

Synopsis
Diamond Blackfan anemia (DBA) is a genetically and clinically heterogeneous disorder characterized by erythroid failure, congenital anomalies and a predisposition to cancer. Faulty ribosome biogenesis, resulting in pro-apoptotic erythropoiesis leading to erythroid failure, is hypothesized to be the underlying defect. The genes identified to date that are mutated in DBA all encode ribosomal proteins associated with either the small (RPS) or large (RPL) subunit and in these cases haploinsufficiency gives rise to the disease. Extraordinarily robust laboratory and clinical investigations have recently led to demonstrable improvements in clinical care for patients with DBA.
Keywords: Diamond Blackfan anemia (DBA), pure red cell aplasia, ribosome biogenesis, inherited bone marrow failure syndrome (IBMFS), cancer predisposition
Introduction
Diamond Blackfan anemia (DBA; MIM #205900) is one of a rare group of genetic disorders, known as the inherited bone marrow failure syndromes (IBMFS) 1. These disorders have in common pro-apoptotic hematopoiesis, bone marrow failure, birth defects 2 and in the majority a predisposition to cancer3. Interest in these disorders has grown dramatically as the study of each has clarified, or revealed for the first time, new molecular events in development or cellular function. Significant expectations await the complete elucidation of these events. In particular DBA has revealed itself as a “ribosomapathy” (reviewed in 4).
Diamond Blackfan anemia was first reported by Josephs in 1936 5 and more completely described by Diamond and Blackfan in 1938 6. The diagnostic criteria for DBA published in 1976 consist of: presentation of anemia prior to the first birthday with near normal or slightly decreased neutrophil counts, variable platelet counts, reticulocytopenia, macrocytosis and normal marrow cellularity with a paucity of red cell precursors 7. These criteria have, until recently, remained the accepted standard.
In addition to macrocytosis, the presence of elevated fetal hemoglobin levels (HbF) and an elevation in erythrocyte adenosine deaminase enzyme (eADA) activity are important supporting features associated with DBA. The presence of macrocytosis and elevated fetal hemoglobin levels, each felt to be a consequence of “stress erythropoiesis” and skipped erythroid cell divisions is not unique to DBA but is observed in most incidences of bone marrow failure. These features are also found in recovery from anemia such as that caused by iron deficiency when erythropoietin levels are elevated. An explanation for the elevated eADA activity in DBA has remained frustratingly obscure for over 25 years since the original observation 8.
The careful analysis of DBA-affected pedigrees for the presence of members with macrocytosis, elevated HbF levels and/or increased eADA activity (elevated in 85% of patients with DBA) and congenital anomalies strongly suggested a greater number of autosomal dominant cases than previously thought 9. With the discovery of the first gene mutated in DBA 10,11, it became evident that the penetrance of autosomal dominant DBA is quite variable with regard to both hematologic and non-hematologic manifestations. Indeed, as a consequence of mutational analysis in family members of probands the estimated incidence of familial, autosomal dominant DBA has increased from approximately 10–15% to 45% 12. Thus, although the diagnostic criteria for classical DBA remain unchanged, there are numerous patients not meeting these criteria for whom a “non-classical” DBA diagnosis is appropriate 13. Consequently, a diagnosis of DBA may now be suitable, for example, in individuals with little or no anemia, macrocytosis only, a presentation in adulthood 14, a phenotypically normal parent of an affected offspring, and individuals with congenital anomalies or short stature and minimal or no evidence of abnormal erythropoiesis 13. Thus, registry data must be interpreted with an understanding of the ascertainment bias introduced by this clinical heterogeneity. Nevertheless, recent advances in the understanding of DBA, in part as a result of data from international Diamond Blackfan anemia registries 9,15–17 are resulting in more sophisticated diagnostic criteria and improvements in clinical care 13.
Pathophysiology and Genetics: DBA a disorder of ribosome biogenesis and the only known disorder characterized by mutations in structural ribosomal proteins
Evidence supporting DBA as an intrinsic disorder of erythropoiesis rather than the result of immune mediated red cell failure first appeared in 1976 when the group from Toronto 18 suggested that some patients with DBA had decreased numbers of erythroid colony forming units (CFU-E). Investigators in Boston 19 extended this observation and suggested a block in erythroid maturation prior to the less mature erythroid burst forming unit (BFU-E) stage. These findings may not, however, be true in all instances as later studies showed that both BFU-E and CFU-E colonies are present, often in normal numbers, in the marrow of young DBA patients but their differentiation to mature erythrocytes is defective, further supporting DBA to be the result of an intrinsic progenitor defect 20. Interestingly, Chan et al. 21 demonstrated that the growth in vitro of DBA progenitor cells in semi-solid media could be enhanced by the addition of corticosteroids similar to the in vivo response 19.
Studies exploring the pathophysiology of DBA had, until recently, been hampered by the fact that there were no available animal models for this disease. RPS19 was the first DBA gene identified, and mutations in this gene account for approximately 25% of DBA cases. The initial RPS19 knockout mouse was homozygous lethal and the hemizygote lacked a DBA phenotype 22. Moreover, no naturally occurring animal models of DBA were known to exist as the anemic and macrocytic W/Wv and Sl/Sld mice did not respond to steroids and had mutations in genes for c-kit and kit ligand (now known not to be involved in the molecular pathology of DBA), respectively23. In the past year however, two groups have been able to knockdown rps19 in zebrafish using antisense morpholino oligonucleotides, each recapitulating the hematologic phenotype as well as producing malformations 24,25. Uechi et al 25 extended this observation to other ribosomal proteins demonstrating defective erythropoiesis in 3 of 20 knockdowns, one being rpl35a, recently described to be mutated in humans with DBA 26. Also within the past year, McGowan and colleagues 27 identified rps19 and rps20 as genes mutated in mice with a dark skin phenotype. Further studies revealed that the rps19Dsk3 mouse recapitulated the human DBA phenotype insofar as a hypoproliferative, pro-apoptotic anemia with growth retardation. Based on available data it is now widely accepted that DBA results from an intrinsic cellular defect in which erythroid progenitors and precursors are highly sensitive to death by apoptosis20,28–30. The observation that phenotypes observed in zebrafish and mouse models of DBA can be partially or fully rescued by mutations in p53 strongly suggests that p53 stabilization and activation plays an important role in the pro-apoptotic phenotype of cells with ribosomal protein haploinsufficiency.
