

Abstract
Epithelial-mesenchymal-transition (EMT) tumorigenesis in the mouse was first described over 100 years ago using various terms such as carcinosarcoma and without any comprehension of the underlying mechanisms. Such tumors have been considered artifacts of transplantation and of tissue culture. Recently, EMT tumors have been recognized in mammary glands of genetically engineered mice. This review provides a historical perspective leading to the current status in the context of some of the key molecular biology. The biology of mouse mammary EMT tumorigenesis is discussed with comparisons to human breast cancer.
Keywords: Epithelial-mesenchymal-transition, Pathology, Mouse mammary tumors, Spindle cell phenotype, Heterogeneity, Genetically engineered mice, E-Cadherin, Snail, Invasion, Metastasis
EMT and Neoplastic Progression: Lessons from the Mouse
As is frequently the case in breast cancer research, much of the fundamental biology begins with the mouse. In fact, what are now called EMT tumors were first described as carcinosarcomas in the mouse mammary tumor literature of the early 1990’s [1, 2]. The first report of a mouse mammary tumor using microscopic examination, in 1854, concludes that the tumor was “scirrhous” [3]. This descriptive term implies “hard” but may have had a different meaning in the 1850s. Livinggood, who described and illustrated five mouse mammary tumors in 1896, felt obliged to comment that they were not sarcomas as believed by others [4]. The terminology in that era was a bit of a slippery slope. For example, the authoritative opinion of the times was that the mouse tumors were “Familiare Endotheliome” (Family Endotheliomas) [5]. This interpretation was probably based on Virchow and Connheim’s “Blastema” hypothesis that all cancers arose from embryonic rests in the connective tissue coupled with a misinterpretation of the microacinar pattern as being endothelial lining [2]. This debate was finally closed 20 years later [6].
Some of the earliest microscopic illustrations of mouse mammary cancers included spindle cell “carcinosarcomas” [1, 7, 8]. Apolant’s classic descriptions of mouse mammary tumors in 1906 included carcinosarcomas with a drawing of a spindle cell tumor in chapter D. entitled “Die Entwickelung des Sarkomas auf dem Boden transplantierter Carcinome.” (The development of sarcomas from primary transplanted carcinomas.”) [1] (Fig. 1a). Apolant described transplantation experiments with tumors as “Sarkom mit Carcinomrest” [1]. His later paper, with Paul Ehrlich, illustrates a tumor with a combination of glands and spindle cells [9]. Haaland, who provided the first descriptive classification of mouse mammary tumors in English in 1911, discusses carcinosarcomas [8]. Woglom, in a massive 1913 review of the extant scientific literature, defended the then prevalent view that carcinosarcomas were primarily artifacts of transplantation [6].
Figure 1.

Historical. Figure 1 depicts copies of illustrations of mammary “Spindelzollensarkom” from Apolant 1907 (a) “carcinosarcoma” from Dunn 1945 (b), mouse mammary “EMT-type” tumor with immunohistochemical stains for cytokeratin 8/18 (c) and smooth muscle actin (d) from White et al. [47] 2001 and a “triple negative” human breast cancer with undifferentiated cells that stain for vimentin (e) and cytokeratin 8/18 (f). Apolant associated his spindle cell and mixed tumors with transplantation [1]. Dunn associated her mixed tumor with tissue culture and explantation [23]. White found spontaneous EMT-type tumors with loss of the ILK1 transgene and up regulation of Snail [47]. The dual staining, triple negative phenotype in human breast cancer is not recognized as a specific subset or with a specific diagnostic term.
The context of these observations needs to be understood. In 1882, alcohol was the major “hardening” agent and formalin is not on the list [10]. Formaldehyde did not become generally available until 1896 [11]. Livinggood stated that he used alcohol fixation [4]. However, celloiden and paraffin sectioning were available and microtomes produced relatively thin sections. By 1913 Mallory provides recipes for almost all of the stains used today [12]. Perhaps, the major limitation was in the available microscopes and photographic equipment to document observations. However, the artwork available provides impressively accurate artistic reproductions [13]. Later, many of the early papers were clearly illustrated using camera lucida [8].
Of course, inbred mice were not yet available during this early era in breast cancer research and transplant histocompatibility was unknown. Mice were usually purchased from mouse fanciers and animal husbandry practices were somewhat poor to non-existent. In 1913, Mallory’s section on mice recommends that mice be kept in glass cages with holes for air, be bedded in cotton balls and be fed bread and cheese [12]. Round worm infestations were prevalent, leading Borell and others to speculate that mouse mammary cancers were caused by round worms [14, 15].
However, some investigators kept detailed transplant records and found that some tumors could be maintained in serial transplantation within families of mice [16]. Jensen developed the first mouse mammary tumor transplant line which could be maintained in serial section [17]. The Walker Prize given by the Royal College of Surgeons to Jensen for his discovery was vigorously, and somewhat rudely, contested by Williams who considered Jensen’s work artifactual [17, 18]. Before discounting the work of these early pioneers, the reader is encouraged, at a minimum, to read the table of contents to the 1911 report of the Imperial Cancer Fund [19]. The pioneers were quintessential natural historians. They asked the right questions while laying the foundations of experimental mammary cancer biology [2, 20].
