it was observed that 24 h treatment of glial cells with different cocaine concentrations (2–6 mM) caused significant (P < 0.01) decrease in membrane potential from 3 mM in a dose dependent manner (Fig. 5). The effective dose, where 50% mitochondrial membrane potential decreased due to cocaine treatment, was 4.0 mM. The data of fluorometric analysis rhodamine- 123 study clearly indicate that cocaine has profound effect on mitochondria and causes cell toxicity by decreasing the membrane potential of mitochondria.
Fig. 5.
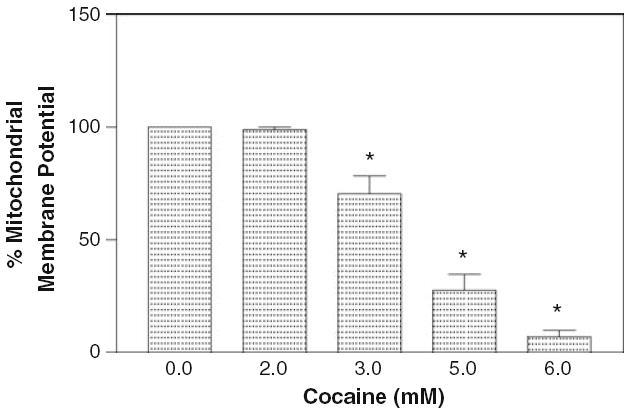
Effect of cocaine on mitochondrial membrane potential. The glial cells (2 × 104 per well) were seeded in 96 well plates with complete RPMI 1640 media containing 10% FBS and were treated with various concentrations of cocaine (2–6 mM) for 24 h. Membrane potential was evaluated fluorometrically as a measure of rhodamine- 123 uptake. Data were represented as mean ± SEM (n = 12, *P < 0.01, highly significant in comparison to control, One-way ANOVA, Dunnett’s multiple comparison test)
Dual G0/G1 and G2/M Phase Inhibition
Cell proliferation is a highly controlled event in mammalian cells. To investigate the effect of cocaine on different phases of the cell cycle, glial cells were treated at 3, 4 and 5 mM cocaine for 24 and 48 h, and analyzed by flow cytometry. Figure 6 shows the effects of increasing concentration of cocaine on glial cell progression through G0/G1, S and G2/M phases. It was observed that in comparison to control, cocaine treatment at various concentrations for 24 h resulted in significant cell cycle arrest in G0/G1 and a reduction in number of cells in S phase. This demonstrates that cocaine arrests cell cycle progression at the G0/G1 to S transition. The accumulation of cells was significant at 4 and 5 mM cocaine (P < 0.05) correlating with a subsequent significant decrease in S phase cells at 4 and 5 mM cocaine (P < 0.05). The cells at G2/M almost remained the same at all doses of cocaine (P > 0.05).
Fig. 6.
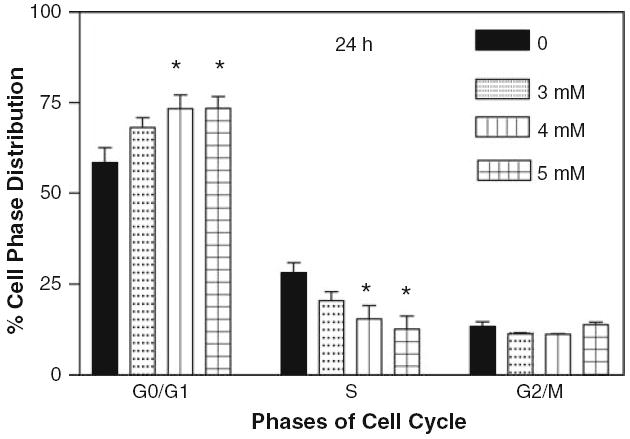
Effect of cocaine on cell cycle after 24 h. The glial cells with starting density of 1.3 × 106 per dish in complete RPMI 1640 medium containing 10% FBS were treated with 0, 3, 4 and 5 mM of cocaine for 24 h. Cells were harvested and stained by propidium iodide staining solution for 1 h in dark and analyzed by flow cytometry. Data were represented as mean ± SEM (n = 5, *P < 0.01, highly significant in comparison to control)
It is interesting to observe that cocaine at 4 and 5 mM concentrations for 48 h induced significant cell cycle arrest at G2/M phase (Fig. 7). The percentage of accumulation of cells at G2/M increased gradually and significantly at 4 and 5 mM cocaine (P < 0.05). On the other hand, in comparison to control, the number of cells at G0/G1 significantly decreased (P < 0.05) at 5 mM treatment, while the cells at S phase almost remained the same at all doses of cocaine (P > 0.05). These results suggest that the particular phase of cell cycle arrest by cocaine depends on the length of incubation.
