

Abstract
Hamartomatous polyposis syndromes are a rare group of hereditary autosomal dominant disorders that comprise less than 1% of all hereditary colorectal cancers. Hamartomatous polyps, in and of themselves, are benign entities; however, these hamartomatous polyposis syndromes have a malignant potential for the development of colorectal cancer as well as extracolonic cancers. Early detection and proper surveillance are vital to minimizing the risk of carcinoma. This article provides a critical review of the clinical presentation, pathology, genetics, and screening and surveillance guidelines of juvenile polyposis syndrome, PTEN hamartoma tumor syndrome, and Peutz-Jeghers syndrome.
Keywords: Hamartomatous polyposis syndromes, juvenile polyposis syndrome, PTEN hamartoma tumor syndrome, Peutz-Jeghers syndrome
Hamartomatous polyposis syndromes are a rare group of hereditary autosomal dominant disorders that comprise less than 1% of all hereditary colorectal cancers.1-3 Hamartomatous polyps, in and of themselves, are benign entities comprised of cells that are indigenous to the area in which they are found (ie, all cell layers with a mesenchymal predominance). However, these hamartomatous polyposis syndromes have a malignant potential for the development of colorectal cancer as well as extracolonic cancers. The progression of hamartomatous polyps to carcinoma is still being elucidated. Unlike adenomatous polyps, in which malignant transformation progresses through the adenomacarcinoma sequence via a gatekeeper or caretaker defect, in hamartomatous polyps, a proposed hamartoma-carcinoma sequence hypothesis involves a landscaper defect in which stromal elements create a local environment that promotes epithelial dysplasia and ultimately leads to carcinoma.4
The hamartomatous polyposis syndromes include juvenile polyposis syndrome (JPS); PTEN hamartoma tumor syndrome, which includes Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome (BRRS); and Peutz-Jeghers syndrome (PJS). Due to the rarity of these conditions, a thorough understanding of their clinical presentation, including extraintestinal manifestations (gross and histopathologic), and genetics is important. For pediatric gastroenterologists, understanding how to recognize and establish the appropriate diagnosis and cancer risk and following appropriate screening and surveillance guidelines is crucial for early detection to minimize the risk of carcinoma as children reach adulthood.
Juvenile Polyposis Syndrome
Clinical Presentation
Juvenile polyps are the most common type of pediatric gastrointestinal polyps. Solitary juvenile polyps can develop at any age, though they appear most frequently in preschool children and have an incidence of 2% in children under 10 years of age. Solitary polyps are generally located in the rectosigmoid area and are usually considered to be a separate entity from JPS, which has an incidence of 1 in 100,000–160,000 individuals.5,6 A family history of juvenile polyps is found in 20–50% of patients with JPS, with an autosomal dominant inheritance pattern of variable penetrance.1,7-10
In JPS, affected individuals develop multiple gastrointestinal juvenile polyps, predominantly in the colon, though the condition may also affect the rest of the gastrointestinal tract.11-13 JPS has been phenotypically classified into 3 categories14: juvenile polyposis coli, in which polyp growth affects only the colon; the rare and often fatal form of JPS called juvenile polyposis of infancy, which is characterized by diarrhea, protein-losing enteropathy, bleeding, and rectal prolapse15; and generalized juvenile polyposis, in which polyp growth can affect the colon, stomach, and small bowel. As opposed to solitary juvenile polyps, which occur in children most commonly at 4–5 years of age, JPS presents in the first or second decade of life, with an average age of 18.5 years at the time of diagnosis.12,16 Typical presenting symptoms include rectal bleeding, anemia, abdominal pain, diarrhea, intussusception, obstruction, and polyp prolapse, though many JPS patients may be asymptomatic.8,17,18
Extraintestinal Manifestations
Multiple extraintestinal manifestations have been reported with JPS, including heart defects, polydactyl, clubbing, intestinal malrotation, Meckel diverticulum, hydrocephalus, macrocephaly, hypertelorism, cleft lip, cleft palate, double renal pelvis and ureter, bifid uterus and vagina, undescended testes, and supernumery teeth.13,16,19 These abnormalities have been reported in approximately 11–20% of cases,8,19,20 mainly in case reports. However, due to the overlap of JPS, CS, and BRRS, the true incidence of extraintestinal manifestations based upon these case reports is difficult to interpret. JPS and hereditary hemorrhagic telangiectasia (HHT) have also been reported together.21 Further credence to this association has been made with the recent report of 2 JPS patients having a germline mutation in the ENG gene, which is the causative gene in HHT.22
Diagnosis
The diagnosis of JPS is clinically established based upon the presence of at least 1 of the following criteria18,23,24: at least 3–10 polyps detected on colonoscopy; polyps located outside of the colon; and any number of polyps in a patient with a family history of juvenile polyps. The number of polyps needed to establish the diagnosis of JPS varies in the literature. Sachatello and associates proposed 10 polyps as the benchmark; however, this number was reduced to 5 polyps by Jass and colleagues and then to 3 polyps by Giardiello and coworkers.18,23,24 There is currently no clear consensus on the number of polyps to use for diagnosis,hence the range of 3–10 listed above.
Gastrointestinal Pathology
The gross appearance of a juvenile polyp is spherical to slightly lobular in shape, and most are pedunculated with long stalks.25 In patients with JPS, polyps may have a multilobulated appearance of a villiform or papillary shape.8 Jass and colleagues reported that approximately 20% of polyps have the latter appearance.18 Polyp size can range from several millimeters to 3 cm (Figure 1A). These polyps are typically very vascular, with a smooth and glistening appearance on the surface; however, they may also have an ulcerated surface from auto-infarction.
Figure 1.

Resection of a colon from a patient with juvenile polyposis syndrome containing multiple juvenile polyps (A).
Reproduced from Demetris AJ, Finkelstein SD, Nalensnik MA, et al. Slide carousel of GI pathology course for medical students. Available at: http://itchforum.net/content/index_eng.html. © Department of Pathology, University of Pittsburgh School of Medicine.
Histologic image of a juvenile polyp showing its characteristic large cystic spaces and a lamina propria with an inflammatory cell component (B).
Reproduced from Mulholland M, Lillemoe K, Doherty G, Maier R, Upchurch G. Greenfield's Surgery: Scientific Principles and Practice. 4th ed. Philadelphia, Pennsylvania: Lippincott Williams & Wilkins; 2006.
