

Abstract
The extent to which a drug inhibits a target responsible for a therapeutic effect is a more rational primary endpoint for dose-finding studies of more selective anticancer drugs than the conventional endpoint of dose-limiting toxicity (DLT) used for cytotoxic agents. An adaptive phase I trial design incorporating maximum target inhibition as the primary endpoint was developed to define the optimal dose of talabostat, a dipeptidyl peptidase (DPP) inhibitor, in children with relapsed or refractory solid tumors. The relationship between dose and effect (percent inhibition of serum DPP-4) was assessed using a maximum effect model. Maximum target inhibition was defined as greater than 90% DPP-4 inhibition in five or more of six patients 24 hours post-dose. If DLT was to occur, the trial would adapt to a traditional phase I design with a more conservative dose escalation. At the 600 μg/m2 dose level, serum DPP-4 inhibition at 24 hours was 85%. No talabostat-related DLT occurred. The maximum effect model predicted that 1200 μg/m2 of talabostat would maximally inhibit DPP-4. This adaptive trial design appears to be feasible, safe, and efficient and warrants further evaluation for development of molecularly targeted agents.
CONTEXTS AND CAVEATS
Prior knowledge
The conventional primary endpoint in dose-finding studies is dose-limiting toxicity (DLT). However, newer more selective anticancer drugs may require a different primary endpoint defined by the extent to which a drug inhibits a therapeutic target. Talabostat mesylate inhibits fibroblast activation protein (FAP), which may play a role in tumorigenesis and tumor stromal remodeling.
Study design
A phase I trial design incorporating maximum target inhibition as the primary endpoint was used to find the optimal dose of talabostat in children with relapsed or refractory solid tumors. Inhibition of dipeptidyl peptidase-4 (DPP-4) was used as a surrogate for FAP inhibition. The trial was designed to revert to a traditional phase I trial if DLT were to occur.
Contribution
DPP-4 activity was completely inhibited at doses lower than that predicted by the maximum effect model. There were no grade 3 or 4 toxic effects or talabostat-related DLT.
Implications
Maximum target inhibition is a rational primary endpoint for selective anticancer drugs. An adaptive trial design incorporating this model can be a feasible and safe means of dose finding for molecularly targeted agents.
Limitations
The trial was stopped because clinical development of talabostat was discontinued, so the effects could not be investigated in a larger sample of patients. DPP-4 was used as a surrogate for FAP because of ease of sampling, so FAP inhibition was not measured directly.
From the Editors
Conventional anticancer drug dose-finding (phase I) trials define the recommended dose as the maximum tolerated dose (MTD) based on the incidence of dose-limiting toxicity (DLT). DLT may not be the optimal endpoint for new classes of more selective, potentially less toxic molecularly targeted drugs because the MTD may substantially exceed the dose required to achieve maximum target inhibition (MTI) (1–6). Determining optimal dose by quantifying target modulation is a rational alternative but depends on identification of the appropriate drug target, availability of a validated real-time assay for quantifying target modulation, tissue selection (tumor or surrogate) for analysis, and timing of tissue sampling relative to drug administration.
We developed an adaptable trial design that incorporates MTI as the primary endpoint to define optimal dose but that can also define MTD if DLT is observed before reaching a dose that achieves MTI (Figure 1). We applied this design to define the optimal dose or MTD of the dipeptidyl peptidase (DPP) inhibitor talabostat mesylate (Point Therapeutics, Inc, Boston, MA) (7,8), which was administered in combination with temozolomide (150 mg/m2/d × 5 days) or carboplatin (adaptively dosed based on renal function to achieve an area under the curve [AUC] of 7 mg•min/mL over 2 days), in children with refractory solid tumors. Talabostat was given orally once daily for 14 days beginning days 7–9 of each 28-day treatment cycle. The starting dose was 100 μg/m2/d with planned dose escalations to 200, 350, 600, and 900 μg/m2/d. Intrapatient dose escalation was permitted on the second and subsequent treatment cycles in patients who did not experience DLT.