Thus considerable attention, facilitated by new zebrafish and mouse models of DBA, has been directed toward this “ribosomal stress hypothesis” reviewed recently by Dianzani and Loreni 4 in which decreased ribosome protein synthesis activates p53, inducing downstream events resulting in cell cycle arrest or apoptosis. This in turn results in the DBA phenotype of anemia, poor growth and congenital malformations. Furthermore, the relationship of the nucleolus and defective ribosome synthesis to p53-mediated apoptosis suggests a role for interdicting mutations in p53 and distal pathways in oncogenesis in DBA 31.
In addition to RPS19, five other ribosomal protein genes have been shown to be mutated in patients with DBA 26,32–34. Mutations in these genes account for approximately 50% of DBA cases. Work being done by investigators in collaboration with the NHLBI Resequencing and Genotyping Service (http://rsng.nhlbi.nih.gov/scripts/about.cfm) to sequence each of the 80 ribosomal protein genes in patients from the North American DBA registry will no doubt identify additional DBA genes. The genes identified to date encode proteins of both the 40S ribosomal subunit (RPS17, RPS19, and RPS24) and the 60S subunit (RPL5, RPL11, and RPL35A). Many of these proteins have been shown to be required for the maturation of their respective ribosomal subunits in both yeast and mammalian cells 26,34–37
There are several potential mechanisms whereby abortive ribosome assembly or nucleolar stress could signal to p53 activation. One of the more intriguing of these mechanisms from the perspective of DBA pathophysiology involves the interaction of certain ribosomal proteins with MDM2 (Murine Double Minute), a potent regulator of p53 levels and activity. MDM2 is a RING finger ubiquitin ligase that interacts with and promotes the degradation of p53. Three proteins of the 60S subunit, RPL5, RPL11, and RPL23 have been shown to bind to MDM2 reducing its ubiquitin ligase activity, which in turn results in p53 stabilization 38–42. The interaction of these proteins with MDM2 has been studied most frequently in the context of drugs that either inhibit RNA polymerase I transcription or have a general effect on the assembly of both 40S and 60S ribosomal subunits. Under these treatments, ribosome assembly is inhibited at very early stages of a complex hierarchical process whereby proteins that bind directly to rRNA facilitate the subsequent binding of other ribosomal proteins, a process facilitated in eukaryotic cells by a host of accessory factors 43,44. Disruption of ribosome assembly early in the process results in the diversion of ribosomal proteins from ribosomal subunits to other fates within cells. In many cases it is thought that free ribosomal proteins are rapidly degraded to avoid the presumed toxicity associated with the release of small highly basic proteins into the cell 45. In recent years, however, it has become clear that ribosomal proteins diverted from their normal fates as components of the ribosome can be important signaling molecules as we see in these studies linking abortive ribosome assembly to p53 activation and stabilization.
There are a number of caveats associated with trying to apply the pathway outlined above linking abortive ribosome assembly to p53 activation to the context of the ribosomal protein haploinsufficiency in DBA. In contrast to the studies using drugs that inhibit the assembly of both ribosomal subunits, a ribosomal protein deficiency primarily affects only its corresponding ribosomal subunit 26,36. Therefore, while there are good candidates for ribosomal protein signaling molecules derived from the 60S subunit, only one protein of the 40S subunit, RPS7, has been shown to interact with MDM2 and this interaction is much weaker than that observed for the large ribosomal subunit proteins mentioned 46. Also, curiously, two recently identified “DBA genes”, encode the signaling ribosomal proteins RPL5 and RPL11 34. What makes this observation puzzling is that RPL5 and RPL11 have been shown to bind synergistically to MDM2, perhaps as a complex with 5S rRNA 47, and so each protein must be present for maximal signaling through MDM2 (Fig. 1). The finding of RPL5 and RPL11 as DBA genes may suggest that this signaling pathway may not need to operate at maximal efficiency to elicit the pro-apoptotic phenotype associated with ribosomal protein haploinsufficiency. Finally, this pathway does not provide a ready rationale for the acute sensitivity of the erythroid lineage to the effects of ribosomal protein haploinsufficiency. One could, however, argue that abortive assembly in the context of the large demands for ribosome synthesis during erythroid development could unleash a relatively high concentration of wayward ribosomal proteins to trigger this and other signaling pathways leading to growth arrest or apoptosis.
Fig. 1. Hypothetic model for abortive ribosome assembly and p53 activation relevant to DBA.

Panel A, 60S subunit assembly in healthy individuals; panel B, DBA patients haploinsufficient for RPL35A; panel C, DBA patients haploinsufficient for RPL5. Ribosomal RNAs are shown with twisted lines and ribosomal proteins are shown with circles. Relevant ribosomal proteins are indicted with their numerical designation in the center. The effects of ribosomal proteins on the level or activity of MDM2 and p53 are shown below the assembly pathways. The solid bars indicate that the 5S rRNA-Rpl5-Rpl11 ternary complex may bind more strongly to MDM2, and have a more substantial effect on p53 stabilization that Rpl11 alone (shown with dashed bars).
As noted, there are other pathways by which nucleolar stress may signal to p53 activation and therefore play a role in DBA pathophysiology. In this context, the tumor suppressor protein ARF is also known to interact with MDM2 to promote p53 stabilization 48. ARF is also known to bind nucleophosmin (B23), a protein involved in ribosome biogenesis and export 49. The relative stoichiometry of ARF and its binding partners MDM2 and B23 is thought to play a critical role in linking cell cycle regulation with ribosome synthesis 50. The extent to which this pathway cooperates with the pathway involving liberated ribosomal proteins in nucleolar stress signaling has not been determined. It is possible that the contribution of each pathway to p53 stabilization and activation may vary depending on the nature of the nucleolar stress.
