

Abstract
The authors identified a missense mutation in the FTL gene (474G>A; A96T) in a 19-year-old man with parkinsonism, ataxia, corticospinal signs, mild nonprogressive cognitive deficit, and episodic psychosis. This mutation was also present in his asymptomatic mother and younger brother, who had abnormally low levels of ferritin in the serum. The patient and his mother displayed bilateral involvement of the pallidum.
Neuroferritinopathy, an autosomal dominant disease caused by mutations in the ferritin light polypeptide gene (FTL),1 has been reported as a movement disorder with heterogeneous presentations starting in the fourth to sixth decades, with either predominating chorea, dystonia, or a bradykinetic-rigid syndrome (table). The cases of neuroferritinopathy previously described1–5 were associated with the insertion of an adenine in position 460–461 of the FTL gene, or a CT dinucleotide in position 498–499, both of which caused frameshifts, originating FTL polypeptides in which the C-terminal amino acid residues were changed and the length of the protein was extended. We describe a novel missense mutation in the FTL gene, associated with an earlier onset of disease.
Table.
Clinical, imaging and neuropathology findings in families with autosomal dominant neuroferritinopathy
| Family origin | Cumbria, North West, England1 | France3 | France5 |
|---|---|---|---|
| No. of patients/family members with mutation | 2/5 | 7/7 | 6/7* |
| Age at onset, y | 40–55 (1 on 3rd decade) | 24–58 (2 on 3rd decade) | 20 |
| Mode of onset | Choreoathetosis + dystonia | Dystonia | Tremor |
| Disease duration, y | − | − | 38 |
| Cognitive impairment | Frontal lobe impairment (1)† | Frontal syndrome (2); dementia (1) | Frontal syndrome; dementia |
| Parkinsonism | + | + | + |
| Dystonia | ++; blepharospasm 1† with palatal tremor, buccolingual dyskinesia and generalized dystonia | +; blepharospasm | +; buccolingual dyskinesia, hand and feet dystonia |
| Choreoatetosis | ++ | + | − |
| Pyramidal signs | + | − | + |
| Cerebellar signs | − | + | ++ |
| Neuropathy | − | − | − |
| Ferritin serum levels | 4–16 μg/L (5 with mutation) | − | − |
| Imagery | BG cavitations | BG cystic changes (3) | Severe cerebellar atrophy; BG T2 hypo, T1 hyperintensities |
| Neuropathology | Reddish discoloration of the BG; iron positive deposits in GP; spherical inclusions positive for ferritin | − | Greyish discoloration of C and P; small cavities in P; intranuclear bodies in neurons and glia positive for ferritin |
| Mutation | FTL 460–461InsA | FTL 460–461InsA | FTL 498–499InsTC |
Only one patient described.
Reference 2.
AD = autosomal dominant; BG = basal ganglia; GP = globus pallidus; C = caudate; P = putamen; + = present; ++ = most impressive feature; − = absent.
Methods
A 19-year-old man of gypsy ancestry with mild non-progressive mental retardation (IQ = 60), showed no evidence of anoxia at birth and developed gait disturbances by age 13. He was admitted at the ages of 14 and 18 to different institutions, with episodes of acute psychosis responsive to neuroleptics. In the latest admission he was diagnosed with Wilson disease and received treatment with d-penicillamine, valproic acid, and trazodone. Eleven months later, he was admitted to our hospital in a state of confusion with apathy. Neurologic examination showed severe ataxia, bradykinetic-rigid syndrome, and plantar extensor responses. Laboratory findings included normal liver function, plasma ceruloplasmin of 26 ng/mL, copper of 76 ng/mL, and urine copper of 8.1 ng/mL. Kayser–Fleischer rings were not present. Abdominal ultrasound was normal. Serum ferritin was 16 ng/mL (N = 20 to 300), serum iron was 174 μg/mL (N = 65 to 175,) and IBC was 376 μg/mL (N = 250 to 450). MRI showed slightly asymmetric pallidal hyperintensity in T2 and fluid-attenuated inversion recovery images and hypointensity in T1. A diffuse atrophy was also evident, predominating in the frontoparietal and vermian regions (figure 1). Upon withdrawal of all medication the patient experienced a dramatic improvement, resuming his daily routine. After 1 year, neurologic examination revealed mild bradykinesia, symmetric cog-wheel rigidity, slight ataxia, and bilateral Babinski sign. No behavioral or cognitive deterioration were observed during this follow-up period.