Fig. 7.
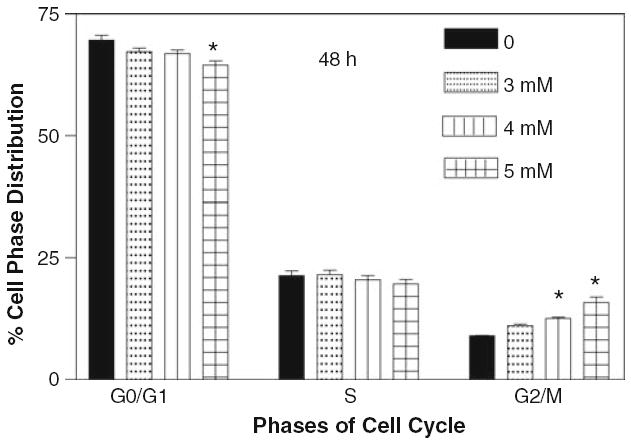
Effect of cocaine on glial cell cycle after 48 h. The glial cells with starting density of 0.7 × 106 per dish in complete RPMI 1640 medium containing 10% FBS were treated with 0, 3, 4 and 5 mM of cocaine for 48 h. Cells were harvested and stained by propidium iodide staining solution for 1 h in dark and analyzed by flow cytometry. Data were represented as mean ± SEM (n = 9, *P < 0.01, highly significant in comparison to control)
Discussion
Insufficient research on the quantification of cocaine sublethal dose under more defined conditions on glial cell toxicity and cell cycle inhibition prompted the present investigation. One of the initial problems in this study was determining the period of cocaine exposure on cells. Reflecting the absence of a standardized protocol for duration of cocaine treatments on cells, most previous in vitro toxicity evaluations of cocaine were performed arbitrarily for 1 day [6, 24, 28], or 6 days [9] or 7 days [29], resulting in diverse outcomes. For example, a longer period of exposure to cocaine typically causes more cell death than a shorter period, and the potency of cocaine in the former case is questionable due to a lack of standard treatment period. Population cell doubling time (PCDT) studies enable us to estimate how long the cell cycle phase-dependent drugs, like cocaine, need to be tested on cells. It is well known that the toxicity of cell cycle-dependent drug inhibitions inversely correlates with the length of cell cycle. For instance, the shorter the cell doubling period, the higher the toxicity and the lower the LC50 value. For this reason, PCDT was used as the criteria to establish incubation periods in our studies. It was observed that after seeding of cells, the lag period lasted almost 24 h, and thereafter cells proliferated linearly with an average doubling period of 19.87 h (Fig. 1). Based on cell doubling studies, 24 and 48 h treatment periods were selected for cocaine toxicity and cell cycle analyses.
Another common problem encountered in culture studies relates to the serum concentration in the media. The question of how much serum protein should be used is often overlooked by researchers even though serum is an important component in culture media. Thus, due to lack of standardized methods in cell culture studies, many laboratories arbitrarily employ reduced serum [10, 30-32] or serum-deprived media [9, 33, 34] with the objective of minimizing cocaine interaction with serum proteins during cytotoxic studies. It is important to understand that serum contains a number of trophic factors [35] and α globulins [36] that are required for adherence and optimum growth of cells. Serum deprivation or reduction in cultures during the drug evaluation period may render the cells more susceptible for toxicity, presumably due to increased intracellular proteolysis [37]. Sometimes complete or partial serum withdrawal may also potentiate the cytotoxic activities of test compounds, as shown earlier [38, 39]. Similarly, significant reduction of serum proteins in the medium may change the fatty acid composition of the plasma membrane [40]. These events may result in high cellular toxicity by the compound in culture studies. Such toxicity observations may be interpreted as a result of the potent nature of the test compound itself, resulting in conclusions reporting that the test compound is very active, when in fact the toxicity may be affected by the susceptible nature of cells under serum-reduced or deprived conditions.