In 1913, Tyzzer and his graduate student C.C. Little, the founder of The Roscoe B. Jackson Memorial Laboratory (JAX), set out to develop inbred strains of mice for the purpose of studying the genetic basis for breast cancer [21]. The inbred laboratory mouse became the primary experimental model in cancer research and led to, amongst many other biologically significant observations, the discovery of histocompatibility antigens [2]. The 1920s and 1930s saw the development of numerous strains of mice inbred for various traits. Biochemistry and tissue culture were developed and added to our scientific tool box. By 1945 Thelma Dunn was able to survey an extensive literature on the pathology of mouse mammary tumors and provided a new classification of mouse mammary tumors that included carcinosarcomas (Fig. 1b). Her conclusion was that carcinosarcomas were primarily artifacts of tissue culture [22, 23]. This effectively ended any debate about mouse mammary carcinosarcomas. The rare spindle cell tumors occurring spontaneously were subsequently ignored or attributed to newly discovered sarcoma viruses.
The Interim
However, those pesky spindle cells did not disappear. Attempts to develop mouse mammary cell lines met with great difficulty. Most cell cultures became “fibroblastic” and were “sarcomatous” upon transplantation. In the 1960s and 1970s investigators devised techniques to rid the cultures of “fibroblasts” [24]. Occasionally, investigators were able to develop sustainable epithelial mouse mammary cell lines [25, 26]. For example, a method was developed for maintaining primary cultures in low passage using high density cultures and light trypsinization to limit growth of “fibroblasts” [24]. Using time-lapse cinematography, Young et al. demonstrated that the epithelial cells in these “dome cultures” divided into two types of daughter cells. One daughter cell became an epithelial cell while the second daughter migrated to the periphery of the dome to become a “fibroblastic” spindle cell.
In 1989, a series of experiments using vectors carrying the H-Ras gene, Strange et al. found that unrestricted transfection led to undifferentiated spindle cell tumors while transfection with a single copy of H-Ras resulted in an epithelial tumor [27]. These experiments performed in the “modern era” were, in retrospect, clues about the plasticity of mouse mammary cells and the role of oncogenes.
During this time, as is well documented in this issue, developmental biologists were discussing epithelial-mesenchymal-transition. In 1989, Thiery’s observations with malignant tranformation in tissue culture introduced the concept of epithelial-mesenchymal-transition (EMT) tumors [28]. Thiery found that EMT was associated with SRC (SRC sarcoma oncogene) and FGF (fibroblast growth factor) [28–30]. While the concept had impact in the experimental pathology community, it had little or no impact or serious discussion in the diagnostic community.
The major emphasis within the tumor biology community has been on the events inside tumors and host-tumor interactions [28, 31]. The work of Condeelis and others has provided dramatic and convincing evidence of EMT within epithelial cancers. Condeelis and Pollard demonstrated, using in-vivo microscopy, that EMT-type of transitions occur in Tg(MMTV:PyVmT) mouse mammary tumors on the level of a single tumor cell [32–36]. These tumor cells “break away” from their adjacent tumor cells and are escorted along collagen fibers by tumor-associated macrophages (TAM) to blood vessels. Cellular analysis of these cells by Condeelis has detailed the structural and molecular changes occurring in these migratory cells [33]. The synergic co-migration of TAM and invading breast cancer cells can be explained by a direct paracrine loop by which carcinoma cells secrete specific macrophage growth factors (i.e. CSF-1 (colony-stimulating factor-1)), that, in turn, activate macrophages to secrete epithelial growth factors and chemokines (i.e. EGF(epidermal growth factor), HGF (hepatocyte growth factor), SDF-1 (stromal cell-derived factor-1)) responsible for stimulating carcinoma cell invasion and intravasation [33, 35, 37]. The crosstalk between TAM and tumor cells also affects the activity of actin regulators, such as WASP (Wiskott–Aldrich syndrome protein), resulting in the generation of the fundamental migratory and invasive “tools” in macrophages and invadopodia in tumor cells [33, 38].
Thompson’s 2007 review article on EMT tumorigenesis elicited sharp replies from the clinical community [39, 40]. Thompson and others envision the type of intratumoral EMT event documented by Condeelis. Tarin took the viewpoint that EMT had never been observed in human breast cancer [39]. One could argue that the two opinions were separated by experiences in experimental biology and clinical pathology. The author of the current review took an intermediate view based on his experiences in experimental and surgical pathology [41]. Namely, the clinical practicioners had not looked with the proper tools. The rest of this review will recount the evidence from genetically engineered mouse models.