The histologic appearance of juvenile polyps shows multiple large mucus-filled glands lined with columnar epithelium (Figure 1B). The lamina propria usually has an inflammatory cell component.18 Smooth muscle components are not present in juvenile polyps, which distinguishes them from PJS polyps, but not from polyps of the other hamartomatous polyposis syndromes.26,27
Genetics
A family history of JPS is found in 20–50% of patients with JPS, with an autosomal dominant inheritance pattern of variable penetrance.17,18 Three genes have been associated with JPS: SMAD4, BMPR1A, and ENG, all of which are part of the transforming growth factor-b(TGF-b) superfamily of proteins.27 The PTEN gene mutation in patients with juvenile polyposis is a controversial topic. It is generally thought that patients with the PTEN gene mutation likely represent CS or BRRS patients who have not yet expressed the extraintestinal clinical features of these conditions.27-29
The SMAD4 gene, located on chromosome 18q21.1, was first identified by Howe using gene linkage analysis on affected families with JPS.30,31 Germline mutations in the SMAD4 gene have a prevalence of 20% in JPS patients.32 Patients with the SMAD4 mutation are more likely to have upper gastrointestinal polyps. A subset of JPS patients also has HHT, which has been linked to patients with SMAD4 mutations. Multiple types of mutations have been reported in the SMAD4 gene, including missense, nonsense, deletions, and insertions; however, the most common mutation is the one originally discovered by Howe (the 4-base pair deletion in exon 9).33 SMAD4 is an intracellular mediator in the TGF-βsignal-ing pathway. It binds to other members of the SMAD family and is involved in transcriptional activation and nuclear localization.26,34,35 In JPS, SMAD4 is thought to act as a tumor suppressor gene, and the inactivation of SMAD4 in the unaffected allele is an important step in polyp development.36 These data are supported by the SMAD4 knockout mouse model.37-39
The bone morphogenetic protein receptor type IA (BMPR1A) gene is located on chromosome 10q22-23 and was reported by Howe and associates in 2001.40-42 Germline mutations in the BMPR1A gene have a prevalence of 20% in JPS patients.32 BMPR1A is a type 1 serine/threonine kinase receptor protein that is bound to a type II serine/threonine kinase receptor protein.34 This receptor complex is also involved in the TGF-βsignaling pathway upstream of SMAD4. It phosphorylates SMAD proteins that then bind to SMAD4.43,44 BMPR1A knockout mice are homozygous lethal, whereas heterozygous mice appear normal with no polyp development.45
ENG has recently been shown to be present in 2 patients with JPS; however, its role as a predisposition gene still requires additional confirmation.22,46 The ENG gene is located on chromosome 9q34.1.47 ENG encodes the protein endoglin, which is an accessory protein of the TGF-bsignaling pathway.48 Endoglin signaling is initiated by the binding of TGF-bin combination with TBR-II and results in a series of activation steps leading to transcriptional activity. Endoglin inhibition appears to have an inhibitory effect on TGF-bin endothelial cells.48,49 ENG knockout mice are homozygous lethal, and heterozygous mice exhibit large numbers of irregular, dilated, and thinner-walled vessels and serve as a model for HHT. There is no reported polyp growth in this mouse model.50
Cancer Risk
Individuals with JPS are at risk for the development of colorectal, gastric, small intestinal, and pancreatic cancers. The risk of developing colorectal cancer from solitary juvenile polyps is thought to be negligible or nonexistent. However, individuals with JPS are at risk for developing adenomatous change and carcinoma. The incidence of colorectal cancer has been reported by Jass and associates to be 20.7%, with a mean age of 34 years (age range, 15–59 years) and an estimated cumulative colorectal cancer risk of 68% by 60 years of age.18 Coburn and colleagues reported a colorectal cancer risk of 17%, with a mean age of 35 years (age range, 15–59 years) at diagnosis.17 Howe and coworkers reported a colorectal cancer risk of 38% and a cumulative risk of gastrointestinal cancer of 55%, with a mean age of 43 years (age range, 17–68 years).51 There have been multiple case reports documenting gastric adenocarcinomas and pancreatic adenocarcinomas.52-54 In an Iowa kindred, Howe and colleagues reported 4 gastric carcinomas, 1 duodenal carcinoma, and 1 pancreatic carcinoma, with an overall risk of upper gastrointestinal cancer of 21% in patients with JPS.51 In a review of JPS patients, Coburn and associates reported 1 gastric carcinoma and 1 duodenal carcinoma.17
Screening and Management
There are no standardized screening and surveillance guidelines for the management of JPS. Two proposed guidelines have been published, one by Howe and coworkers and another by Dunlop.55,56 Howe and coworkers suggested that genetic testing be performed in patients at risk for JPS. According to their guidelines, a complete blood count (CBC), upper endoscopy, and colonoscopy should be performed for JPS in all at-risk patients when symptoms present or in asymptomatic patients by 15 years of age. If no polyps are found, a repeat colonoscopy should be performed every 3 years. If a genetic mutation is present, screening with upper endoscopy, colonoscopy, and CBC should also be performed every 3 years; however, if no genetic mutation is present and no polyps are found at the initial endoscopy, a repeat endoscopy should be performed every 10 years until 45 years of age. If no polyps are found by 45 years of age, standard colorectal cancer screening should be used (Figure 2).55 Wirtzfeld and colleagues critiqued the every-10-year screening intervals for at-risk JPS patients without genetic mutations by claiming that, due to the genetic heterogeneity of JPS, a 10-year interval may be too long of a period of time to go without screening.3
Figure 2.

Proposed algorithm for endoscopic surveillance of juvenile polyposis syndrome.
Dunlop's proposed guidelines for JPS screening starts at 15–18 years of age for colonoscopy in asymptomatic atrisk patients and at 25 years of age for upper endoscopy. If a genetic mutation is found and no polyps are detected at the time of the initial endoscopy, screening should continue until 70 years of age; however, if no genetic mutation is found and no polyps are detected at the initial endoscopy, repeat endoscopy should be performed every 1–2 years until 35 years of age.56
Both sets of authors agree that any found polyps should be removed endoscopically. Endoscopy should then be repeated yearly until no polyps are detected, at which point screening should return to every 3 years, according to Howe and coworkers, or every 1–2 years, according to Dunlop.