Cocaine potency may also depend on the quantity and variability of the serum proteins in the cell culture media reported in earlier studies [9, 10, 30-34], making it difficult to estimate cocaine’s true potency on cells. It was observed that glial cells proliferated better in 10% FBS compared to lower concentrations, like 2.5% or less (unpublished observations). Thus, all studies in this report were performed in the presence of 10% serum proteins in the media. It is expected that this concentration may facilitate similar types of interactions normally observed [41] between serum proteins and cocaine in vivo.
The selected concentration range (2–7 mM) for cocaine toxicity in this study was based on the response of glial cells to cocaine under our experimental conditions. For instance, cocaine treatments for 24 h at 1 mM or less failed to cause significant glial cell death (data not shown), an observation consistent with a previous report [24]. On the other hand, it was observed that cocaine at 7 mM caused almost 100% cell death. Based on these initial observations, cocaine concentrations were adjusted in the range of 2–7 mM for toxicity studies (Fig. 3). This range is consistent with previous studies, where cocaine toxicity was evaluated in cultures up to 10 mM [24]. The concentration range in our study has clinical significance similar to that reported in studies on cells outside of the central nervous system [14, 42]. Furthermore, cocaine concentrations used in this study, approximate common concentrations found in the blood of drug addicts, when the underestimation due to hydrolysis by esterases is taken into account [43].
It may be argued that the time course and cocaine dose in this study were too high to represent in vivo cocaine usage. It is important to understand that both in vitro and in vivo are different thermodynamically and are not comparable, because in vitro is an isolated and closed system, while in vivo is a more complicated open system. Comparing in vitro results to the in vivo situation is like comparing apples to oranges. All initial drug evaluations are carried out on cell cultures under in vitro conditions, because they provide reliable and rapid information on the behavior of cells at the molecular level. When viewed with this perspective, the time course and cocaine concentrations used in this study fall within the parameters of most previous reports [24].
We observed that cocaine treatments at 3 mM or higher for 24 or 48 h (Fig. 2) caused significant alterations in glial cell morphology. At 24 h treatment, the cells were elongated (3 mM). The intercellular gaps expanded with increasing cocaine concentration (4–6 mM). And finally, the cells became smaller and round, clumped together, and lost anchorage property (6 and 7 mM). At 48 h treatment, changes occurred even at 2 mM cocaine concentration. The cells became slender with many intercellular gaps. With increasing cocaine concentration (3–7 mM), the cells became smaller and round, and clumped together.
Under our experimental conditions, the lethal dose, where 50% of the cells were killed by cocaine was determined to be 4.307 mM at 24 h. Most previous studies [2, 3, 9, 14] did not quantify cocaine sublethal toxic dose on cell viability. Only one study quantified cocaine toxicity in rat cortical neurons after 24 h [24]. The lethal dose value in our study (LC50 = 4.307 mM) is highly consistent with that report (LC50 = 4.3 mM). Since our studies were performed in the presence of 10% FBS, consistent with earlier studies [24, 27, 44], the high LC50 values at 24 and 48 h may suggest serum attenuated cocaine toxicity. This serum concentration level brings us a step closer to approximating in vivo serum-cocaine interactions [41] and thus may represent a better in vivo simulation for the predictive cocaine dose in preclinical trials.
Studies on various cell cultures have indicated that cocaine toxicity was associated with cell vacuolation [45, 46]. Consistent with these reports, our dose dependent toxicity studies at 24 and 48 h suggest that cell death may be due to massive vacuolation in glial cells (Fig. 4). In fact, it was observed that within 1 h of cocaine treatment several vacuoles were noticed in the cytoplasm, confirming previous results on different cells [27]. Because the acidic environment can induce vacuolization in some cell types [47], the stocks as well as all working stock solutions of cocaine hydrochloride in our studies were dissolved in PBS. The samples were added to glial cells in minimum volume (5 μl/well/200 μl final vol.) corresponding to 2.5% to prevent pH changes of the media. The final pH after addition of cocaine solution from different working stocks was found to be around 7.4. Employment of this method ensures that vacuole induction in glial cells was due to cocaine exposure rather than pH fluctuations.