Recent Observations
In 1984, murine breast cancer biology was turned upside down by the introduction of genetic engineering [42]. In vivo experimental genomics now became a reality. Suddenly, specific genes could be cloned and inserted behind mammary-specific promotors to produce gene-specific tumor phenotypes [43, 44] and pathway specific phenotypes [43, 44]. Conversely, knocking out tumor suppressor genes led to heterogeneous tumor types that could be attributed to the initiating oncogene [45]. The p53 null mice developed numerous types of cancers including mammary cancers [46]. About 50% of the mammary cancers from these knock-out mice were “spindle cell” tumors and classified as “fibrosarcomas” or “angiosarcomas”. When mammary-specific conditional knock-outs continued to produce heterogeneous tumors with unknown initiating oncogenes [45], the “occasional” spindle cell component was ignored or under-reported.
For us, the first real clue about these pesky, but rare, spindle cells occurred in 2001 when White reported mammary tumors induced in Tg(Ilk-1) (integrin-linked kinase) mice had a long latency, incomplete penetrance and lost of expression of the initiating oncogenic transgene [47] (Fig. 1c, d). These tumors all had a spindle cell component mixed with dysplastic glands. They over-expressed the Snail transcription factor associated with epithelial-mesenchymal-transition [47]. The speculation, at that time, was that these were the E-M Transitions first described in cultured tumor cells by Thiery et al. in 1989 [29, 30]. Later, spindle cell tumors were also found within populations of persistent and recurrent doxycycline-induced tumors [48]. Some of the inducible mouse lines produced more spindle cell tumors than others. Further analysis showed these spindle cell tumors to be associated with loss of p53 [49]. Since the original observations in GEM mammary tumors, an increasing number of examples have come to light in a wide variety of mouse models [48–58].
The EMT Type Tumor
Yes, these types of spindle cell tumors of the mammary gland have been around for a long time. What is new, is that the investigator now has immunohistochemistry to document that the spindle cell population in these “mixed tumors” expresses epithelial (Fig. 2) and mesenchymal markers (Fig. 3). We also have mechanistic explanations for the spindle cell phenotype. As we learn more about how phenotypes reflect molecules, our classifications should include molecular mechanisms. Thus, EMT type tumor becomes a synonym for carcinosarcoma and includes spindle cell populations that are only recognized using immunohistochemistry and molecular analysis. Spindle cell tumors appear as primary mammary tumors in some genetically engineered mice with a loss of the expression of the initiating oncogenes or escape from oncogene addiction [33, 36, 59, 60]. Although EMT tumorigenesis is emperically linked with the loss of oncogene expression, the mechanisms are unknown. At the present time, the transition to mesenchymal phenotypes appears yet another mechanism creating tumor plasticity.
Figure 2.
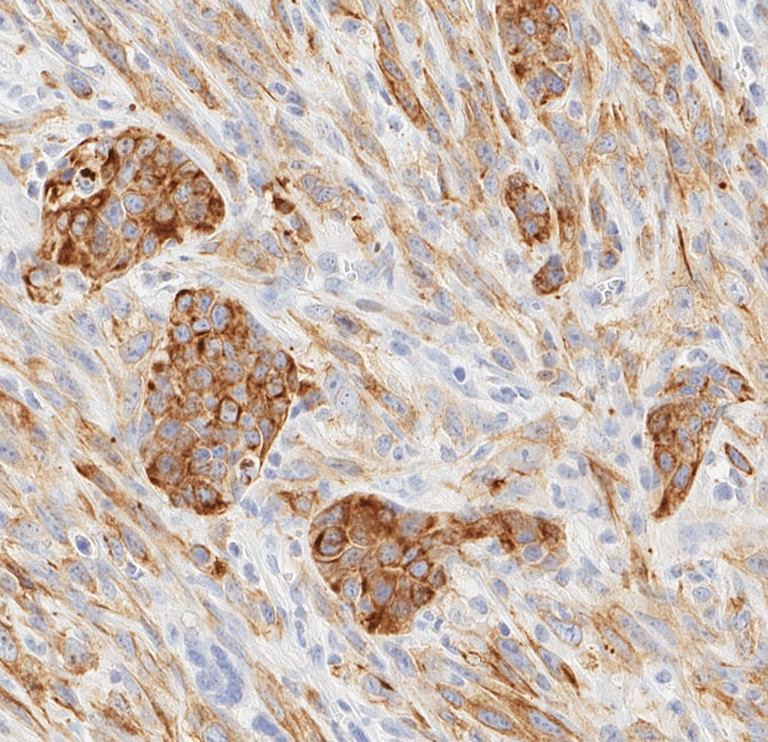
Spindle cell tumor. Figure 2 illustrates the expression of cytokeratin 8/18 detected by immunocytochemistry. Note that the epithelial clusters have the most intense staining pattern. Futhermore, the spindle cell population of fusiform cells with polar cytoplasm also expresses the epithelial antigen. Compare this pattern with staining patterns in Figs. 1 and 3.
Figure 3.