The indications for surgery include a large polyp burden that cannot be managed endoscopically, polyps with adenomatous change, severe diarrhea with hypoprotenemia, or gastrointestinal bleeding with anemia. There are no standards for the type of surgery. Surgical options include subtotal colectomy with ileorectal anastomosis and total colectomy with J-pouch ileoanal anastomo-57,58 Regardless of the surgery performed, endoscopic surveillance is recommended after surgery for polyp recurrence in the pouch.57,58
Gastric polyps should be removed endoscopically; however, this guideline may be challenging due to the number of polyps found. Indications for total or subtotal gastrectomy include severe anemia due to chronic bleeding,adenomatous change,or adenocarcinoma.26
PTEN Hamartoma Tumor Syndrome
PTEN hamartoma tumor syndrome is the term that has been used recently to describe both CS and BRRS.59 These disorders are both caused by mutations in the PTEN gene and are both characterized by extraintestinal manifestations more than intestinal polyposis. The clinical presentations for the diseases will be discussed separately in this paper, though it should be noted that the differences between them are likely due to the variable phenotypic expression seen in PTEN gene mutations.59-62
Cowden Syndrome
Clinical Presentation CS is a rare autosomal dominant syndrome, with a reported incidence of 1 in 200,000 individuals.63 This syndrome is characterized by macrocephaly, mucocutaneous lesions (such as facial trichilemmoma), acral keratosis, and papillomatous papules (Figure 3). It is also associated with thyroid, breast, and endometrial manifestations, including cancer in all of these areas.26,27,64 CS has been linked to Lhermitte-Duclos disease, which is characterized by hamartomas of the cerebellum.65 Hamartomatous polyps throughout the gastrointestinal tract are associated with this syndrome but are not as common as the extraintestinal findings associated with the syndrome. The incidence of gastrointestinal polyps in CS varies in the literature, ranging anywhere from 30% 64,66,67 It is generally thought that the incidence of gastrointestinal polyps in CS is less than that of BRRS, though this belief is debated in the literature.66 Another gastrointestinal manifestation of CS is glycogenic acanthosis of the esophagus (Figure 4), which involves large benign glycogen-filled epithelial cells that are gray to white in color.68
Figure 4.
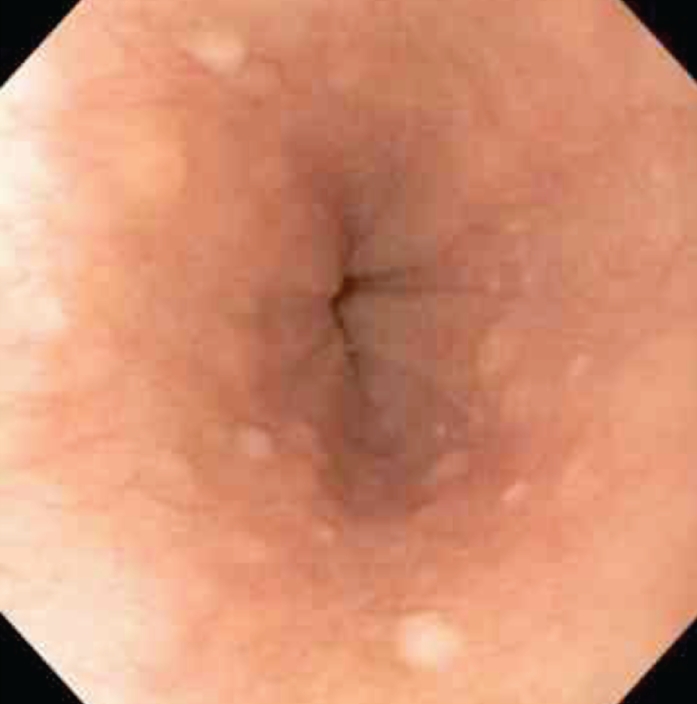
Glycogenic acanthosis Table 1. Extraintestinal Manifestations of Cowden Syndrome of the esophagus in a patient With Their Diagnostic Criteria with Cowden syndrome.
Figure 3.

Multiple skin-colored facial warty papules representing tricholemmonas (A). Multiple reddish confluent papules on the oral mucosa revealing a cobblestone appearance (B).
Both figures are reproduced from Wolff K, Johnson RA, Fitzpatrick TB. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, New York: McGraw-Hill Medical; 2009.
Extraintestinal Manifestations As stated above, extraintestinal manifestations are the hallmark of the syndrome and summarized in Table 1.
Table 1.
Extraintestinal Manifestations of Cowden Syndrome With Their Diagnostic Criteria
| Manifestation | Type of criteria |
|---|---|
| Facial tirchilemmomas | Pathognomonic |
| Acral keratoses | Pathognomonic |
| Papillomatous papules | Pathognomonic |
| Lhermitte-Duclos disease | Pathognomonic |
| Breast adenocarcinoma | Major |
| Fibrocystic breast disease | Minor |
| Thyroid multinodular goiter | Minor |
| Thyroid adenoma | Minor |
| Thyroid carcinoma | Major |
| Uterine leiomyomas | Minor |
| Endometrial carcinoma | Major |
| Bicornutae uterus | Minor |
| Macrocephaly | Major |
| Developmental delay | Minor |
| Lipomas | Minor |
| Fibromas | Minor |
| Melanoma | Minor |
Diagnosis The diagnosis of CS is based upon pathognomonic, major, and minor criteria. These criteria are updated by the National Comprehensive Cancer Network (NCCN) and can be accessed via the Web site www.nccn.org.27 A working diagnosis can be established by fulfilling 1 pathognomonic criterion; 2 major criteria; 1 major and 3 or more minor criteria; or 4 or more minor criteria. These criteria are listed in Table 1.
Bannayan-Riley-Ruvalcaba Syndrome
Clinical Presentation The term BRRS was first used by Gorlin and coworkers to describe 3 clinically similar autosomal dominant syndromes: Riley-Smith syndrome, Ban-nayan-Zonana syndrome, and Ruvalcaba-Myhre-Smith syndrome.69 BRRS is characterized by macrocephaly; developmental delays; pigmented speckling of the penis; lipomas; and hamartomatous polyps of the intestine.69,70 The incidence of gastrointestinal polyps in BRRS has been reported to be 45%.69 Extraintestinal Manifestations As with CS, the extraintestinal manifestations of this syndrome are its hallmark and are summarized in Table 2.
Table 2.
Extraintestinal Manifestations of Bannayan-Riley-Ruvalcaba Syndrome
|
Diagnosis Due to the rarity of this syndrome, there are no formal diagnostic criteria for BRRS. The diagnosis of BRRS should be considered when a patient exhibits 1 or more extraintestinal manifestations with or without polyps, or has a family history of BRRS or CS.