The dose dependent decrease in mitochondrial membrane potential (Fig. 5) in glial cells in our study demonstrates that cocaine interacts with mitochondria and impairs energy metabolism. This impairment may lead to an unusual glucose oxidation, commonly observed in certain CNS associated diseases such as schizophrenia and Alzheimer’s disease [48]. Loss in mitochondrial potential was also shown in cells outside of CNS. For example, cocaine was shown to cause dose and time dependent loss in the mitochondrial membrane potential in rat cardiomyocytes primary cultures [14]. These observations may suggest that mitochondria are the primary target of cocaine toxicity in various cells.
Comparing the LC50 and ED50 values of cytotoxicity and mitochondrial membrane potential in our study, it appears that loss of mitochondrial membrane potential was the primary event related to cytotoxicity. This observation is consistent with earlier studies [14]. Since cell viability reflects a function of mitochondrial status, the close LC50 (4.703 mM) and ED50 (4.0 mM) values in the 24 h study corroborate this relationship.
Even though cocaine toxicity in wide range of cells is well documented, only one report [6] showed the effect of cocaine on cell cycle phases. We observed that cocaine arrested cell cycle progression at both G0/G1 and G2/M at 24 and 48 h, respectively (Figs. 6, 7). This observation is consistent with rat neural progenitor (AF5) cells, which also show G1/S arrest by cocaine after 24 h [6]. Our results further suggest that cell cycle phase arrest by cocaine may depend on length of exposure as shown above. Since astrocytes in the mature brain proliferate even in adulthood [16], the present findings suggest that cocaine consumption likely inhibits astroglia cells at G0/G1 or G2/M cell cycle phases, triggering their death.
On the other hand, as mature neurons in the brain do not proliferate, the results of cell cycle analyses in this study suggest that glial cells may be the earliest targets in cocaine abusers. This needs confirmation. The mechanism of action of cell death was not the objective of study in this investigation. However, significant collapse of mitochondrial membrane potential (Fig. 5) and increased cell cycle arrest at G0/G1 and G2/M phases (Figs. 6, 7) by cocaine may indicate that glial cell death could be due to apoptosis, an observation consistent with our preliminary study (data not shown). Since DNA damage induces cell cycle block at G1 and G2 phases [49], we speculate that cocaine treatment in our studies resulted in more DNA damage in glial cells leading to arrest at these phases. This, however, needs further study to confirm.
Results of cell viability (Fig. 3), mitochondrial membrane potential (Fig. 5), and cell cycle analyses (Figs. 6, 7) clearly demonstrate a gradual and dose dependent response with cocaine treatment. These observations suggest that cocaine toxicity is not of non-specific type.
This is the first report to demonstrate cocaine-induced morphological alterations in glial cells and to quantify the sublethal dose of cocaine toxicity based on cell doubling studies in the presence of serum proteins. In vitro cell culture studies invariably have disadvantages due to deviation from the normal context of all body organs of in vivo, which is highly complex. Nevertheless, in vitro studies provide useful information on drug dose estimation for in vivo studies. Earlier studies demonstrated that cocaine interacts with serum proteins in the blood [41]. Similar interactions may be possible under in vitro conditions when the media is supplemented with serum proteins. Because the extent of these interactions may depend on the amount of serum in media, using at least 10% serum in the media for cocaine-related studies may help to identify a better predictive dose for acute toxic studies in preclinical trials. We conclude that the loss of mitochondrial membrane potential, vacuolation, and cell cycle arrest under our experimental conditions represent the early onset of some key events in cocaine-induced toxicity in glial cells.
Acknowledgments
This study was supported by NCRR/RCMI G12 RR03020, NIGMS/MBRS/SCORE GM08111, and HRSA SD34HP0 4018 of the US.