A Clinically Heterogeneous Syndrome: Perhaps Diamond Blackfan “anemia” is a misappellation and it should be designated as Diamond Blackfan syndrome (but alas it is probably too late for that)
Congenital Anomalies
Birth defects have long been known to be a feature of DBA. A distinct facial appearance and triphalangeal thumbs have been classically described in DBA as the Cathie facies 51 and Aase syndrome 52, respectively. A cute snub nose and wide-spaced eyes as originally described by Cathie and other craniofacial anomalies, some quite severe, are the most common physical anomalies described in DBA. Abnormal thumbs are classic 53. In all, congenital anomalies were found in 30–47% of the patients in the Italian 16, French 15, UK 12, and North American registries 54 Additional anomalies of the upper limb and hand, genitourinary system and heart each are described in as many as 30–40 % of these patients 54. The prevalence of genitourinary and cardiac anomalies may be underestimated when abdominal/pelvic and cardiac ultrasonography is not routinely performed in asymptomatic patients. More than one anomaly is described in about a quarter of all patients. Short stature is clearly constitutional, a function of the ribosomapathy, in many patients. However an accurate assessment of the etiology of linear growth retardation is complicated in patients who may be anemic, iron-overloaded or taking corticosteroids, all from a very young age. A representative table of malformations has been published 15.
A disorder of craniofacial morphogenesis linked to diminished ribosome biogenesis, Treacher Collins syndrome (TCS; MIM #154500), provides additional evidence linking DBA to defective ribosome synthesis. TCS is characterized in classical cases by, “bilateral downslanting palpebral fissures, frequently accompanied by colobomas of the lower eyelids and a paucity of eyelashes medial to the defect, abnormalities of the external ears, atresia of the external auditory canal and bilateral conductive hearing loss, hypoplasia of the zygomatic complex and mandible and cleft palate” 55. As in DBA the disorder is inherited as an autosomal dominant trait. The incidence is of the same order of magnitude at about 1 in 50,000 live births for TCS 56 and 1 in 100,000 for DBA 15. Case ascertainment of mild phenotypes in both disorders is clearly a cause for underreporting. Additional similarities between TCS and DBA exist. Orofacial clefts have been reported in 3% of cases of DBA in the literature 57 and in 5.7% of cases from the Diamond Blackfan Anemia Registry 9. Gripp et al. 2 identified first cousins, male children of sisters, with bilateral microtia and cleft palate consistent with TCS, but with hematologic abnormalities consistent with DBA. The proband had been previously enrolled in the Diamond Blackfan Anemia Registry of North America (DBAR), a database of now over 500 patients 9,54 while his cousin with similar facial anomalies had not developed classic DBA at the time of the report. The cousin’s relevant hematologic parameters were significant for macrocytosis, elevated HbF level and increased eADA activity, and therefore consistent with a non-classical hematologic DBA phenotype. Physical and hematologic evaluations of the patients’ mothers were normal. Therefore, both mothers appear to be obligate heterozygotes for this dominantly inherited disorder, with neither demonstrating any hematologic evidence of DBA or even minimal congenital anomalies. This multiplex family demonstrates the extremely variable penetrance and expressivity of this DBA gene. In all, the described proband and 2 other of the 21 patients with DBA and a cleft palate reported to the DBAR were originally diagnosed with “classical” TCS. However, none of these three had mutations in TCOF1 (5q32–q33.1), the gene mutated in 90% of patients with TCS 58. Indeed, in the absence of hematologic manifestations a number of the DBA patients would likely be considered as non-classical TCS. Thus a distinct minority of DBA patients are phenocopies of TCS (Figure 2). All the patients met the criteria for a diagnosis of DBA (either classical or non-classical) however none of those with craniofacial anomalies had RPS19 mutations (unpublished, North American DBA Registry). In addition to these observations from the DBAR, no other investigators 12,59, have identified patients with cleft palate/microtia and an RPS19 mutation 12,59. Intriguingly, a possible genotype-phenotype correlation has recently been identified when a “TCS-like” phenotype in DBA, not associated with mutations in RPS19 or TCOF1, was identified in 4 of 7 patients mutated at RPL5 34. The discovery of the remaining DBA genes will enable further genotyping of this important subset of patients. Other patients with DBA and craniofacial anomalies have not yet been evaluated for TCOF1 mutations, nor have extensive hematologic evaluations of patients with TCS been reported. Thus it is probable that a substantial minority of patients with TCS will have a DBA genotype.
Fig. 2.
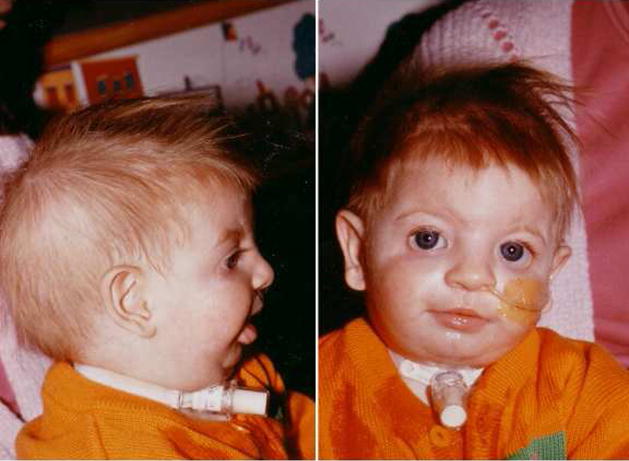
Patient with Diamond Blackfan anemia originally diagnosed with Treacher Collins syndrome. Note absent lower eyelashes, down slanting palpebral fissures, deformed external ears, malar hypoplasia, and micrognathia.