Cowden Syndrome and Bannayan-Riley-Ruvalcaba Syndrome Gastrointestinal Pathology The appearance of polyps within the gastrointestinal tract in both CS and BRRS resembles the gross and histologic appearance of juvenile polyps.69 Therefore, when juvenile polyps are detected, physicians should evaluate the patient for possible extraintestinal manifestations of CS and BRRS.
Cowden Syndrome and Bannayan-Riley-Ruvalcaba Syndrome Genetics CS and BRRS have an autosomal dominant inheritance pattern with variable penetrance. Both syndromes have been associated with the PTEN gene, which is located on chromosome 10q22-23. CS was linked to chromosome 10q22-23 by Nelen and associates, and the PTEN gene was later identified at this locus by Liaw and colleagues, and confirmed by Nelen and coworkers, as well as Lynch and associates.63,71-73 Marsh and colleagues then found an association between PTEN and BRRS.74 The PTEN gene is found in 80% of CS patients and 60% of BRRS patients.62,75 CS and BRRS are allelic diseases in which a mutation of the PTEN gene is found in all exons except 1, 4, and 9 in CS. In BRRS, mutations occur preferentially in exons 6 and 7 and are also associated with balanced translocations and 62,63,71,72,74 It has been hypothesized that the differential expression of the PTEN gene correlates with the different phenotypes seen in CS and BRRS.76
The PTEN gene is a tumor suppressor gene that is also a tyrosine phosphatase that dephosphorylates tyrosine, serine, and threonine.77 PTEN is a negative regulator of the Akt/PKB signaling pathway,77,78 which controls the levels of phosphoinositol triphosphate. PTEN is also involved in regulating cell cycle, apoptosis, and angiogenesis.78,79
Cancer Risk Individuals with CS are at risk for developing breast, thyroid, and endometrial cancers. The risk of adenocarcinoma of the breast has been reported to range from 30% to 50% in women with CS.27,64,66 In addition, there are reports of breast cancer in men with CS.80 Individuals with CS are also subject to benign conditions of the breast such as fibrocystic disease.64 Thyroid abnormalities such as multinodular goiter and thyroglossal duct cysts are associated with this syndrome, as well as a 10% risk of thyroid cancer. CS patients also have a risk of leiomyomas, as well as an up-to-10% risk of endometrial cancer.64,81 Renal cell cancer has also been associated with CS.64 The risk of developing gastrointestinal carcinoma in CS is unclear at this point. It has been reported by some studies that there is no increased risk of gastrointestinal cancer; however, there are multiple case reports of gastric and colorectal cancer.64,82-84
In BRRS, the cancer risk is unclear. The limited number of patients with this disease makes it difficult to determine the risk; however, there have been case reports of breast and endometrial cancer.62,85 With additional evidence supporting the idea that CS and BRRS are variable phenotypic expressions in the PTEN gene, it is therefore recommended that individuals with BRRS be considered at risk for malignancy, as with CS.
Screening and Management Individuals with CS and BRRS who have mutations in the PTEN gene should be screened according to the CS guidelines summarized in Table 3 1,5,59,68,86 These guidelines include breast cancer screening with self and clinical breast examinations, in addition to annual mammography and breast magnetic resonance imaging for breast cancer beginning at 25 years of age or 5–10 years earlier than the youngest case in the family. Thyroid cancer screening involves clinical examinations and yearly thyroid ultrasounds starting at 18 years of age. Endometrial cancer screening with endometrial biopsies should be performed at 35–40 years of age or 5 years earlier than the youngest case in the family. Renal cell cancer screening can be performed with yearly urinalysis and ultrasound.1,5,68,86 Updated guidelines for CS screening can be found on the NCCN Web site.59
Table 3.
Cowden and Bannayan-Riley-Ruvalcaba Syndrome Management
|
Peutz-Jeghers Syndrome
Clinical Presentation
PJS, as with the other hamartomatous syndromes, is an autosomal dominant syndrome that is typified by its characteristic mucocutaneous pigmentation and intestinal hamartomatous polyps. The incidence of PJS is reported to be 1 in 150,000 to 200,000 individuals.86,87 Pigmentation is seen around the vermilion border of the lips in over 95% of cases, with the buccal mucosa being the second most common site (80%; Figure 5).88,89 Other areas of pigmentation include the hands, feet, genitals, and around the nose and eyes. Pigmentation typically presents in early childhood and starts to fade with age usually after the start of puberty.90,91
Figure 5.

Multiple dark-brown lentigines on the vermillion border of the lip and buccal mucosa in a patient with Peutz-Jeghers syndrome.
Reproduced from Swartz MH. Textbook of Physical Diagnosis: History and Examination. 4th ed. Philadelphia, Pennsylvania: WB Saunders; 2002:295.
Hamartomatous polyps in PJS are commonly found in the small intestine; however, they are also found in the stomach and colon. The number of polyps in the intestine may range from 1 to a complete carpeting of the gastrointestinal tract (Figure 6).89,92 The most common presentation of PJS is abdominal pain secondary to intussusception. Other clinical presentations include anemia, melena, hematochezia, hematemesis, and obstruction. Approximately one third of PJS patients present in the first decade of life, with up to 60% presenting by the second or third decade.90,93,94
Figure 6.

Polyps from a patient with Peutz-Jeghers syndrome are pedunculated and tend to be large and multilobulated.
Reproduced from Mulholland M, Lillemoe K, Doherty G, Maier R, Upchurch G. Greenfield's Surgery: Scientific Principles and Practice. 4th ed. Philadelphia, Pennsylvania: Lippincott Williams & Wilkins; 2006.
Extraintestinal Manifestations
In PJS, polyps have been reported outside of the gastrointestinal tract, including areas involving the nose, bronchi, renal pelvis, and bladder.95 Polyps have also been reported in the gallbladder and bile ducts; however, polyps in the gallbladder have been histologically adenomatous and not hamartomatous.96 Bile duct polyps have presented with jaundice secondary to obstructive symptoms.97,98 PJS is also associated with various malignancies that will be described below.
Diagnosis
The diagnosis of PJS is clinically established on the presence of histologic tissue that is consistent with hamartomatous polyps and 2 of the following criteria99: a family history of PJS; the presence of mucocutaneous pigmentation;and the presence of small-bowel polyps.
Pathology
The gross appearance of a polyp in PJS is typically pedunculated with long stalks. These polyps tend to be multilobulated and have a villiform or papillary shape. Polyp size can range from 0.5 cm to 5 cm in diameter. The polyp has a characteristic histologic appearance consisting of smooth muscle proliferation in a tree branch, a process that is called arborization (Figure 7).100 The epithelium that covers the smooth muscle can become displaced into the submucosa and muscularis propria, giving a pseudocarcinoma appearance of a mucinousa denocarcinoma.101
Figure 7.