Figure 1.

Trial schema. Algorithm of the adaptable trial design used to define an optimal dose of talabostat based on the degree of target (dipeptidyl peptidase-4 [DPP-4]) inhibition. Maximum target inhibition (MTI) is defined as a greater than 90% decrease in DPP-4 activity relative to baseline, 24 hours after the first dose of talabostat. Two patients are enrolled at the starting dose. If dose-limiting toxicity (DLT) is not observed (boxes with a single line border) and one or both do not achieve MTI, the dose would be escalated. If MTI is achieved in both, the dose level would be expanded to six patients. If fewer than five of the six have MTI, the dose would be escalated. If five or more achieve MTI, the dose would be escalated one additional dose level to ensure that the optimal dose is on the plateau of the dose–response curve. If DLT is observed in one patient at any point, the dose escalation would switch to a traditional phase I (3 + 3 design), and a more conservative 40% dose escalation would be used (boxes with double line border), but DPP-4 inhibition will continue to be monitored. If two or more patients at a dose level experience a DLT, a maximum tolerated dose (MTD) would be defined.
Talabostat competitively and reversibly inhibits fibroblast activation protein (FAP; inhibition constant Ki = 5 nM), which may play a role in tumorigenesis and tumor stromal remodeling. Talabostat also inhibits DPP-4 (Ki = 0.18 nM), present in tissues, including plasma, which is readily accessible (9–12). We used DPP-4 inhibition as a surrogate for FAP inhibition and defined the optimal talabostat dose as the dose inhibiting more than 90% of serum DPP-4 enzyme activity at 24 hours post-dose in five or more of six patients. Serum DPP-4 activity at baseline, 1 hour, and 24 hours after the first talabostat dose of each cycle was quantified using a validated fluorometric assay based on cleavage of Ala-Pro-7-amino-4-trifluoromethylcoumarin conjugate (Bachem, King of Prussia, PA) to a fluorescent product by DPP-4 (13). The assay detects a 90% decrease in enzyme activity with intra- and interassay coefficient of variation of 1.4% and 13.3%, respectively.
Plasma concentrations of talabostat were measured after the first dose on cycle 1 using liquid chromatography with turbo ion-spray tandem mass spectrophotometric detection (Applied Biosystems, Foster City, CA). The assay has a lower limit of quantification of 0.6 ng/mL.
Statistical properties of the adaptable trial design were investigated by calculating probabilities of the three possible outcomes (MTD exceeded, MTI achieved in five or more of six patients, and dose escalation) using a range of probabilities of DLT and greater than 90% DPP-4 inhibition (Supplemental Table 1, available online). At a given dose level, if the true probability of MTI is 0.90 and true DLT probability is 0.05, the probability of exceeding the MTD is 0.03, of achieving MTI in five or more of six patients is 0.76, and of dose escalation is 0.21. With a true probability of MTI of 0.90 and an unacceptable DLT probability of 0.33, the outcome probabilities become 0.61, 0.29, and 0.10, respectively, whereas if the DPP-4 inhibition probability is 0.50 with a 0.05 DLT probability, outcome probabilities are 0.02, 0.10, and 0.88, respectively. Thus, the design has a reasonable chance of correctly identifying the proper dose for attaining MTI.
Six patients, median age 15 years (range 4.5–18 years), were enrolled at doses of 100 (n = 2), 200 (n = 2), and 350 (n = 2) μg/m2/d (Figure 2). Two patients who received two cycles and one who received three cycles had intrapatient talabostat dose escalation for a total of 10 cycles and a maximum talabostat dose of 600 μg/m2/d. No grade 3 or 4 toxic effects and no talabostat-related DLT occurred. The trial stopped before completion because clinical development of talabostat was discontinued, but data from these six patients illustrate the utility of this adaptable phase I trial design.
Figure 2.