Heterogeneity. This panel illustrates some types of heterogeneity found in EMT tumorigenesis. Panels a and b show a subcutaneous EMT tumor from a Tm(p53±xHas±) female. a is stained with hematoxylin and eosin illustrating the spindle cell morphology of EMT tumors. b is stained using immunohistochemistry (IHC) for Cytokeratin 8/18 demonstrating that the spindle cells are uniformly postive of K8/18. The rectangle shown in the thumbnail overviews indicate the approximate area viewed in the high magnification. Compare this pattern with the IHC patterns in the Tg(Myc)-associated EMT tumor in panels C-F. c is stained for K8/18, d for Vimentin, e K14 and f for Smooth Muscle Actin (SMA). Different areas in the tumor have differential staining for each antigen. For example, the densest SMA stain is at the upper tip of the upper sample (f) but K14 staining is more intense around this area (e). This tumor has many tumor giant cells that are lacking in a and b. The scale bar (d) indicates 50 μm magnification of all the higher magnification images. Compare these patterns with the cover illustration that shows well-defined nests of epithelial cells in a sea of spindle cells.
These phenomena required reconsideration of EMT tumorigenesis as a process and as a diagnostic category [47, 48, 54, 61]. These tumors were initially categorized as “spindle-cell” tumors but subsequent immunohistochemical analysis demonstrated the presence of CKs, vimentin and SMA. These molecules suggest a mixed lineage or a myoepithelial cell origin [54]. Some cohorts have up-regulation of the Snail transcription factor associated with other types of EMT [48]. Others have mutations in p53. These attributes justified the application of the term “EMT” to these tumor types. Dual staining with epithelial (cytokeratin) and mesenchymal (vimentin) biomarkers is a reliable diagnostic criterion [51]. But dual staining should not exclude non-staining tumors.
Epithelial Origin
Since most of the GEM tumors arose in animals with epithelial-specific targeted transgenes or silenced tumor suppressor genes, the tumors likely arose from the mammary epithelium. Direct experimental evidence of epithelial origin comes from tumors derived directly from an epithelial cell line following in-vitro manipulation [62]. Another spindle cell phenotype tumor arose directly from cultured epithelial cells after in vivo transfection with a Snail construct [48]. Remember that “carcinosarcomas” were regarded by early investigators as artifacts of transplantation and tissue culture. These experiments are reminiscent of these early observations but provide some mechanistic proof.
Studies in GEM demonstrate that spindle cell tumors arising from the mammary epithelium, in spite of the diverse initial oncogenic stimuli, have a common morphological end-point. While the spindle cell populations dominate these tumor types, they have a range of epithelial phenotypes. Some populations have no epithelial clusters. Others have small clusters of epithelial cells. Some tumors retain very distinct epithelial populations with minor dual staining populations. These cohorts illustrate the range of possibilities in the EMT phenotype in the mouse mammary gland.
Immunohistochemistry studies demonstrated that dual staining for the two intermediate filaments, vimentin and CKs, staining for SMA and a loss of E-Cadherin staining are common immunophenotypes in most spindle cell tumors in all cohorts thus far studied. Simultaneous immunohistochemical staining of representative samples demonstrated colocalization of the two intermediate filaments in the same fusiform cells. Thus, immunohistochemistry provides convenient, reliable and specific criteria for EMT-phenotype in mouse mammary tumors. The retention of IHC staining epithelial biomarkers also suggests retention of epithelial characteristics.
We have recommended that the diagnosis of EMT tumor be restricted to those tumors whose biological and experimental history is known or those tumors with dual staining cells [51, 57]. The variants that appear within EMT cohorts or of unknown origin should be considered EMT-Type tumors until definitive proof of lineage is provided.
EMT-Type Tumor Heterogeneity
The supposition that any spindle cell population originates from epithelium can be challenged. Not all spindle cell tumors in the mouse mammary glands are “EMT Type Tumors”. Other tumor types may have a spindle cell phenotype but are not dual staining [51]. The spindle cell tumors associated with splenic myeloid hyperplasia and dermal sarcomas are an example [63]. Pure sarcomas do exist and may appear in the mammary fat pad. As documented above, some EMT Type Tumors do not express dual biomarkers [51]. Other EMT tumors will vary in the intensity of antigenic staining from region to region (Figs. 1, 2 and 3). Thus, a dilemma occurs when tumors resemble the spindle cell tumors of epithelial origins but do not stain for intermediate filaments. The diagnosis in these cases resides in the molecular phenotype as discussed below.
As intimated above, the usual dual-staining spindle cell tumor from the mouse mammary gland has a mixture of gland-like structures, tumor giant cells and dominant spindle cell populations (compare the tumors in Figs. 1, 2 and 3). The amount of and differentiation of, the pure epithelial component varies. In some cases, “epithelial” differentiation can only be identified with IHC stains for keratins (See Fig. 3a, b). In other cases, “almost normal” epithelial ducts are present. It is not always clear whether these well differentiated structures are entrapped normal host or a part of the neoplastic population. In some extreme examples, a large, if not major, component of the tumor is clearly epithelial, thus creating the appearance of a “collision” tumor. The variation of tumors within any given experimental cohort demonstrates the heterogeneity (Fig. 3c–f).