Histologic image of a Peutz-Jeghers po lyp showing its characteristic histologic appearance, which consists of smooth (a process that is called arborization) (A). Enlargement of a Peutz-Jeghers polyp showing smooth muscle proliferation (B).
Both images reproduced from Boland CR. The colon, rectum, and anus. In: Feldman M, ed. Gastroenterology and Hepatology: The Comprehensive Visual Reference. Philadelphia, Pennsylvania: Churchill Livingstone; 1996.
Genetics
PJS, as with the other hamartomatous syndromes, has an autosomal dominant pattern of inheritance with both familial and sporadic transmission.102 The gene associated with PJS is a serine-threonine kinase that is located on chromosome 19p13.3.103,104 Hemminki and coworkers and Jenne and associates independently identified the gene in this region as LKB1/STK11.105,106 This gene has been reported in 80% of patients with PJS.107 Common mutations are frameshift and nonsense mutations in exons 1–6; however, large deletion mutations missed by direct sequencing have been recently described using multiple ligation probes.107
LKB1/STK11 is a tumor suppressor gene that encodes a serine-threonine kinase that phosphorylates and activates members of the AMPK-related subfamily of protein kinases.108 LKB1/STK11 has an essential role in G1 cell cycle arrest, cell polarity, p53-dependent apoptosis, and cellular energy levels.109,110 LKB1 (+/-) mice develop gastrointestinal polyps with histologic characteristics resembling those of human PJS polyps.111
Cancer Risk
Individuals with PJS are at risk for the development of colorectal, gastric, small intestinal, esophageal, and pancreatic cancers. PJS patients are also at risk for extraintestinal cancer such as lung, breast, ovarian, testicular, and endometrial cancers.26,27,90 A meta-analysis showed that the risk of developing any type of cancer by 64 years of age was 93% (relative risk of 15).112 Lim and colleagues evaluated the St. Mark's polyp registry and found that the relative risk of developing any type of cancer was 47% by 65 years of age in PJS patients with known genetic mutations in LKB1/STK11.113 More recently, a study looking at 419 PJS patients, 297 of whom had documented mutations, showed the risk of cancer to be 60% by 60 years of age and 85% by 70 years of age.114 This same study reported the risks of developing gastrointestinal cancer (31%), breast cancer (31%), gynecologic cancer (18%), pancreatic cancer (7%), and lung cancer (13%) by 60 years of age.114 Individuals with PJS are also at risk for developing rare sex cord tumors. Women are at risk for sex cord tumors with annular tubules that are benign, and men are at risk for developing Sertoli cell tumors, which result in feminization. Both of these tumors arise from the same embryonic tissue.115,116
Screening and Management
Individuals at risk for PJS should be evaluated at birth for pigmented spots, precocious puberty, and testicular tumors. Asymptomatic at-risk individuals should undergo genetic testing for the affected member's mutation in the STK11/LKB1 gene at 8 years of age.90 If the at-risk family member does not have a genetic mutation, they are not thought to have PJS. However, genetic testing is only useful if the family member with PJS has a known STK11/LKB1 mutation. If the family member with PJS has a mutation that is unidentifiable, endoscopic surveillance, as apposed to genetic testing, should be performed in at-risk family members. These asymptomatic at-risk individuals should, at the bare minimum, undergo small-bowel contrast studies every 2 years until 25 years of age. Alternatively, this group of at-risk patients should undergo upper endoscopy, colonoscopy, and small-bowel contrast study every 6 years starting at 12 years of age until 24 years of age. Any at-risk patients with pigmentation should undergo the PJS surveillance guidelines, as outlined below.
The PJS surveillance guidelines proposed by Giardiello and Trimbath90 (Table 4) include upper endoscopy and small-bowel contrast study starting at 8 years of age every 2–3 years. Starting at 18 years of age, colonoscopy should be performed every 2–3 years in coordination with upper endoscopy and small-bowel contrast study. Pancreatic screening should begin at 25–30 years of age, with endoscopic ultrasound every 1–2 years, with possible computed tomography scan and CA 19-9 cancer marker. For women, breast cancer screening with self and clinical breast examinations should begin at 18 years of age. Annual mammography and/or breast magnetic resonance imaging should begin at 25 years of age or 5–10 years earlier than the youngest case in the family. Other genitourinary cancers should be evaluated starting at 21 years of age, with yearly transvaginal ultrasound and CA-125 cancer markers, as well as yearly pelvic examinations and pap smears. Men should undergo yearly testicular examinations starting at birth and testicular ultrasounds every 2 years up until 12 years of age.27,93 Capsule endoscopy may replace small-bowel contrast studies in the future.
Table 4.
Peutz-Jeghers Syndrome Surveillance Recommendations
| Cancer screening by organ | Disease | Diagnostic testing | Screening interval | Age interval to be screened |
|---|---|---|---|---|
| Testes | Sertoli tumor |
|
|
Birth to age 12 |
| Ovary | Ovarian carcinoma |
|
|
Start at age 25 |
| Cervix and uterus | Cervical and uterine carcinoma |
|
1 year | Start at age 21 |
| Breast | Breast carcinoma |
|
|
|
| Pancreas | Pancreatic carcinoma |
|
|
|
| Gastrointestinal tract |
|
|
|
|
Polyps found in the stomach and colon should be removed at the time of surveillance endoscopy. Polyps detected in the small bowel that are 1–1.5 cm or larger in size should also be removed.90,117,118 In the past, removal was performed by push enteroscopy or laparotomy by either polypectomy or, in the case of very large polyps, bowel resection. However, advances in enteroscopy with the use of double- and single-balloon technology, and now spiral enteroscopy, allow for more of the small bowel to be reached without requiring laparotomy.