Dose–effect curve and pharmacokinetic parameters for oral talabostat at doses ranging from 100 to 600 μg/m2. Talabostat effect is percent inhibition of serum dipeptidyl peptidase-4 (DPP-4) enzyme activity measured 24 hours after the first dose of talabostat. A maximum effect model, 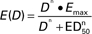 , where E(D) is the observed effect at a given dose D, Emax is the maximum effect (100% inhibition), ED50 is the dose achieving 50% of the Emax, and n is the slope, was fit to the dose–effect data with MLAB (Civilized Software, Silver Spring, MD; http://www.civilized.com/). A) The curve represents the fitted maximum effect model (n = 1.0, ED50 = 130 μg/m2). The maximum effect model predicts that the maximum target inhibition would be achieved at doses exceeding 1200 μg/m2. Each symbol represents an individual patient. B) Pharmacokinetic parameters for each patient and dose. Talabostat pharmacokinetic parameters include Cmax, maximum concentration; Tmax, time to peak concentration; AUC0–8, area under the concentration × time curve extrapolated to infinity; CL/F, apparent clearance.
, where E(D) is the observed effect at a given dose D, Emax is the maximum effect (100% inhibition), ED50 is the dose achieving 50% of the Emax, and n is the slope, was fit to the dose–effect data with MLAB (Civilized Software, Silver Spring, MD; http://www.civilized.com/). A) The curve represents the fitted maximum effect model (n = 1.0, ED50 = 130 μg/m2). The maximum effect model predicts that the maximum target inhibition would be achieved at doses exceeding 1200 μg/m2. Each symbol represents an individual patient. B) Pharmacokinetic parameters for each patient and dose. Talabostat pharmacokinetic parameters include Cmax, maximum concentration; Tmax, time to peak concentration; AUC0–8, area under the concentration × time curve extrapolated to infinity; CL/F, apparent clearance.
AUC0–8 of talabostat increased in proportion to dose (mean AUC0-8 was 7.0 ng•h/mL at 100 μg/m2, 20 ng•h/mL at 200 μg/m2, and 34 ng•h/mL at 350 μg/m2). Mean half-life of talabostat was 2.8 hours. DPP-4 activity was completely inhibited (median = 98%) 1 hour after the first dose of talabostat on nine of the 10 treatment cycles at doses ranging from 100 to 600 μg/m2. One patient experienced nausea and delayed gastric emptying, as evidenced by an undetectable plasma concentration 1 hour post-dose. Plasma talabostat concentration 1 hour post-dose on cycle 1 (100–350 μg/m2) ranged from 0.64 to 10.1 ng/mL (n = 5). At the 600 μg/m2 dose level, serum DPP-4 inhibition was 85% on two cycles administered to one patient (Figure 2). Talabostat plasma concentration 24 hours post-dose (C24 h) was less than 0.6 ng/mL in five of the six patients. One patient, who received 350 μg/m2, had a C24 h of 0.86 ng/mL. The maximum effect model predicted that a dose of 1200 μg/m2 would be required to achieve MTI.
Characterization of the dose–effect relationship by application of basic pharmacodynamic principles was the basis of this dose-finding study. A surrogate tissue (serum) and target (DPP-4) were selected as the endpoint because of the ease of sampling and similar Ki for FAP and DPP-4. To assess whether the target was maximally inhibited throughout the dosing interval, we measured DPP-4 inhibition 24 hours post-dose. The maximum effect model predicted that 1200 μg/m2 would be required to achieve MTI on a once-daily schedule. This dose was not tolerable in adults (14); therefore, a change to twice-daily dosing was planned. The mean plasma talabostat concentration 10 hours post-350 μg/m2 was 0.86 ng/mL, which should be inhibitory (15).
Intrapatient dose escalation with DPP-4 inhibition measured on every treatment cycle provides additional valuable dose–effect data characterizing the dose–effect curve within individual patients as well as the population to more efficiently evaluate multiple dose levels. One limitation of the study was that FAP inhibition was not directly measured. A second limitation was early closure of the study because of drug availability. However, treatment of six patients on four dose levels provided sufficient data to project optimal dose. This adaptable trial design appears to be feasible, safe, and efficient. Further evaluation of this trial design in the development of molecularly targeted agents with validated biomarkers is warranted.