References
- 1.National Institute of Drug Abuse. What is the scope of cocaine use in the United States? NIDA Research Report Series; Bethesda, Maryland: 1999. p. 2. [Google Scholar]
- 2.He J, Xiao Y, Zhang L. Cocaine-mediated apoptosis in bovine coronary artery endothelial cells: role of nitric oxide. J Pharmacol Exp Ther. 2001;298:180–187. [PubMed] [Google Scholar]
- 3.Li G, Xiao Y, Zhang L. Cocaine induces apoptosis in fetal rat myocardial cells through the p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J Pharmacol Exp Ther. 2005;312:112–119. doi: 10.1124/jpet.104.073494. [DOI] [PubMed] [Google Scholar]
- 4.Ndikum-Moffor F, Schoeb TR, Roberts SM. Liver toxicity from norcocaine nitroxide, an N-oxidative metabolite of cocaine. J Pharmacol Exp Ther. 1998;284:413–419. [PubMed] [Google Scholar]
- 5.Zachor D, Cherkes JK, Fay CT, Ocrant I. Cocaine differentially inhibits neuronal differentiation and proliferation in vitro. J Clin Invest. 1994;93:1179–1185. doi: 10.1172/JCI117071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee C, Chen J, Hayashi T, Tsai Sanchez JF, Errico SL, Amable R, Su T, Lowe RH, Huestis MA, Shen J, Becker KG, Geller HM, Freed WJA. Mechanism for the inhibition of neural progenitor cell proliferation by cocaine. PLoS Med. 2008;5(6):e117. doi: 10.1371/journal.pmed.0050117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Candel ER, Schwartz JH, Jessel TM. Principles of neural science. Appleton and Lange; Norwalk: 1991. p. 20. [Google Scholar]
- 8.Lloyd SA, Faherty CJ, Smeyne RJ. Adult and in utero exposure to cocaine alters sensitivity to the Parkinsonian toxin 1- methyl-4-phenyl-1, 2, 3, 6-trahydropyridine. Neuroscience. 2006;137:905–913. doi: 10.1016/j.neuroscience.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 9.Gu J, Yassini P, Goldberg G, Zhu W, Konat GW, Wiggins RC. Cocaine cytotoxicity in serum-free environment: C6 glioma cell culture. NeuroToxicology. 1993;14:19–22. [PubMed] [Google Scholar]
- 10.Barg J, Belcheva MM, Zimlichman R, Levy R, Saya D, McHale RJ, Johnson FE, Coscia CJ, Vogel Z. Opioids inhibit endothelin-mediated DNA synthesis, phosphoinositide turnover, and Ca2+ mobilization in rat C6 glioma cells. J Neurosci. 1994;14(5):858–5864. doi: 10.1523/JNEUROSCI.14-10-05858.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broadwell RD, Sofroniew MV. Serum proteins bypass the blood–brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol. 1993;120:245–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Looney D, Taub D, Chang SL, Way D, Witte MH, Graves MC, Fiala M. Cocaine opens the blood brain barrier to HIV-1 invasion. J Neurovirol. 1998;4:619–626. doi: 10.3109/13550289809114228. [DOI] [PubMed] [Google Scholar]
- 13.Someda K, Kageyama N. Observation of serum proteins in the edematous brain tissue by fluorescein-antibody technique. Acta Neuropathol. 1968;10:347–355. doi: 10.1007/BF00690709. [DOI] [PubMed] [Google Scholar]
- 14.Yuan C, Acosta D., Jr Cocaine-induced mitochondrial dysfunction in primary cultures of rat cardiomyocytes. Toxicology. 1996;112:1–10. doi: 10.1016/0300-483x(96)03341-0. [DOI] [PubMed] [Google Scholar]
- 15.Yu RC, Lee T, Wang TC, Li J. Genetic toxicity of cocaine. Carcinogenesis. 1999;20:1193–1199. doi: 10.1093/carcin/20.7.1193. [DOI] [PubMed] [Google Scholar]
- 16.Raff MC, Abney ER, Miller RH. Two glial cell lineages diverge prenatally in the rat optic nerve. Dev Biol. 1984;106:53–64. doi: 10.1016/0012-1606(84)90060-5. [DOI] [PubMed] [Google Scholar]