The observation of great heterogeneity, within the same cohort and the same tumor, raises the possibility that not all of the spindle cell tumors are the result of EMT. For example, they could arise from mesenchymal subpopulations and be mixed cell populations or bi-phasic tumors. The demonstration that two intermediate filaments colocalized in the same cells supports the hypothesis that they arose from the same lineage but does not exclude the possibility of fusion of two cell types or recruitment from other cell populations such as a putative stem cell. For the time being, it seems reasonable to assume that dual staining in spindle cell populations is evidence of an epithelial-mesenchymal-transition. However, in most cases, one is limited to observation of the terminal events without rigorous experimental evidence of origins [51].
Common and Rare Phenotypes Associated with EMT
As indicated in the historical review, spindle cell tumors have been observed in the mammary glands of laboratory mice for more than a century. They are frequently observed in the context of transplantation and explants from tissue culture. As discussed above they are also seen in the context of genetic engineered mice. Now that we have specific criteria for the recognition of EMT tumorigenesis, one wonders whether such tumors occur in other previously overlooked situations. Sixty-four ageing wild type FVB/N females were studied to establish the prevalence and phenotype of spontaneous mammary and pituitary tumors [56]. Of fourteen mammary tumors, five mice with pituitary tumors had mammary masses consistent with EMT-phenotype tumors. They all showed immunohistochemical expression of CK8/18, vimentin and SMA.
The rare and unexpected observation was that ERα-expression in these EMT-phenotype tumors was observed in the cytoplasm [56]. A retrospective analysis of selected mouse mammary EMT tumors from all of the EMT cohorts previously analyzed also showed cytoplasmic expression of ERα. A marked ERα nuclear expression without cytoplasmic signal was found only in those scattered neoplastic glandular structures suggesting that cytoplasmic ERα is closely related to the EMT phenotype. These empirical observations illustrate an association but do not distinguish between cause and effect. However, anyone with experience with human breast cancer will testify that cytoplasmic staining for ER is frequently observed and rigorously ignored. Is this another one of those phenomena ignored until validated in the mouse [41]?
EMT Tumor Biology
EMT has been consistently associated with poor prognosis in human tumors. The Epithelial to Mesenchymal Transition has been suggested as one of the mechanisms that allowed primary tumors to invade and colonize other tissue, forming metastases and giving a poorer prognoses. In human breast cancer, Snail is being used as a surrogate biomarker for EMT. Over expression of Snail, a transcriptional repressor, has been associated with an increased incidence of metastases in human literature [64]. The Condeelis and Pollard experiments are consistent with this notion because they demonstrate that macrophages are recruited into Tg(PyVmT) tumors and “escort” individual tumor epithelial cells to vascular spaces and into the circulation as metastatic cells. However, mouse EMT tumors, with increased expression of Snail and it’s downstream pathway, are associated with decreased metastases [51, 53]. Further, the spindle cell tumors from other cohorts, although more locally invasive, have had no documented metastases (Fig. 4). In fact, metastatic cells in most mouse models travel to the lungs via intravascular tumor emboli composed of clusters of epithelial cells [45, 65, 66]. Lungs with a combination of micrometastases and tumor emboli may demonstrate that both mechanisms can contribute to the metastatic phenotype.
Figure 4.

Biology. This panel illustrates the expansile, pushing margin between tumor and host that generally occurs in mouse mammary tumors (a) (image captured at 4×). Compare image 4A with the typical infiltrating margin of an spindle cell EMT-type tumor (b) which infiltrates between the fat cells in the field depicted rather than pushing and compressing the host apidocytes (scale bar is 100 um). Both images depict Tg(Neu) tumors. Image 4A is a Neu-induced primary tumor. Image 4B is a recurrent tumor in a doxycycline-induced Neu host.
The EMT tumors of mouse mammary glands are locally aggressive, invading local tissues and growing through fascia and muscles. Thus far, we have not observed a single distant metastasis from spontaneous or tissue culture explants. However, tail vein injections of spindle cell tumors will colonize the mouse lungs. Therefore, while EMT is associated with more aggressive types of neoplasia in humans, the same is not true in mice.
Several hypotheses can explain the different biological behaviors. The most obvious is that the mouse is not human and the same phenomena could have different species-related outcomes. While a tempting explanation, it is not a testable hypothesis. One testable hypothesis would be that the spindle cell tumors of the mouse are a biological dead-end with terminal differentiation. The terminal differentiation is not metastatic. This hypothesis would suggest that tumors we see in mice have lost the plasticity required for EMT and its reciprocal, MET. Perhaps, if human EMT tumors could be encouraged to terminally differentiate, their populations would not metastasize.