TCOF1 encodes the nucleolar phosphoprotein treacle 58–60, mutated in TCS. Analogous to RPL5 in DBA 34, TCS results most likely from treacle haplo-insufficiency 61. Treacle has been reported to interact with upstream binding factor (UBF), a known transcription regulator of RNA polymerase I (Pol I) which participates in the initiating event in ribosomal DNA (rDNA) transcription, generating the 47S pre-ribosomal RNA (pre-rRNA) 62. Treacle is also involved in other early steps in ribosome synthesis, linking rDNA transcription to pre-rRNA post-transcriptional methylation 63. Thus treacle haplo-insufficiency would inhibit early events in the ribosome biosynthetic pathway, having a profound effect upon ribosome synthesis. Comparable to pro-apoptotic erythropoiesis in DBA, the mechanism for the disruption of craniofacial structures in TCS appears to be increased apoptosis in the neural crest cells of the prefusion neural folds just prior to fusion during embryogenesis 64. Intriguingly, just as in animal models of DBA, the “neurocristopathy” in the mouse model of TCS can be prevented by inhibition of p53 65.
Thus, the molecular underpinnings of DBA and TCS share certain common features, just as do their clinical presentations. The remarkable observation that a subset of DBA patients with orofacial abnormalities cluster together with mutations in RPL5 demonstrates that not all ribosomal protein genes behave identically in disrupting craniofacial development. Identification of the potential ties that bind TCOF1 with RPL5 and other currently unknown DBA genes that disrupt orofacial development promises to be a fascinating area of investigation. These studies suggest that there may be other genotype/phenotype relationships buried within the DBA registries. Moreover, as with TCS other syndromes with congenital anomalies shared with DBA patients may be shown to have ribosome biogenesis defects as their molecular bases. In fact a recent survey of patients who were diagnosed or had undergone repair of common DBA-associated anomalies, namely congenital heart disease (atrial septal defect, ventricular septal defect, coarctation of the aorta and multiple defects), radial ray anomalies (bifid, subluxed, triphalangeal, or hypoplastic thumbs and flat thenar muscles) and orofacial clefts, demonstrated approximately 2–3% had otherwise unexplained macrocytosis (North American DBAR, unpublished observations). Although this was not a controlled study it is probable that unrecognized cases of DBA and perhaps other IBMFS exist undiagnosed in the general population. Furthermore there is a reasonable possibility that other mutations in the complex process of ribosome assembly not related to either ribosomal proteins or hematopoiesis may account for a portion of these malformations.
Cancer: DBA is a cancer predisposition syndrome
At least 30 cases of cancer in patients with DBA have been reported in the literature. Fifteen were hematopoietic malignancies. Of these, 10 were cases of acute myeloid leukemia (AML), 3 Hodgkin disease, 1 non-Hodgkin lymphoma and 1 acute lymphoblastic leukemia. Three additional cases of myelodysplastic syndrome (MDS) were not included in this total. Fifteen solid tumors have been reported; 7 osteogenic sarcoma, 2 breast cancer, 2 hepatocellular carcinoma and one each of colon carcinoma, gastric carcinoma, vaginal melanoma and malignant fibrous histiocytoma (updated from Alter 66 and Yaris et al. 67 ). A number of cases have been reported from international registries and a large institutional cohort 15,54,68. Of over 500 patients registered in the DBAR at the time of the most recent analysis, there were 12 patients who were found to have a total of 14 malignancies. Some of these patients also appear as case reports in the literature. Three patients were diagnosed with osteogenic sarcoma, one with acute myeloid leukemia, 3 with myelodysplastic syndrome ( including myelofibrosis with myeloid metaplasia), 2 with colon cancer, 2 with breast cancer, one with a soft tissue sarcoma, and 2 with squamous cell carcinoma (oral and vaginal). There is an individual who is a DBA affected relative of a registered patient with melanoma, who is not yet enrolled in the DBAR. Although cancer is relatively rare in DBA as compared to Fanconi anemia, the incidence appears to be well in excess of what would be expected for the age group represented. In particular, cases of breast and colon cancer have been described in very young adults 3,15. The prognosis for patients with DBA and cancer is poor. This is due in part to severe chemotherapy induced myelosuppression 3. The presence of these malignancies and the young age at diagnosis of many of these cancers appears to define DBA as a cancer predisposition syndrome. However, before a true cancer incidence is determined, data from international DBA registries will need to undergo a careful analysis as has been applied to Fanconi anemia 69 and severe congenital neutropenia 70.
A mechanism leading to AML and MDS in Fanconi anemia has been postulated 71 and has been recently confirmed in a murine model of clonal selection 72. In this model, outlined in more detail in Chapter 13, the existence of pro-apoptotic hematopoiesis exerts a selective pressure on the myeloid compartment. This results in the emergence of clones with apoptosis-contravening mutations, resulting in leukemia. It seems reasonable to suggest that a similar mechanism could exist for DBA. Several recent studies, including work already cited, have shown that nucleolar stress can induce p53-dependent cell cycle arrest and/or apoptosis 73,74. Mutations in the p53 gene and MDM2 could potentially subvert this process providing a growth advantage to clones harboring p53 mutations, which at the same time could favor malignant outgrowth. Other mechanisms also exist that could explain the increased cancer incidence in DBA patients. Since DBA patients characterized to date have one remaining active ribosomal gene, mutations that enhance ribosomal protein gene expression could potentially compensate for the inactive allele. The oncogene, c-Myc, is known to up-regulate many components of the translational machinery in its role in stimulating cell growth and proliferation 75,76. Activating mutations in c-Myc could therefore compensate for a ribosomal protein deficiency allowing for the emergence of clones with a survival advantage predisposed to malignancy. Since many signaling pathways promoting cell growth and division regulate ribosome synthesis as a downstream target, numerous other targets also exist to explain the increased cancer incidence in DBA patients 77. Finally, heterozygous mutations in 11 different ribosomal protein genes in zebrafish have recently been found in fish with peripheral nerve sheath tumors 78. These dominantly inherited mutations appear to result from loss of function, raising the specter that under certain circumstances ribosomal protein genes may act as tumor suppressor loci (Figure 3).
Fig. 3. Proposed mechanism for cancer predisposition in Diamond Blackfan anemia.