References
- 1.Attard TM, Young RJ. Diagnosis and management of gastrointestinal polyps: pediatric considerations. Gastroenterol Nurs. 2006;29:16–22. doi: 10.1097/00001610-200601000-00003. quiz 3-4. [DOI] [PubMed] [Google Scholar]
- 2.Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- 3.Wirtzfeld DA, Petrelli NJ, Rodriguez-Bigas MA. Hamartomatous polyposis syndromes: molecular genetics, neoplastic risk, and surveillance recommendations. Ann Surg Oncol. 2001;8:319–327. doi: 10.1007/s10434-001-0319-7. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 5.Burt RW, Bishop DT, Lynch HT, Rozen P, Winawer SJ. Risk and surveillance of individuals with heritable factors for colorectal cancer. WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ. 1990;68:655–665. [PMC free article] [PubMed] [Google Scholar]
- 6.Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20:1634–1640. doi: 10.1111/j.1440-1746.2005.03865.x. [DOI] [PubMed] [Google Scholar]
- 7.Alhan E, Unuvar E, Gumustekin E. Juvenile polyps of the colon and rectum. Turk J Pediatr. 1988;30:99–103. [PubMed] [Google Scholar]
- 8.Desai DC, Neale KF, Talbot IC, Hodgson SV, Phillips RK. Juvenile polyposis. Br J Surg. 1995;82:14–17. doi: 10.1002/bjs.1800820106. [DOI] [PubMed] [Google Scholar]
- 9.Pillai RB, Tolia V. Colonic polyps in children: frequently multiple and recurrent. Clin Pediatr (Phila) 1998;37:253–257. doi: 10.1177/000992289803700406. [DOI] [PubMed] [Google Scholar]
- 10.Reed K, Vose PC. Diffuse juvenile polyposis of the colon: a premalignant condition? Dis Colon Rectum. 1981;24:205–210. doi: 10.1007/BF02962337. [DOI] [PubMed] [Google Scholar]
- 11.Sachatello CR, Pickren JW, Grace JT., Jr. Generalized juvenile gastrointestinal polyposis. A hereditary syndrome. Gastroenterology. 1970;58:699–708. [PubMed] [Google Scholar]
- 12.Coffin CM, Dehner LP. What is a juvenile polyp? An analysis based on 21 patients with solitary and multiple polyps. Arch Pathol Lab Med. 1996;120:1032–1038. [PubMed] [Google Scholar]
- 13.McColl I, Busxey HJ, Veale AM, Morson BC. Juvenile polyposis coli. Proc R Soc Med. 1964;57:896–897. [PubMed] [Google Scholar]
- 14.Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75:107–114. [PubMed] [Google Scholar]
- 15.Ruymann FB. Juvenile polyps with cachexia. Report of an infant and comparison with Cronkhite-Canada syndrome in adults. Gastroenterology. 1969;57:431–438. [PubMed] [Google Scholar]
- 16.Veale AM, McColl I, Bussey HJ, Morson BC. Juvenile polyposis coli. J Med Genet. 1966;3:5–16. doi: 10.1136/jmg.3.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coburn MC, Pricolo VE, DeLuca FG, Bland KI. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol. 1995;2:386–391. doi: 10.1007/BF02306370. [DOI] [PubMed] [Google Scholar]
- 18.Jass JR, Williams CB, Bussey HJ, Morson BC. Juvenile polyposis—a precancerous condition. Histopathology. 1988;13:619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 19.Desai DC, Murday V, Phillips RK, Neale KF, Milla P, Hodgson SV. A survey of phenotypic features in juvenile polyposis. J Med Genet. 1998;35:476–481. doi: 10.1136/jmg.35.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bussey HJ, Veale AM, Morson BC. Genetics of gastrointestinal polyposis. Gastroenterology. 1978;74:1325–1330. [PubMed] [Google Scholar]
- 21.Inoue S, Matsumoto T, Iida M, Hoshika K, Shimizu M et al. Juvenile polyposis occurring in hereditary hemorrhagic telangiectasia. Am J Med Sci. 1999;317:59–62. doi: 10.1097/00000441-199901000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Sweet K, Willis J, Zhou XP, Gallione C, Sawada T et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294:2465–2473. doi: 10.1001/jama.294.19.2465. [DOI] [PubMed] [Google Scholar]
- 23.Giardiello FM, Hamilton SR, Kern SE, Offerhaus GJ, Green PA et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66:971–975. doi: 10.1136/adc.66.8.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sachatello CR. Polypoid diseases of the gastrointestinal tract. J Ky Med Assoc. 1972;70:540–544. [PubMed] [Google Scholar]
- 25.Horrilleno EG, Eckert C, Ackerman LV. Polyps of the rectum and colon in children. Cancer. 1957;10:1210–1220. doi: 10.1002/1097-0142(195711/12)10:6<1210::aid-cncr2820100619>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008;88:779–817. doi: 10.1016/j.suc.2008.05.002. vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4:492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 28.Eng C. To be or not to BMP. Nat Genet. 2001;28:105–107. doi: 10.1038/88802. [DOI] [PubMed] [Google Scholar]
- 29.Eng C, Ji H. Molecular classification of the inherited hamartoma polyposis syndromes: clearing the muddied waters. Am J Hum Genet. 1998;62:1020–1022. doi: 10.1086/301847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe JR, Ringold JC, Summers RW, Mitros FA, Nishimura DY, Stone EM. A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am J Hum Genet. 1998;62:1129–1136. doi: 10.1086/301840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 32.Howe JR, Sayed MG, Ahmed AF, Ringold J, Larsen-Haidle J et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484–491. doi: 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merg A, Howe JR. Genetic conditions associated with intestinal juvenile polyps. Am J Med Genet C Semin Med Genet. 2004;129C:44–55. doi: 10.1002/ajmg.c.30020. [DOI] [PubMed] [Google Scholar]
- 34.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 35.Massague J. TGFbeta signaling: receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- 36.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 37.Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107–119. doi: 10.1101/gad.12.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 39.Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 41.Kim IJ, Park JH, Kang HC, Kim KH, Kim JH et al. Identification of a novel BMPR1A germline mutation in a Korean juvenile polyposis patient without SMAD4 mutation. Clin Genet. 2003;63:126–130. doi: 10.1034/j.1399-0004.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhou XP, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M et al. Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet. 2001;69:704–711. doi: 10.1086/323703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehra A, Wrana JL. TGF-beta and the Smad signal transduction pathway. Biochem Cell Biol. 2002;80:605–622. doi: 10.1139/o02-161. [DOI] [PubMed] [Google Scholar]