What constitutes EMT Tumorigenesis? Does such a tumor type exist in human breast cancer [39, 40, 67]? The current status of the field leaves us with diagnostic and biological dilemmas. In the clinical arena, reports of EMT in human tumors are based largely on expression microarrays emphasizing the loss of expression of “epithelial” genes such as e-Cadherin and up-regulation of genes associated with mesenchymal differentiation (Snail, Twist and Slug). Images of the histopathology show disorganized epithelial tumors that do not have the spindle cell differentiation found in the mouse tumors [68] These observations raise the question of appropriate diagnostic criteria of “EMT” tumors. What are the limits of EMT? Does a range of tumors, such as found in the mouse, exist that fit one or more criteria for EMT? Or, should the diagnostic use of the term be limited only to tumors that have all of the criteria? Are we justified in categorizing breast tumors on the basis of their expression microarrays? A huge literature now exists that sorts human breast cancer using expression microarrays or immunohistochemistry to correlate certain expression patterns with clinical outcome [69, 70].
As in the past, the biology of neoplasia might be better understood from the perspective of the mouse. If the observation in GEM is correct and universal, EMT tumorigenesis involves the loss of expression of the initiating oncogene with concomitent silencing of the entire oncogenic pathway. What are the mechanisms that trigger this dramatic change in oncogenic stratigies? Is the phenomena epigenetic as implied by the MET (mesenchymal-epithelial-transition) implied by the morphology of the metastaic foci [39, 40]. We have postulated here that the spindle cell phenotype that is so characteristic of the mouse EMT tumors, is an end point in the neoplastic progression. If so, the intermediate stages need to be identified. Again the mechanistic implications of each stage need definition.
By any criteria, the emerging data correlating over expression of mesenchymal and EMT transcription markers with poor prognosis demands our attention. Our diagnostic and therapuetic approaches will be based on a better understanding of the origins and mechanisms of EMT tumorigenesis.
Acknowledgements
The research reported here was sponsored by grants from the Congressionally Directed Medical Program, Cancer Research Program Breast Cancer Center of Excellence grant #BC043200, the National Cancer Institute’s Mouse Models of Human Cancers Consortium grant #U01 CA105490-01 and the NIH NCRR Mutant Mouse Regional Resource Center grant #U42 RR14905. The author also appreciates the contributions of Drs. Patrizia Damonte and Enrico Radaelli whose research and publications serve as a framework for this essay.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- EMT
epithelial-mesenchymal-transition
- MET
mesenchymal-epithelial-transition
- K
cytokeratin
- CK
cytokeratin
- IHC
immunohistochemistry
- SMA
smooth muscle actin
- Erα
estrogen receptor alpha
- GEM
genetically engineered mice
References
- 1.Apolant H. Die epithelialen Geschwülste der Maus. Arbeiten ad Koniglchn Inst FExptTher zu Frankfurt aM. 1906;1:7–68. [Google Scholar]
- 2.Cardiff RD, Kenney N. Mouse mammary tumor biology: a short history. Adv Cancer Res. 2007;98:53–116. doi: 10.1016/S0065-230X(06)98003-8. [DOI] [PubMed] [Google Scholar]
- 3.Crisp E. Malignant tumor on the pectoral muscle of a mouse (M. musculus) Trans Pathol Soc Lond. 1854;5:348. [Google Scholar]
- 4.Livinggood LE. Tumors in the mouse. Johns Hopkins Hosp Bul. 1896;7:177–8. [Google Scholar]
- 5.Eberth CJ, Spude CJ. Familiare endotheliome. Arch f Path Anat. 1898;153:60. doi: 10.1007/BF01947720. [DOI] [Google Scholar]
- 6.Woglom WH. The study of experimental cancer research. A review. New York: Columbia University Press; 1913. [Google Scholar]
- 7.Morau H. Recherches esperimentalis sur la transmissibilite de certain acoplasmes (epithliomas cylindirques) Arch de Med Expl et d’ anat Path. 1894;6:677. [Google Scholar]
- 8.Haaland. Spontaneous tumours in mice. London; 1911.
- 9.Ehrlich P, Apolant H. Ueber spontane Mischtumoren der Maus. Berliner Klinische Wochenschrift. 1907;1907(44):1–3. [Google Scholar]
- 10.Satterthwaite TE. A manual of histology. 2. New York: Wood; 1882. [Google Scholar]
- 11.Fox CH, Johnson FB, Whiting J, Roller PP. Formaldehyde fixation. J Histochem Cytochem. 1985;33(8):845–53. doi: 10.1177/33.8.3894502. [DOI] [PubMed] [Google Scholar]
- 12.Mallory FB, Wright JH. Textbook of pathologic techniques. Philadelphia: Saunders; 1913. [Google Scholar]
- 13.Dunham EK. Histology; normal and morbid. New York: Lea; 1898. [Google Scholar]
- 14.Borrel A. Epithelioses infecticuses of epitheliomas. Ann de L’Inst Pasteur. 1903;17:81–118. [Google Scholar]
- 15.Borrel A. Parasitisme et tumours. Ann de L’Inst Pasteur. 1910;24:778. [Google Scholar]
- 16.Murray JA. Cancerous ancestry and the incidence of cancer in mice. London: Imperial Cancer Research Fund; 1911. [Google Scholar]
- 17.Jensen CO. Experimentelle Untersuchungen uber Krebs bei Mausen. Centbl F Bakt. 1903;34:28 and 122.