Funding
American Society of Clinical Oncology Young Investigator Award (H.M.); the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (F.M.B., A.A., P.W., R.F.M., S.M.S., B.C.W., E.F.).
Supplementary Data
Supplementary data can be found at http://www.jnci.oxfordjournals.org/.
Footnotes
The funders did not have any involvement in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
References
- 1.Fox E, Curt GA, Balis FM. Clinical trial design for target-based therapy. Oncologist. 2002;7(5):401–409. doi: 10.1634/theoncologist.7-5-401. [DOI] [PubMed] [Google Scholar]
- 2.Hoekstra R, Verweij J, Eskens FA. Clinical trial design for target specific anticancer agents. Invest New Drugs. 2003;21(2):243–250. doi: 10.1023/a:1023581731443. [DOI] [PubMed] [Google Scholar]
- 3.Hunsberger S, Rubinstein LV, Dancey J, Korn EL. Dose escalation trial designs based on a molecularly targeted endpoint. Stat Med. 2005;24(14):2171–2181. doi: 10.1002/sim.2102. [DOI] [PubMed] [Google Scholar]
- 4.Morabito A, Di Maio M, De Maio E, Normanno N, Perrone F. Methodology of clinical trials with new molecular-targeted agents: where do we stand? Ann Oncol. 2006;17(suppl 7):vii128–vii131. doi: 10.1093/annonc/mdl965. [DOI] [PubMed] [Google Scholar]
- 5.Parulekar WR, Eisenhauer EA. Novel endpoints and design of early clinical trials. Ann Oncol. 2002;13(suppl 4):139–143. doi: 10.1093/annonc/mdf651. [DOI] [PubMed] [Google Scholar]
- 6.Balis FM, Fox E, Widemann BC, Adamson PC. Clinical drug development for childhood cancers. Clin Pharmacol Ther. 2009;85(2):127–129. doi: 10.1038/clpt.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham CC. Talabostat. Expert Opin Investig Drugs. 2007;16(9):1459–1465. doi: 10.1517/13543784.16.9.1459. [DOI] [PubMed] [Google Scholar]
- 8.Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004;64(15):5471–5480. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 9.Cheng JD, Weiner LM. Tumors and their microenvironments: tilling the soil. Commentary re: A.M. Scott et al., A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res., 9: 1639–1647, 2003 [comment]. Clin Cancer Res. 2003;9(5):1590–1595. [PubMed] [Google Scholar]
- 10.Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci U S A. 1990;87(18):7235–7239. doi: 10.1073/pnas.87.18.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Connolly BA, Sanford DG, Chiluwal AK, et al. Dipeptide boronic acid inhibitors of dipeptidyl peptidase IV: determinants of potency and in vivo efficacy and safety. J Med Chem. 2008;51(19):6005–6013. doi: 10.1021/jm800390n. [DOI] [PubMed] [Google Scholar]
- 12.Mentlein R. Cell-surface peptidases. Int Rev Cytol. 2004;235:165–213. doi: 10.1016/S0074-7696(04)35004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sedo A, Krepela E, Kasafirek E. A kinetic fluorometric assay of dipeptidyl peptidase IV in viable human blood mononuclear cells. Biochimie. 1989;71(6):757–761. doi: 10.1016/0300-9084(89)90092-8. [DOI] [PubMed] [Google Scholar]
- 14.Uprichard MJ, Jones BJ. Phase 1 rising multiple-dose study of talabostat (PT-100) in healthy subjects. American Society of Hematology Annual Meeting. 2004 San Diego, CA. [Google Scholar]
- 15.Nemunaitis J, Vukelja SJ, Richards D, et al. Phase I trial of PT-100 (PT-100), a cytokine-inducing small molecule, following chemotherapy for solid tumor malignancy. Cancer Invest. 2006;24(6):553–561. doi: 10.1080/07357900600894732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.