- 17.Sibenaller ZA, Etame AB, Ali MM, Barua M, Braun TA, Casavant TL, Ryken TC. Genetic characterization of commonly used glioma cell lines in the rat animal model system. Neurosurg Focus. 2005;19:1–9. doi: 10.3171/foc.2005.19.4.2. [DOI] [PubMed] [Google Scholar]
- 18.Zimmer DB, Van Eldik LJ. Analysis of the calcium-modulated proteins S100 and calmodulin, and their target proteins during C6 glioma cell differentiation. J Cell Biol. 1989;108:141–151. doi: 10.1083/jcb.108.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badisa RB, Tzakou O, Couladis M, Pilarinou E. Cytotoxic activities of some Greek Labiatae herbs. Phytother Res. 2003;17:472–476. doi: 10.1002/ptr.1175. [DOI] [PubMed] [Google Scholar]
- 20.Davis JM. Basic cell culture: a practical approach. Oxford University Press Inc; New York: 2002. pp. 165–167. [Google Scholar]
- 21.Wujuan Z, Shuqing W, Xingguo C, Zhide H, Hongping X. Determination of nucleic acids with crystal violet by a resonance light-scattering technique. Analyst. 2001;126:513–517. [PubMed] [Google Scholar]
- 22.Badisa RB, Darling-Reed SF, Joseph P, Cooperwood JS, Latinwo LM, Goodman CB. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009;29:2993–2996. [PMC free article] [PubMed] [Google Scholar]
- 23.Ipsen J, Feigl P. Bancroft’s introduction to biostatistics. Harper & Row; New York: 1970. p. 455. [Google Scholar]
- 24.Cunha-Oliveira T, Rego AC, Cardoso SM, Borges F, Swerdlow RH, Macedo T, de Oliveira CR. Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res. 2006;1089:44–54. doi: 10.1016/j.brainres.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 25.Vitullo JC, Karam R, Mekhail N, Wicker P, Engelmann GL, Khairallah PA. Cocaine-induced small vessel spasm in isolated rat hearts. Am J Pathol. 1989;135:85–91. [PMC free article] [PubMed] [Google Scholar]
- 26.Welder AA. A primary culture system of adult rat heart cells for the evaluation of cocaine toxicity. Toxicology. 1992;72:175–187. doi: 10.1016/0300-483x(92)90111-q. [DOI] [PubMed] [Google Scholar]
- 27.Yu RC, Li J, Lee T, Lin L. Characterization of cocaine-elicited cell vacuolation: the involvement of calcium/calmodulin in organelle deregulation. J Biomed Sci. 2008;15:215–226. doi: 10.1007/s11373-007-9213-z. [DOI] [PubMed] [Google Scholar]
- 28.Garg UC, Turndorf H, Bansinath M. Effect of cocaine on macromolecular syntheses and cell proliferation in cultured glial cells. Neuroscience. 1993;57:467–472. doi: 10.1016/0306-4522(93)90079-u. [DOI] [PubMed] [Google Scholar]
- 29.Hu S, Cheeran MC-J, Sheng WS, Ni HT, Lokensgard JR, Peterson PK. Cocaine alters proliferation, migration, and differentiation of human fetal brain-derived neural precursor cells. J Pharmacol Exp Ther. 2006;318:1280–1286. doi: 10.1124/jpet.106.103853. [DOI] [PubMed] [Google Scholar]
- 30.Syapin PJ, Militante JD, Garrett DK, Ren L. Cytokine-induced iNOS expression in C6 glial cells: transcriptional inhibition by ethanol. J Pharmacol Exp Ther. 2001;298:744–752. [PubMed] [Google Scholar]
- 31.Chin T, Hwang H, Chueh S. Distinct effects of different calcium-mobilizing agents on cell death in NG 108- 15 neuroblastoma X glioma cells. Mol Pharmacol. 2002;61:486–494. doi: 10.1124/mol.61.3.486. [DOI] [PubMed] [Google Scholar]
- 32.Ohta K, Kuno S, Mizuta I, Fujinami A, Matsui H, Ohta M. Effects of dopamine agonists bromocriptine, pergolide, cabergoline, and SKF-38393 on GDNF, NGF, and BDNF synthesis in cultured mouse astrocytes. Life Sci. 2003;73:617–626. doi: 10.1016/s0024-3205(03)00321-7. [DOI] [PubMed] [Google Scholar]
- 33.Carrasco E, Werner P. Selective destruction of dopaminergic neurons by low concentrations of 6-OHDA and MPP+: protection by acetylsalicylic acid (aspirin) Parkinsonism Relat Disord. 2002;8:407–411. doi: 10.1016/s1353-8020(02)00022-6. [DOI] [PubMed] [Google Scholar]