- 18.Williams WR. What is Jensen’s tumor? Lancet. 1906;1906:1285–6. [Google Scholar]
- 19.Bashford EF. Introduction. In: Fourth Scientific Report on the Investigations of The Imperial Cancer Research Fund. London: Taylor and Francis; 1911.
- 20.Ewing J. Neoplastic diseases: a text-book on tumors. Saunders; 1919.
- 21.Little CC, Tyzzer EE. Further experimental studies on the inheritance of susceptibility to a transplantable carcinoma (JA) of the Japanese waltzing mouse. J Med Res. 1916;33:393–427. [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn TB. Morphology of mammary tumors in mice with and without the agent. Acta Unio Int Contra Cancrum. 1951;7(2):234–7. [PubMed] [Google Scholar]
- 23.Dunn TB. Morphology and histogenesis of mammary tumors. In: Moulton FRe, editor. Symposium on mammary tumors in mice. Washington: Am Assoc Adv Sci; 1945:13–38.
- 24.Young LJ, Cardiff RD, Ashley RL. Long-term primary culture of mouse mammary tumor cells: production of virus. J Natl Cancer Inst. 1975;54(5):1215–21. doi: 10.1093/jnci/54.5.1215. [DOI] [PubMed] [Google Scholar]
- 25.Yagi MJ. Characteristics of mammary tumor cultures from four mouse strains infected with mammary tumor virus. Cancer Res. 1975;35(2):370–3. [PubMed] [Google Scholar]
- 26.Parks WP, Scolnick EM. Murine mammary tumor cell clones with varying degrees of virus expression. Virology. 1973;55(1):163–73. doi: 10.1016/S0042-6822(73)81018-9. [DOI] [PubMed] [Google Scholar]
- 27.Strange R, Aguilar-Cordova E, Young LJ, Billy HT, Dandekar S, Cardiff RD. Harvey-ras mediated neoplastic development in the mouse mammary gland. Oncogene. 1989;4(3):309–15. [PubMed] [Google Scholar]
- 28.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Thiery JP, Tucker GC, Boyer B, Valles AM, Gavrilovic J, Moens G, et al. Mechanisms of induced epithelium-fibroblast conversion in a line of vesical cancer in the rat. Pathol Biol (Paris) 1989;37(9):1034. [PubMed] [Google Scholar]
- 30.Boyer B, Tucker GC, Valles AM, Gavrilovic J, Thiery JP. Reversible transition towards a fibroblastic phenotype in a rat carcinoma cell line. Int J Cancer Suppl. 1989;4:69–75. doi: 10.1002/ijc.2910440719. [DOI] [PubMed] [Google Scholar]
- 31.Micalizzi DS, Ford HL. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009;5(8):1129–43. doi: 10.2217/fon.09.94. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed F, Wyckoff J, Lin EY, Wang W, Wang Y, Hennighausen L, et al. GFP expression in the mammary gland for imaging of mammary tumor cells in transgenic mice. Cancer Res. 2002;62(24):7166–9. [PubMed] [Google Scholar]
- 33.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. 2006;235(12):3222–9. doi: 10.1002/dvdy.20972. [DOI] [PubMed] [Google Scholar]
- 35.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64(19):7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 36.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 37.Karreth F, Tuveson DA. Twist induces an epithelial-mesenchymal transition to facilitate tumor metastasis. Cancer Biol Ther. 2004;3(11):1058–9. doi: 10.4161/cbt.3.11.1302. [DOI] [PubMed] [Google Scholar]
- 38.Kedrin D, van Rheenen J, Hernandez L, Condeelis J, Segall JE. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12(2–3):143–52. doi: 10.1007/s10911-007-9046-4. [DOI] [PubMed] [Google Scholar]
- 39.Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65(14):5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- 40.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65(14):5991–5. doi: 10.1158/0008-5472.CAN-05-0616. [DOI] [PubMed] [Google Scholar]
- 41.Cardiff RD. Epithelial to mesenchymal transition tumors: fallacious or snail’s pace? Clin Cancer Res. 2005;11(24 Pt 1):8534–7. doi: 10.1158/1078-0432.CCR-05-2250. [DOI] [PubMed] [Google Scholar]
- 42.Cardiff RD. Genomic pathology for genomic biology. Pathologica. 2008;100(1):3–5. [PMC free article] [PubMed] [Google Scholar]
- 43.Cardiff RD, Sinn E, Muller W, Leder P. Transgenic oncogene mice. Tumor phenotype predicts genotype. Am J Pathol. 1991;139(3):495–501. [PMC free article] [PubMed] [Google Scholar]
- 44.Rosner A, Miyoshi K, Landesman-Bollag E, Xu X, Seldin DC, Moser AR, et al. Pathway pathology: histological differences between ErbB/Ras and Wnt pathway transgenic mammary tumors. Am J Pathol. 2002;161(3):1087–97. doi: 10.1016/S0002-9440(10)64269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cardiff RD, Munn RJ, Galvez JJ. The tumor pathology of genetically engineered mice: a new approach to molecular pathology. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The mouse in biomedical research: experimental biology and oncology. 2. New York: Elsevier, Inc; 2006. pp. 581–622. [Google Scholar]