Figure 1.

MRI study of the patient (a through e) and his mother (f). A bilateral pallidal hyperintensity can be observed in axial T2-weighted (a), fluid-attenuated inversion recovery (b), and coronal T2 images; no significant surrounding hypointensities were noted in these images. A diffuse atrophy of the parenchyma could also be appreciated, including the vermian region (d). Similar T2-weighted images of the patient (e) and his mother (f) revealed the same pattern of pallidal involvement, although the extent of signal alterations was smaller in the mother’s study, where no concomitant parenchimal atrophy was noted.
We observed nine family members (figure 2a) and performed biochemical and genetic studies in all and MRI studies in the patient, his mother, and younger brother.
Figure 2.
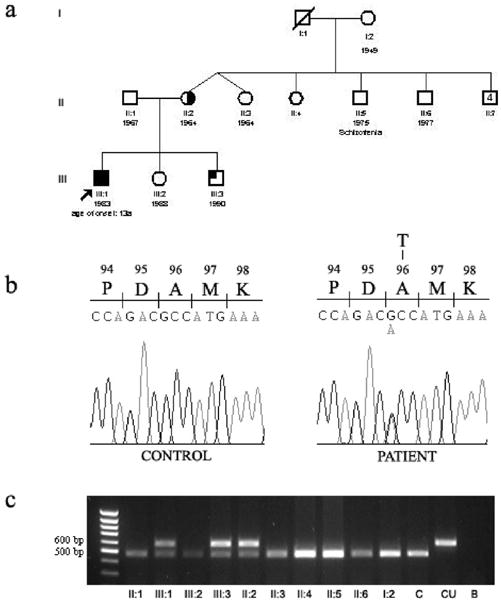
Pedigree of the family in study (a): black square = individual with clinical signs, low ferritin levels, MRI findings and FTL mutation; half-filled circle = low ferritin levels, MRI findings and FTL mutation; low ferritin levels and FTL mutation; circle or square with an n = no FTL mutation. Molecular analysis revealing the heterozygous 474 A>G sequence change in the FTL gene: (b) sequencing chromatogram of the PCR product including exons 3 and 4 of the patients and (c) PCR-RFLP analysis of all family members available for study. C- control individual, CU –undigested PCR product, B-PCR blank.
None of the other relatives observed had signs of neurologic or psychiatric disease, other than an uncle who had been previously diagnosed with schizophrenia. The patient and his mother displayed a similar pattern of pallidal necrosis in MRI (see figure 1).
We extracted DNA from 50 randomly selected and anonymous Guthrie cards obtained from the national neonatal phenylketonuria screening program. This study was authorized by the ethics committee of the Medical Genetics Institute where samples were obtained.
After informed consent was obtained, genomic DNA was extracted from blood samples using the Puregene system (Gentra Systems, Minneapolis, MN) according to the manufacturer’s protocol and from Guthrie cards using Chelex. For analysis of the FTL gene (OMIM 134790), DNA was amplified by PCR and sequenced, using primers FTLgIF, FTL1bF, FTL2R, FTL3F, FTL4R1 and primers FTLp1 (5′TGAGCCACTTCTTCCGCGAAT3′) and FTLp2 (5′TGAGCCACTTCTTCCGCGAAC3′). For confirmation of the G474A mutation, a PCR-RFLP assay was developed, using primers FTL3F/FTL4R for PCR amplification, followed by digestion with BsaHI.
Serum ferritin levels were evaluated using chemiluminescence immunoassay, serum iron by colorimetric analysis without precipitation, and serum transferrin by nephelometry.
Results
Upon sequencing the coding region of the FTL gene, we identified the presence in the proband of a G to A mutation at position 474 of the cDNA (NM_000146), that results in the codon GCC being changed to ACC, thus originating an amino acid substitution in which alanine 96 is replaced by threonine (figure 2b). The same mutation was detected in the 40-year-old mother, who is asymptomatic but displays similar MRI findings, and in her 13-year-old unaffected son. The A96T mutation was not present in the control population and in all other family members tested (figure 2c), including the maternal grandmother, suggesting inheritance from the maternal grandfather’s side. This family member had died of accident at the age of 45, and it was impossible to test that branch of the family, so we could not confirm this hypothesis.