Ribosomal protein haploinsufficiency results in the accumulation of partially assembly intermediates that result in nucleolar stress signaling. Nucleolar stress, in turn, leads to p53 activation and either cell cycle arrest or apoptosis. These steps are outlined in red. Interdicting mutations that subvert the p53 pathway and favor the outgrowth of malignant clones are shown in green. Also shown in green are interdicting mutations that could enhance ribosomal protein synthesis and diminish nucleolar stress but because of the oncogenic nature of the genes involved could again favor the outgrowth of malignant clones.
Ribosome Dysfunction and Red Blood Cell Development
Conventional wisdom describes selective red cell hypoplasia as the defining characteristic of DBA, which seems at odds with the fact that ribosomes are a ubiquitous feature of all cell types with the exception of mature erythrocytes. However, this uniquely hematologic prospective disregards the existence of growth retardation and other congenital anomalies in DBA patients. Furthermore, in rare instances other significant hematologic cytopenias are also observed. How then does haploinsufficiency for RPS19, RPS24, RPS17 and RPL35a, RPL5 and RPL11 manifest clinically by selectively affecting only certain tissues and most evidently red cell production? It has been argued that the high demand for ribosome synthesis associated with the proliferation and differentiation of red cell precursors may make these precursors unusually sensitive to the effects of a reduction in ribosome synthesis28,79,80. It is important to consider this point of view from the perspective of distinct features of red cell development between the fetus, the neonatal period, and the transition to adult erythropoiesis. Red cell production in rapidly growing fetuses in the third trimester is reported to be approximately 3–5 times that in the adult steady state 81. If DBA is solely a consequence of the unusually high demands for ribosome synthesis in red cell progenitors, one would predict DBA to manifest initially during fetal development. Although there are instances of early fetal loss (perhaps attributable to erythroid failure) and hydrops fetalis in DBA, the median age at presentation of classical DBA is 8 weeks with ninety three percent of DBA patients presenting during the first year of life.
A number of developmental and physiological changes conspire to decrease red blood cell production shortly after birth. After birth, erythropoietin production decreases in response to high partial pressures of oxygen, a high hemoglobin level and the switch to the lower O2 - affinity adult hemoglobin allowing more oxygen delivery to tissues81. Since erythropoietin provides proliferative, survival and differentiation signals to erythroid progenitors, the fall in its production with birth contributes to a diminished erythron. There is also evidence to suggest that erythroid progenitors become less sensitive to erythropoietin in the transition from the fetal/neonatal state to the adult state82. Together, these changes lead to a transient physiological anemia at 4–8 weeks after birth until a new steady-state of red cell production is reached. The failure to reach a new steady state would result in an anemic presentation of DBA at 8 weeks, the time when new red cell production is required. It is also conceivable that the switch from primitive embryonic and fetal hematopoiesis derived from mesoderm to definitive fetal and adult erythropoiesis derived from liver and then bone marrow repopulating hematopoietic stem cells as described by Palis and Segal 81 is accompanied by more robust translation making them more vulnerable to ribosomal protein insufficiency. Thus, after birth when the demand for red cell production decreases, the demand for ribosome synthesis likely also decreases. Then upon the reestablishment of an erythroid drive, and the establishment of a new steady-state of ribosome synthesis and red cell production the presence of haploinsufficiency for critical ribosomal proteins becomes manifest. An important question relevant to DBA pathophysiology is, after eight weeks of postnatal life, what effect these changes in developmental and physiological signals have on ribosome synthesis and whether, as conditions change beyond this 8 week time point, a state is triggered where mutational inactivation of an allele of any of the implicated ribosomal protein genes limits ribosome synthesis in erythroid progenitors. Ribosome synthesis defects, in turn, could result in p53-dependent cell cycle arrest and enhanced apoptosis thereby explaining the ability of mutations in the p53 genes and compounds that inactivate p53 to rescue phenotypes in animal models of DBA 24,27.
Making the Diagnosis of Diamond Blackfan Anemia
The median age at presentation and diagnosis of classical DBA are 8 weeks and 12 weeks, respectively 54. Ninety three percent of DBA patients present during the first year of life, but like the other IBMFS, DBA may present in adulthood13 when it is often misdiagnosed. The differential diagnosis of DBA in children presenting with red cell failure, anemia, reticulocytopenia and decreased or absent marrow erythroid precursors is limited. However in adults the diagnosis is less common and the differential diagnosis includes a vast array of conditions, many of which rarely, if ever, are seen in children (Table 1). With awareness that mild DBA phenotypes may be missed in children and even more obvious cases misdiagnosed in adults, maintaining an index of suspicion is important. In the presence of a family history and in particular when a mutation is identified, the diagnosis of non-classical DBA may be quite straightforward. Isolated macrocytosis, for example, should not be dismissed after more obvious etiologies (folate or B12 deficiencies) are ruled out. Of utmost importance – patients with characteristic birth defects must be carefully evaluated for DBA and other IBMFS.
Table 1.