- 44.Reddi AH. Bone morphogenetic proteins: an unconventional approach to isolation of first mammalian morphogens. Cytokine Growth Factor Rev. 1997;8:11–20. doi: 10.1016/s1359-6101(96)00049-4. [DOI] [PubMed] [Google Scholar]
- 45.Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- 46.Howe JR, Haidle JL, Lal G, Bair J, Song C et al. ENG mutations in MADH4/BMPR1A mutation negative patients with juvenile polyposis. Clin Genet. 2007;71:91–92. doi: 10.1111/j.1399-0004.2007.00734.x. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez-Ruiz E, St-Jacques S, Bellon T, Letarte M, Bernabeu C. Assignment of the human endoglin gene (END) to 9q34-->qter. Cytogenet Cell Genet. 1993;64:204–207. doi: 10.1159/000133576. [DOI] [PubMed] [Google Scholar]
- 48.Fonsatti E, Del Vecchio L, Altomonte M, Sigalotti L, Nicotra MR et al. Endoglin: an accessory component of the TGF-beta-binding receptor-complex with diagnostic, prognostic, and bioimmunotherapeutic potential in human malignancies. J Cell Physiol. 2001;188:1–7. doi: 10.1002/jcp.1095. [DOI] [PubMed] [Google Scholar]
- 49.Dallas NA, Samuel S, Xia L, Fan F, Gray MJ et al. Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Clin Cancer Res. 2008;14:1931–1937. doi: 10.1158/1078-0432.CCR-07-4478. [DOI] [PubMed] [Google Scholar]
- 50.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751–756. doi: 10.1007/BF02303487. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe A, Nagashima H, Motoi M, Ogawa K. Familial juvenile polyposis of the stomach. Gastroenterology. 1979;77:148–151. [PubMed] [Google Scholar]
- 53.Yoshida T, Haraguchi Y, Tanaka A, Higa A, Daimon Y et al. A case of generalized juvenile gastrointestinal polyposis associated with gastric carcinoma. Endoscopy. 1988;20:33–35. doi: 10.1055/s-2007-1018122. [DOI] [PubMed] [Google Scholar]
- 54.Stemper TJ, Kent TH, Summers RW. Juvenile polyposis and gastrointestinal carcinoma. A study of a kindred. Ann Intern Med. 1975;83:639–646. doi: 10.7326/0003-4819-83-5-639. [DOI] [PubMed] [Google Scholar]
- 55.Howe JR, Ringold JC, Hughes JH, Summers RW. Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery. 1999;126:162–170. [PubMed] [Google Scholar]
- 56.Dunlop MG. Guidance on gastrointestinal surveillance for hereditary nonpolyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut. 2002;51(5):V21–27. doi: 10.1136/gut.51.suppl_5.v21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oncel M, Church JM, Remzi FH, Fazio VW. Colonic surgery in patients with juvenile polyposis syndrome: a case series. Dis Colon Rectum. 2005;48:49–55. doi: 10.1007/s10350-004-0749-y. discussion -6. [DOI] [PubMed] [Google Scholar]
- 58.Scott-Conner CE, Hausmann M, Hall TJ, Skelton DS, Anglin BL, Subra-mony C. Familial juvenile polyposis: patterns of recurrence and implications for surgical management. J Am Coll Surg. 1995;181:407–413. [PubMed] [Google Scholar]
- 59.Zbuk KM, Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat Rev Cancer. 2007;7:35–45. doi: 10.1038/nrc2037. [DOI] [PubMed] [Google Scholar]
- 60.Arch EM, Goodman BK, Van Wesep RA, Liaw D, Clarke K et al. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet. 1997;71:489–493. [PubMed] [Google Scholar]
- 61.Celebi JT, Tsou HC, Chen FF, Zhang H, Ping XL et al. Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN. J Med Genet. 1999;36:360–364. [PMC free article] [PubMed] [Google Scholar]
- 62.Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 63.Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–1387. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 64.Starink TM, van der Veen JP, Arwert F, de Waal LP, de Lange GG et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986;29:222–233. doi: 10.1111/j.1399-0004.1986.tb00816.x. [DOI] [PubMed] [Google Scholar]
- 65.Albrecht S, Haber RM, Goodman JC, Duvic M. Cowden syndrome and Lhermitte-Duclos disease. Cancer. 1992;70:869–876. doi: 10.1002/1097-0142(19920815)70:4<869::aid-cncr2820700424>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 66.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37:828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marra G, Armelao F, Vecchio FM, Percesepe A, Anti M. Cowden's disease with extensive gastrointestinal polyposis. J Clin Gastroenterol. 1994;18:42–47. doi: 10.1097/00004836-199401000-00011. [DOI] [PubMed] [Google Scholar]
- 68.McGarrity TJ, Wagner Baker MJ, Ruggiero FM, Thiboutot DM, Hampel H et al. GI polyposis and glycogenic acanthosis of the esophagus associated with PTEN mutation positive Cowden syndrome in the absence of cutaneous manifestations. Am J Gastroenterol. 2003;98:1429–1434. doi: 10.1111/j.1572-0241.2003.07496.x. [DOI] [PubMed] [Google Scholar]
- 69.Gorlin RJ, Cohen MM, Jr, Condon LM, Burke BA. Bannayan-Riley-Ruval-caba syndrome. Am J Med Genet. 1992;44:307–314. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 70.Ruvalcaba RH, Myhre S, Smith DW. Sotos syndrome with intestinal polyposis and pigmentary changes of the genitalia. Clin Genet. 1980;18:413–416. doi: 10.1111/j.1399-0004.1980.tb01785.x. [DOI] [PubMed] [Google Scholar]
- 71.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 72.Lynch ED, Ostermeyer EA, Lee MK, Arena JF, Ji H et al. Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am J Hum Genet. 1997;61:1254–1260. doi: 10.1086/301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13:114–116. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 74.Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16:333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 75.Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7:507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 76.Sarquis MS, Agrawal S, Shen L, Pilarski R, Zhou XP, Eng C. Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Hum Genet. 2006;79:23–30. doi: 10.1086/504392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 78.Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett. 2006;241:184–196. doi: 10.1016/j.canlet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 80.Fackenthal JD, Marsh DJ, Richardson AL, Cummings SA, Eng C et al. Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet. 2001;38:159–164. doi: 10.1136/jmg.38.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hanssen AM, Fryns JP. Cowden syndrome. J Med Genet. 1995;32:117–119. doi: 10.1136/jmg.32.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carlson GJ, Nivatvongs S, Snover DC. Colorectal polyps in Cowden's disease (multiple hamartoma syndrome) Am J Surg Pathol. 1984;8:763–770. doi: 10.1097/00000478-198410000-00005. [DOI] [PubMed] [Google Scholar]