The serum ferritin levels of the patient were abnormally low (16 ng/mL); his mother and his younger brother also had low plasma ferritin (mother: 13 ng/mL; younger brother: 10 ng/mL), in the presence of normal values of other iron parameters. All other relatives studied had levels of ferritin in plasma within normal limits (see figure 2a).
Multiple sequence alignment revealed that the alanine residue at position 96 of the FTL polypeptide was conserved among distantly related species (figure E-1 on the Neurology Web site at www.neurology.org). Theoretical modeling of the effect of the A96T mutation upon the structure of the FTL polypeptide shows that this residue is located in the same region, concerning tertiary structure, that is affected by the 460–461InsA mutation.
Discussion
Evidence to suggest that the alanine to threonine substitution at position 96 of the FTL polypeptide is the cause of disease in this patient comes from evolutionary data showing conservation of this amino acid and from structural modeling of the polypeptide that predicts an important effect in a functionally relevant region (appendices E-1 and E-2). Additional evidence comes from the association of this mutation with low serum ferritin levels in all individuals tested, and from the consistency of MRI findings in the patient and his mutation carrier mother, suggesting expression of the mutation in specific structures of the CNS, namely the globus pallidus (figure 1). Whereas the patient, once removed from all medication, had a mild clinical presentation, the mother remains asymptomatic, in spite of MRI findings. This difference in age at onset is not unexpected in diseases related to iron accumulation, in which women, given the iron loss associated with menses, are generally less affected.
One particular feature of the neuroferritinopathy cases described in the literature,1–3 (which we have also observed in all the A96T mutation carriers), are the lower than normal plasma ferritin levels, in spite of evidence of iron and ferritin accumulation in the CNS; this may reflect an altered regulation of its secretion process6 or instability and increased turnover of mutant ferritins. Why mutations in FTL lead to a deregulation of iron storage in the CNS remains unclear, but evidently there are very specific target regions, displaying selective vulnerability either to iron accumulation or to the effect of accumulated iron and ferritin. MRI of the brain showed considerable hyperintensity on T2-weighted images in the pallidum, which is consistent with other reports of neuroferritinopathy. Yet, in contrast with previous descriptions, no significant involvement of the putamen, thalamus, substantia nigra and dentate nuclei was found in the patient. As in Wills’ report,2 we also failed to find evidence of altered mineralization of the pallidum. In accordance to that report, a significant brain atrophy was detected in the patient, but not in his mother.
At the clinical level, we cannot affirm that the patient’s psychotic episodes are in definite association with the A96T mutation, since a non-carrying uncle had schizophrenia. This particular FTL mutation does appear to be associated, in this family, with pallidal lesions, in two of the carriers, and, in one, with an early onset slowly progressive form of the disease, leading to minor functional impairment after six years. The identification of two pre-symptomatic individuals of 13 and 40 years provides an excellent opportunity for studying the onset of clinical expression and the disease process in neuroferritinopathy.
Acknowledgments
The authors thank the patient and family for their participation.
Research supported by FCT/FEDER (CBO/33485/99). M.C.C. and M.C. received FCT scholarships.
Footnotes
Disclosure: The authors report no conflicts of interest.
Additional material related to this article can be found on the Neurology Web site. Go to www.neurology.org and scroll down the Table of Contents for the August 23 issue to find the title link for this article.
References
- 1.Curtis A, Fey C, Morris C, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 2.Wills A, Sawle G, Guilbert P, Curtis A. Palatal tremor and cognitive decline in neuroferritinopathy. J Neurol Neurosurg Psychiatry. 2002;73:86–95. doi: 10.1136/jnnp.73.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chinnery P, Curtis A, Fey C, et al. Neuroferritinopathy in a French family with late onset dominant dystonia. J Med Genet. 2003;40:e69. doi: 10.1136/jmg.40.5.e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wills A. Hereditary neuroferritinopathy presenting with palatal tremor, orofacial dyskinesias, bulbar symptoms, and unsteadiness of gait. Mov Disord. 2003;18:1057. [Google Scholar]
- 5.Vidal R, Ghetti B, Takao M, et al. Intracellular ferritin acumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the Ferritin Light Polypeptide gene. J Neuropathol Exp Neurol. 2004;63:363–380. doi: 10.1093/jnen/63.4.363. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh S, Hevi S, Chuck S. Regulated secretion of glycosilated human ferritin from hepatocytes. Blood. 2004;103:2369–2376. doi: 10.1182/blood-2003-09-3050. [DOI] [PubMed] [Google Scholar]