| Inherited |
| Diamond Blackfan anemia |
| Acquired PRCA |
| Congenital |
| Pearson syndrome |
| Primary |
| Autoimmune (includes Transient Erythroblastopenia of Childhood [TEC] |
| Preleukemic |
| Idiopathic |
| Secondary, associated with: |
| Thymoma |
| Hematologic malignancies |
| Chronic lymphocytic leukemia |
| Large granular lymphocytic leukemia (Tγ lymphoproliferative disorder) |
| Chronic myelocytic leukemia |
| Acute lymphoblastic leukemia |
| Hodgkin disease |
| Non-Hodgkin lymphomas |
| Multiple myeloma |
| Waldenström macroglobulinemia |
| Myelofibrosis with myeloid metaplasia |
| Essential thrombocythemia |
| Solid Tumors |
| Carcinoma of the stomach |
| Adenocarcinoma of the breast |
| Adenocarcinoma of bile duct |
| Squamous cell carcinoma of the lung |
| Epidermoid carcinoma of the skin |
| Carcinoma of the thyroid |
| Renal cell carcinoma |
| Carcinoma of unknown primary site |
| Kaposi sarcoma |
| Infections |
| Human B19 parvovirus |
| Human immunodeficiency virus (HIV) |
| T-cell leukemia-lymphoma virus |
| Epstein-Barr virus (infectious mononucleosis) |
| Viral hepatitis |
| Mumps |
| Cytomegalovirus |
| Atypical pneumonia |
| Meningococcemia |
| Staphylococcemia |
| Leishmaniasis |
| Chronic Hemolytic Anemias (usually associated with B19 parvovirus) |
| Collagen Vascular Diseases |
| Systemic lupus erythematosus |
| Rheumatoid arthritis |
| Mixed Connective Tissue disease |
| Sjögren syndrome |
| Drugs and Chemicals |
| Pregnancy |
| Severe renal failure |
| Severe nutritional deficiencies |
| Miscellaneous |
| Post-ABO incompatible bone marrow transplantation |
| Angioimmunoblastic lymphadenopathy |
| Autoimmune multiple endocrine gland insufficiency |
| Autoimmune hypothyroidism |
| Autoimmune chronic hepatitis |
| Anti-EPO antibodies post-treatment with EPO |
Modified from, Dessypris EN, Lipton, JM: Red cell aplasia. In: Wintrobe’s Clinical Hematology, 11th ed., JP, Greer, J, Foerster, JN, Lukens, GM, Rogers, F, Paraskevas, BE, Glader (eds.), Lippincott, Williams & Wilkins, Philadelphia, 2003, pp. 1421–1437
The differential diagnoses that need to be more commonly considered in children are highlighted in bold
In children the more common disorder, transient erythroblastopenia of childhood (TEC) must be ruled out. TEC is an acquired, short-lived failure of red cell production usually of a month or so in duration. As in DBA, children with TEC often present with profound anemia. TEC, most likely a post-infectious, transient autoimmune IgG-mediated disorder 83 characteristically occurs in toddlers probably as the result of infections acquired through contact with playmates. However as more infants are placed in day care settings, TEC seems to be presenting more often in children younger than 1 year of age. Table 2 summarizes the salient features distinguishing the two disorders. Most important, a positive family history and congenital anomalies are not characteristic of TEC. Elevated fetal hemoglobin levels and macrocytosis, common presenting features in DBA, are only seen in TEC upon recovery as a consequence of so-called “stress erythropoiesis”. Of note macrocytosis is often obscured in the newborn period by residual fetal erythrocytes and also can be masked by concomitant thalassemia minor or iron deficiency and other cytopenias are found in both disorders. Although the mechanism is unknown, an elevated erythrocyte adenosine deaminase activity is found in about 85% of patients with DBA and none with TEC 13. Thus the two disorders can usually be distinguished. The use of limited packed red cell transfusions to achieve a hemoglobin level that will not inhibit erythroid recovery is recommended, particularly when the diagnosis is unclear. Once the diagnosis is made definitive treatment for DBA can commence.
Table 2.
Differential Diagnosis of DBA versus TEC*
| Diamond Blackfan Anemia (DBA) | Transient Erythroblastopenia of Childhood (TEC) | |
|---|---|---|
| Pure red cell aplasia | Present | Present |
| Age | Younger than 1 year | Older than 1 year |
| Inheritance | Sporadic and dominant inheritance. Mutation analysis as available. | Not inherited |
| Congenital anomalies | Present | Absent |
| Mean Corpuscular Volume (MCV) | Elevated | Normal (may be elevated upon recovery) |
| Fetal hemoglobin | Elevated | Normal (may be elevated upon recovery) |
| erythrocyte ADA (eADA) activity | Elevated | Normal |
| Other cytopenias | May be present | May be present |
All RBC characteristics except eADA activity are helpful only when tested in a reticulocytopenic child. During recovery from TEC, a transient wave of fetal-like erythropoiesis with elevated MCV and fetal hemoglobin may be detected.
Modified from, Dessypris EN, Lipton, JM: Red cell aplasia. In: Wintrobe’s Clinical Hematology, 11th ed., JP, Greer, J, Foerster, JN, Lukens, GM, Rogers, F, Paraskevas, BE, Glader (eds.), Lippincott, Williams & Wilkins, Philadelphia, 2003, pp. 1421–1437
When DBA is clinically diagnosed, or in cases where a DBA diagnosis is equivocal based on clinical presentation, mutational analysis of known genes should be performed to confirm the diagnosis. All immediate family members of the proband should be evaluated with a history and physical examination looking for evidence of a transient or mild to moderate chronic anemia and birth defects. In addition a complete blood count with red cell indices, HbF levels and eADA activity should be performed in search of a mild phenotype. When the proband has a mutation in a known DBA gene, family members, even with normal evaluations, should be similarly tested. This information is of value with regard to reproductive choices and in the consideration of family members as potential hematopoietic stem cell donors. In the future the identification of a specific mutation may direct cancer-screening strategies or other clinical decisions. The deliberations of a consensus conference describing, in great detail, the current approach to diagnosis and treatment of DBA has recently been published 13.