- 46.Cui XS, Donehower LA. Differential gene expression in mouse mammary adenocarcinomas in the presence and absence of wild type p53. Oncogene. 2000;19(52):5988–96. doi: 10.1038/sj.onc.1203993. [DOI] [PubMed] [Google Scholar]
- 47.White DE, Cardiff RD, Dedhar S, Muller WJ. Mammary epithelial-specific expression of the integrin-linked kinase (ILK) results in the induction of mammary gland hyperplasias and tumors in transgenic mice. Oncogene. 2001;20(48):7064–72. doi: 10.1038/sj.onc.1204910. [DOI] [PubMed] [Google Scholar]
- 48.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8(3):197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD, et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev. 2003;17(4):488–501. doi: 10.1101/gad.1051603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrechek ER, Cardiff RD, Chang JT, Gatza ML, Acharya CR, Potti A, et al. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc Natl Acad Sci U S A. 2009;106(38):16387–92. doi: 10.1073/pnas.0901250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Damonte P, Gregg JP, Borowsky AD, Keister BA, Cardiff RD. EMT tumorigenesis in the mouse mammary gland. Lab Invest. 2007;87(12):1218–26. doi: 10.1038/labinvest.3700683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Debies MT, Gestl SA, Mathers JL, Mikse OR, Leonard TL, Moody SE, et al. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16 Ink4a loss. J Clin Invest. 2008;118(1):51–63. doi: 10.1172/JCI33320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jessen KA, Liu SY, Tepper CG, Karrim J, McGoldrick ET, Rosner A, et al. Molecular analysis of metastasis in a polyomavirus middle T mouse model: the role of osteopontin. Breast Cancer Res. 2004;6(3):R157–69. doi: 10.1186/bcr768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene. 2001;20(25):3247–57. doi: 10.1038/sj.onc.1204411. [DOI] [PubMed] [Google Scholar]
- 55.Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D, Andrulis IL, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106(31):12903–8. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radaelli E, Arnold A, Papanikolaou A, Garcia-Fernandez RA, Mattiello S, Scanziani E, et al. Mammary tumor phenotypes in wild-type aging female FVB/N mice with pituitary prolactinomas. Vet Pathol. 2009;46(4):736–45. doi: 10.1354/vp.08-VP-0280-R-FL. [DOI] [PubMed] [Google Scholar]
- 57.Radaelli E, Damonte P, Cardiff RD. Epithelial-mesenchymal transition in mouse mammary tumorigenesis. Future Oncol. 2009;5(8):1113–27. doi: 10.2217/fon.09.93. [DOI] [PubMed] [Google Scholar]
- 58.McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, et al. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009;119(9):2663–77. doi: 10.1172/JCI37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit? Cancer Res. 2008;68(9):3081–6. doi: 10.1158/0008-5472.CAN-07-5832. [DOI] [PubMed] [Google Scholar]
- 60.Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A. 2006;103(44):16266–71. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, et al. Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Cripto-1 transgenic mice. J Cell Physiol. 2004;201(2):266–76. doi: 10.1002/jcp.20062. [DOI] [PubMed] [Google Scholar]
- 62.Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, et al. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22(1):47–59. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- 63.Cardiff RD, Leder A, Kuo A, Pattengale PK, Leder P. Multiple tumor types appear in a transgenic mouse with the ras oncogene. Am J Pathol. 1993;142(4):1199–207. [PMC free article] [PubMed] [Google Scholar]
- 64.Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21(20):3241–6. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- 65.Oshima RG, Lesperance J, Munoz V, Hebbard L, Ranscht B, Sharan N, et al. Angiogenic acceleration of neu induced mammary tumor progression and metastasis. Cancer Res. 2004;64(1):169–79. doi: 10.1158/0008-5472.CAN-03-1944. [DOI] [PubMed] [Google Scholar]
- 66.Sugino T, Kusakabe T, Hoshi N, Yamaguchi T, Kawaguchi T, Goodison S, et al. An invasion-independent pathway of blood-borne metastasis: a new murine mammary tumor model. Am J Pathol. 2002;160(6):1973–80. doi: 10.1016/S0002-9440(10)61147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66(17):8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 68.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11(24 Pt 1):8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 69.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 70.Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5(6):607–16. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]