- 83.Hamby LS, Lee EY, Schwartz RW. Parathyroid adenoma and gastric carcinoma as manifestations of Cowden's disease. Surgery. 1995;118:115–117. doi: 10.1016/s0039-6060(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 84.Kato M, Mizuki A, Hayashi T, Kunihiro T, Nagata H et al. Cowden's disease diagnosed through mucocutaneous lesions and gastrointestinal polyposis with recurrent hematochezia, unrevealed by initial diagnosis. Intern Med. 2000;39:559–563. doi: 10.2169/internalmedicine.39.559. [DOI] [PubMed] [Google Scholar]
- 85.Longy M, Coulon V, Duboue B, David A, Larrègue M et al. Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J Med Genet. 1998;35:886–889. doi: 10.1136/jmg.35.11.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boardman LA. Heritable colorectal cancer syndromes: recognition and preventive management. Gastroenterol Clin N Amer. 2002;31:1107–1131. doi: 10.1016/s0889-8553(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 87.Kutscher AH, Zegarelli EV, Rankow RM, Slaughter TW. Incidence of Peutz-Jeghers syndrome. Am J Dig Dis. 1960;5:576–577. doi: 10.1007/BF02233059. [DOI] [PubMed] [Google Scholar]
- 88.Traboulsi EI, Maumenee IH. Periocular pigmentation in the Peutz-Jeghers syndrome. Am J Ophthalmol. 1986;102:126–127. doi: 10.1016/0002-9394(86)90229-1. [DOI] [PubMed] [Google Scholar]
- 89.Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71–82. [PubMed] [Google Scholar]
- 90.Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4:408–415. doi: 10.1016/j.cgh.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 91.Kyle J. Peutz-Jeghers syndrome. Scottish Med J. 1961;6:361–367. doi: 10.1177/003693306100600802. [DOI] [PubMed] [Google Scholar]
- 92.Westerman AM, Wilson JH. Peutz-Jeghers syndrome: risks of a hereditary condition. Scand J Gastroenterol. 1999;230:64–70. doi: 10.1080/003655299750025561. [DOI] [PubMed] [Google Scholar]
- 93.Brosens LA, van Hattem WA, Jansen M, de Leng WW, Giardiello FM, Offerhaus GJ. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29–46. doi: 10.2174/156652407779940404. [DOI] [PubMed] [Google Scholar]
- 94.Tovar JA, Eizaguirre I, Albert A, Jimenez J. Peutz-Jeghers syndrome in children: report of two cases and review of the literature. J Pediatr Surg. 1983;18:1–6. doi: 10.1016/s0022-3468(83)80262-0. [DOI] [PubMed] [Google Scholar]
- 95.Dormandy TL. Gastrointestinal polyposis with mucocutaneous pigmentation (Peutz-Jeghers syndrome) N Engl J Med. 1957;256:1186–1190. doi: 10.1056/NEJM195706202562506. concl. [DOI] [PubMed] [Google Scholar]
- 96.Foster DR, Foster DB. Gall-bladder polyps in Peutz-Jeghers syndrome. Post. grad Med J. 1980;56:373–376. doi: 10.1136/pgmj.56.655.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gentile AT, Bickler SW, Harrison MW, Campbell JR. Common bile duct obstruction related to intestinal polyposis in a child with Peutz-Jeghers syndrome. J Pediatr Surg. 1994;29:1584–1587. doi: 10.1016/0022-3468(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 98.Parker MC, Knight M. Peutz-Jeghers syndrome causing obstructive jaundice due to polyp in common bile duct. J R Soc Med. 1983;76:701–703. doi: 10.1177/014107688307600814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 100.Jass JR. Gastrointestinal polyposes: clinical, pathological and molecular features. Gastroenterol Clin North Am. 2007;36:927–946. doi: 10.1016/j.gtc.2007.08.009. viii. [DOI] [PubMed] [Google Scholar]
- 101.Shepherd NA, Bussey HJ, Jass JR. Epithelial misplacement in Peutz-Jeghers polyps. A diagnostic pitfall. Am J Surg Pathol. 1987;11:743–749. doi: 10.1097/00000478-198710000-00001. [DOI] [PubMed] [Google Scholar]
- 102.Bartholomew LG, Moore CE, Dahlin DC, Waugh JM. Intestinal polyposis associated with mucocutaneous pigmentation. Surg Gynecol Obstet. 1962;115:1–11. [PubMed] [Google Scholar]
- 103.Hemminki A, Tomlinson I, Markie D, Järvinen H, Sistonen P et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- 104.Mehenni H, Blouin JL, Radhakrishna U, Bhardwaj SS, Bhardwaj K et al. Peutz-Jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet. 1997;61:1327–1334. doi: 10.1086/301644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 106.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 107.Volikos E, Robinson J, Aittomäki K, Mecklin JP, Järvinen H et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006;43 doi: 10.1136/jmg.2005.039875. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S et al. Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005;14:1283–1292. doi: 10.1093/hmg/ddi139. [DOI] [PubMed] [Google Scholar]
- 109.Forcet C, Billaud M. Dialogue between LKB1 and AMPK: a hot topic at the cellular pole. Sci STKE. 2007 doi: 10.1126/stke.4042007pe51. 2007:pe51. [DOI] [PubMed] [Google Scholar]
- 110.Marignani PA. LKB1, the multitasking tumour suppressor kinase. J Clin Pathol. 2005;58:15–19. doi: 10.1136/jcp.2003.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miyoshi H, Nakau M, Ishikawa TO, Seldin MF, Oshima M, Taketo MM. Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 2002;62:2261–2266. [PubMed] [Google Scholar]
- 112.Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 113.Lim W, Hearle N, Shah B, Murday V, Hodgson SV et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer. 2003;89:308–313. doi: 10.1038/sj.bjc.6601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 115.Cantu JM, Rivera H, Ocampo-Campos R, Bedolla N, Cortés-Gallegos V et al. Peutz-Jeghers syndrome with feminizing sertoli cell tumor. Cancer. 1980;46:223–228. doi: 10.1002/1097-0142(19800701)46:1<223::aid-cncr2820460137>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 116.Scully RE. Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer. 1970;25:1107–1121. doi: 10.1002/1097-0142(197005)25:5<1107::aid-cncr2820250516>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 117.Hinds R, Philp C, Hyer W, Fell JM. Complications of childhood Peutz-Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004;39:219–220. doi: 10.1097/00005176-200408000-00027. [DOI] [PubMed] [Google Scholar]
- 118.McGrath DR, Spigelman AD. Preventive measures in Peutz-Jeghers syndrome. Fam Cancer. 2001;1:121–125. doi: 10.1023/a:1013896813918. [DOI] [PubMed] [Google Scholar]