Treatment and Outcomes
Corticosteroids and Red Cell Transfusions
Corticosteroids and red cell transfusions are the mainstays of therapy for DBA. Since 195184, it has been known that the anemia of DBA can be ameliorated by corticosteroids. The response to corticosteroids perpetuated the erroneous notion of DBA as an autoimmune disease, even when only miniscule doses were required to maintain adequate erythropoiesis. Clinically the almost unlimited use of corticosteroids, even when toxic doses were required, continued into the early 21st century. This appears to be due largely to the persistent fear, in patients and physicians, of transfusion-acquired HIV and Hepatitis C as well as the difficulty encountered in the almost daily subcutaneous administration of the iron chelator, deferoxamine (Desferal®, Novartis) 85. Data from international registries has clarified the efficacy and toxicity of corticosteroid therapy. Data from the DBAR are representative 54. As has been reported in the literature, 79% of patients are initially responsive to steroids, 17% were non-responsive and 4% of patients were never treated with steroids. Not surprising, nearly half of the patients had developed cushingoid features. However with the disturbing finding that 22% and 12% of patients ever treated with steroids developed pathologic fractures and cataracts respectively, the use of steroids has been modified. A snapshot of the DBAR reveals 37% of the patients receiving corticosteroids and 31% receiving red cell transfusions. Of the transfusion dependent patients, 35% were never steroid responsive, 22% became steroid refractory over time and 33% could not be weaned to an acceptable dose. Five per cent never received steroid therapy and 5% are being transfused for unknown reasons. Recently there appears to be an increase in the number of patients judged as “unable to be weaned” as well as an increase in patients who never received steroids. The number of patients never on corticosteroids reflects the youngest cohort for whom corticosteroid administration is being delayed during the critical period of growth and development during the first year of life54. The ability to wean a patient is dependent to some extent upon the side effects that the patient and the physician are willing to tolerate. Corticosteroids should commence with a corticosteroid-equivalent dose of prednisone of 2mg/kg/day. Patients who fail to respond within a month are considered steroid refractory and must receive red cell transfusions. In general, a corticosteroid dose-equivalent of 0.5 mg/kg/day (≤ 1 mg/kg/day every other day) of prednisone is suggested as a maximum “maintenance” dose. Clearly the recognition that the blood supply is safe combined with the recent availability of the oral iron chelator Deferasirox (ICL670, Exjade®, Novartis) 86 seems to have permitted a more liberal use of packed red cell transfusions in patients rather than toxic doses of corticosteroids. Thus patients should be closely monitored for steroid toxicity and the drug discontinued rather than risk significant complications.
Although remissions had been reported in DBA, data on the fraction of patients who were able to sustain erythropoiesis for over 6 months without treatment has been sparse. The DBAR has provided some actuarial data 54. About 20% of patients will enter remission, the majority sustained. Of these about 75% do so prior to their 10th birthday. These data may reflect the bias to a younger age of patients in the DBAR, underestimating the actuarial likelihood of remission, as anecdotal observations suggest a surge in remissions in adolescent males. Proportionally an equal number of patients remit from both transfusion and steroid therapy. However there are very few remitters who had never responded to corticosteroids. Pregnancy 87 and the use of birth control pills contribute to relapse.
Stem Cell Transplantation
Although curative in DBA 15,54,88 hematopoietic stem cell transplantation remains the most controversial aspect of therapy. A recent series from the IBMTR and a compilation from the literature are consistent with findings from the DBAR 89. At the last published evaluation 36 patients had undergone stem cell transplant (SCT); 21 HLA-matched related and 15 alternative donor SCT. The major indication for SCT was transfusion dependence. In addition, two patients had developed severe aplastic anemia and one significant thrombocytopenia. Two patients transplanted using a non-myeloablative conditioning regimen, one receiving a matched related umbilical cord and one receiving unrelated bone marrow, are alive and well. The majority of alternative donor transplants were performed using total body irradiation for conditioning whereas busulfan/cyclophosphamide containing regimens were typical of the matched-related transplants. Sixteen of the 21 HLA-matched sibling donor transplants are alive and red cell transfusion-independent. Of the 15 alternative donor SCT, 2 patients received mismatched related bone marrow, 4 patients unrelated cord blood, 8 unrelated bone marrow and one unrelated peripheral blood stem cells. Four of these 15 patients are alive. Of the 16 deaths 15 were related to infection, graft versus host disease and/or veno-occlusive disease of the liver with only 1 death, in the alternative donor group, occurring as a consequence of graft failure. In contrast to Fanconi anemia, dyskeratosis congenita and Shwachman Diamond syndrome, these deaths do not appear to be the consequence of intolerance to typical transplant conditioning regimens. The survival for allogeneic sibling versus alternative donor transplant is 72.7% ± 10.7% versus 19.1% ± 11.9% at greater than 5 years from SCT (p=0.01) or 17.1% ± 10.8% (including a patient diagnosed with osteogenic sarcoma post transplant, p=0.012). The survival rate for patients under the age of 10 years receiving matched related SCT is greater than 90%. Unpublished data from the DBAR on over 50 patients continues to show a greater than 90% actuarial survival for young DBA patients transplanted using matched allogeneic related donors. This success has led to an increase in families looking to preimplantation genetic diagnosis (PGD) with in vitro fertilization (IVF) to “create” HLA-matched, non-RPS19 mutated sibling donors. A number of patients worldwide have been successfully transplanted using umbilical cord derived stem cells from donors produced in this way. A discussion of the complicated religious, ethical and economic questions generated by this approach is beyond the scope of this review 90,91. Unpublished data from the DBAR show the actuarial survival, since 2000, for all DBA patients transplanted using alternative donors to be approximately 80%, offering this option to carefully selected patients lacking a suitable matched-related donor.
Of the 36 deaths reported to the DBAR at the last evaluation 25 (70%) were treatment-related: 5 from infections (2 pneumocystis jiroveci pneumonia, 1 varicella pneumonia, 1 pseudomonas pneumonia/sepsis, 1 unknown infection), 5 from complications of iron overload, 1 from cardiac tamponade secondary to a vascular access device complication, and 14 from stem cell transplant complications. Only 3 of the deaths related to stem cell transplantation were in patients with life-threatening cytopenias. Recent unpublished data shows an expected increase in reported iron overload-related deaths as the DBAR population matures. This finding is of considerable concern! Just eight deaths were directly related to DBA: 1 from severe aplastic anemia and 7 from malignancy (2 of whom also received a stem cell transplant), and 3 deaths were from undetermined causes. Thus although SCT is potentially curative and reduces the risk of aplastic anemia, AML and MDS, and the outcome for unrelated donor SCT has improved considerably reasonable restraint should be exercised in the use of this modality. The probability of remission must also be taken in to account when high-risk SCT is being considered.
The last reported overall actuarial survival at greater than 40 years of age is 75.1% ± 4.8; 86.7% ± 7.0% for corticosteroid-maintainable patients and 57.2% ± 8.9% for transfusion-dependent patients54. There is a statistically significant survival advantage for steroid-maintainable patients as compared to transfusion dependent patients (p=0.007). Seven transfusion-dependent patients died as a consequence of stem cell transplant-related complications. Clearly the next analysis will reflect both the increase in iron overload-related and the decrease in alternative donor transplant-related deaths, respectively.