- 34.Charles I, Khalyfa A, Kumar DM, Krishnamoorthy RR, Roque RS, Cooper N, Agarwal N. Serum deprivation induces apoptotic cell death of transformed rat retinal ganglion cells via mitochondrial signaling pathways. Invest Ophthalmol Vis Sci. 2005;46:1330–1338. doi: 10.1167/iovs.04-0363. [DOI] [PubMed] [Google Scholar]
- 35.Lieberman I, Ove P. A protein growth factorfor mammalian cells in culture. J Biol Chem. 1958;233:637–642. [PubMed] [Google Scholar]
- 36.Holmes RS. Preparation from human serum of an alpha-one protein which induces the immediate growth of unadapted cells in vitro. J Cell Biol. 1967;32:297–308. doi: 10.1083/jcb.32.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berger JJ, Dice JF. Proteolysis in cultured cells during prolonged serum deprivation and replacement. Am J Physiol. 1986;251:C748–C753. doi: 10.1152/ajpcell.1986.251.5.C748. [DOI] [PubMed] [Google Scholar]
- 38.Cadet JL, Ordonez S. Serum withdrawal potentiates the toxic effects of methamphetamine in vitro. Ann N Y Acad Sci. 2000;914:82–91. doi: 10.1111/j.1749-6632.2000.tb05186.x. [DOI] [PubMed] [Google Scholar]
- 39.Tognon G, Frapolli R, Zaffaroni M, Erba E, Zucchetti M, Faircloth GT, D’Incalci M. Fetal bovine serum, but not human serum, inhibits the in vitro cytotoxicity of ET-743 (Yondelis, trabectedin) Cancer Chemother Pharmacol. 2004;53:89–90. doi: 10.1007/s00280-003-0704-y. [DOI] [PubMed] [Google Scholar]
- 40.Stoll LL, Spector AA. Changes in serum influence the fatty acid composition of established cell lines. In Vitro Cell Dev Biol. 1984;20:1054–5476. doi: 10.1007/BF02618879. [DOI] [PubMed] [Google Scholar]
- 41.Edwards DJ, Bowles SK. Protein binding of cocaine in human serum. Pharm Res. 1988;5:440–442. doi: 10.1023/a:1015992502509. [DOI] [PubMed] [Google Scholar]
- 42.Zaragoza A, Diez-Fernandez C, Alvarez AM, Andres D, Cascales M. Mitochondrial involvement in cocaine-treated rat hepatocytes: effect of N-acetylcysteine and deferoxamine. Br J Pharmacol. 2001;132:1063–1070. doi: 10.1038/sj.bjp.0703909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraker TD, Temesy-Armos PN, Brewster PS, Wilkerson RD. Mechanism of cocaine-induced myocardial depression in dogs. Circulation. 1990;81:1012–1016. doi: 10.1161/01.cir.81.3.1012. [DOI] [PubMed] [Google Scholar]
- 44.Nassogne M, Evrard P, Courtoy PJ. Selective neuronal toxicity of cocaine in embryonic mouse brain cocultures. Proc Natl Acad Sci USA. 1995;92:11029–11033. doi: 10.1073/pnas.92.24.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodammer JE, Murchelano RA. Cytological study of vacuolated cells and other aberrant hepatocytes in winter flounder from Boston Harbor. Cancer Res. 1990;50:6744–6756. [PubMed] [Google Scholar]
- 46.Lazo JS, Schisselbauer JC, Herring GM, Kennedy KA. Involvement of the cellular vacuolar system with the cytotoxicity of bleomycin-like agents. Cancer Commun. 1990;2:81–86. doi: 10.3727/095535490820874687. [DOI] [PubMed] [Google Scholar]
- 47.de Bernard M, Papini E, de Filippis V, Gottardi E, Telford J, Manetti R, Fontana A, Rappuoli R, Montecucco C. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J Biol Chem. 1995;270:23937–23940. doi: 10.1074/jbc.270.41.23937. [DOI] [PubMed] [Google Scholar]
- 48.Blass JP. Glucose/mitochondria in neurological conditions. Int Rev Neurobiol. 2002;51:325–376. doi: 10.1016/s0074-7742(02)51010-2. [DOI] [PubMed] [Google Scholar]
- 49.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]