Conclusions
The utility of patient registries for rare diseases has been clearly demonstrated in DBA. And these valuable resources need to be supported. These databases have, for example, permitted the more precise description of congenital anomalies in DBA revealing the connection between Diamond Blackfan anemia and Treacher Collins syndrome. Similarities between the two diseases were instrumental is recognizing DBA as disorder of ribosome biosynthesis. Other disorders involving defects in ribosome assembly and or function; cartilage hair hypoplasia, Shwachman Diamond syndrome and dyskeratosis congenita have also been described (reviewed in 79) and a new nosology of human disease is emerging. Figure 4 demonstrates the known defects in ribosome biosynthesis and function characterized by this new class of disorders. Motivated by newly recognized clinical relevance the study of ribosome biosynthesis and function is attracting more interested scientists.
Fig. 4. Putative biochemical function for Treacle, Dyskerin, Rps19/Rps24, RMRP, and Sbds in ribosomal biogenesis.

A simplified version of the human pre-rRNA processing pathway is shown. Functions are deduced from data for either yeast or human orthologs. Mature rRNA species (denoted in bold) are liberated from the 45S pre-rRNA primary transcript by a series of processing steps. 5′ and 3′ ETS represent external transcribed sequences found on the 5′ and 3′ ends of the primary transcript, respectively. ITS represents internal transcribed sequences present in the primary transcript that are not retained in mature rRNAs. The 18S rRNA is a component of the 40S ribosomal subunit. The 5.8S and 28S rRNAs, together with the 5S rRNA (not shown), are components of the 60S ribosomal subunit.
Extensive work is ongoing in a number of laboratories utilizing patient databases for DBA gene discovery. The complete genotyping of the patient databases should contribute to the discovery of the majority of the remaining DBA genes. Ultimately the availability of a genetically based DBA diagnosis in the remaining 50% of patients not mutated at the known “DBA genes” will allow the diagnosis of previously undetectable mild phenotypes, facilitating reproductive choices and SCT donor selection 92. The patient substrate provided by these registries will also allow for genotype-phenotype correlations that will shed light on scientific questions and at the same time suggest treatment and surveillance strategies. Within a few years an accurate cancer risk assessment will be available and perhaps cancer-prone genotypes will emerge. Furthermore these registries have provided a clearer understanding of the epidemiology of DBA. Treatment options are already being modified based upon new information regarding the natural history of DBA. Prolonged use of corticosteroids, starting in infancy, has been curtailed as the result of this new knowledge. Packed red cell transfusions, rather than corticosteroids, are being used to avoid steroid-related growth retardation, developmental delay, pathological fractures, cataracts and other significant steroid-related side effects. Analyses of outcomes reveal that the vast majority of patients succumb to complications that may be avoidable. The serious risk of death from transfusion-related iron overload has been clearly established. The extremely poor outcome previously reported with alternative donor hematopoietic stem cell transplantation has improved dramatically. A greater than expected incidence of remission and a lower than expected incidence of aplastic anemia, myelodysplastic syndrome and hematopoietic malignancy, still support restraint in the use of alternative donor stem cell transplants. In contrast, allogeneic matched sibling SCT appears to have an acceptable outcome, justifying its use as frontline therapy. In such a rare disease as DBA controlled stem cell transplant clinical trials are virtually impossible to perform. Thus, emerging therapies such as SCT utilizing reduced intensity conditioning regimens in high-risk DBA patients must be captured by the registries and provide a reasonable evaluation of the efficacy of this approach. As data mature the predisposition to non-hematopoietic malignancy in the context of stem cell transplantation will also become known. The registries also provide the platform for clinical trials. The availability of tumor specimens, obtained from well-characterized patients, will allow for an investigation of the molecular biology of tumorigenesis in DBA. The recognition of non-classical occult DBA phenotypes strongly suggests that patients with characteristic birth defects, certain cancers or unexplained macrocytosis may in fact have DBA. The clarification of these issues is critical with regard to patient follow-up, genetic counseling and reproductive strategies. Preliminary work setting the stage for gene therapy trials is ongoing 93.
Our understanding of the molecular underpinnings of DBA has also pointed the way to new potential therapies. The observation that DBA may be a “ribosome deficiency” disease has led Pospisilova et al. 94 to test the efficacy of nutritional supplementation with the amino acid leucine as a potential therapy for DBA. This approach is based on the documented role of this amino acid in up-regulating components of the protein synthetic machinery in skeletal muscle 95. Positive results have so far been reported in a single patient 94. A clinical trial for the efficacy of leucine supplementation in ameliorating the bone marrow failure in DBA is currently being designed in the U.S. The finding that inhibitors of p53 can reverse phenotypes in mouse models of DBA and TCS also points to new treatment modalities 24,27,65,96. While p53 inhibitors would pose an unacceptable risk for cancer in patient populations, perhaps small molecules inhibitors of the signaling pathways linking abortive ribosome assembly to p53 activation can provide clinical benefit without increasing cancer risk. Finally, the recent observation that the loss of a small subunit ribosomal protein gene, RPS14, is responsible for the refractory anemia in 5q- syndrome has raised the possibility the lenalidomide may have some potential clinical benefit in DBA 97.
The study of DBA, since its first description 70 years ago, has yielded extraordinary opportunities to understand the biology of hematopoiesis, oncogenesis and morphogenesis. As important, significant improvements in patient care have emerged and should continue with a greater knowledge of disease mechanism.
Acknowledgments
This work was supported by grants from the Daniella Maria Arturi Foundation, Diamond Blackfan Anemia Foundation, Pediatric Cancer Foundation, National Institutes of Health R01 HL 079571 and R01HL 079583 and the Feinstein Institute for Medical Research at the NSLIJ General Clinical Research Center M01 RR018535.
There has been considerable progress made in our understanding of Diamond Blackfan anemia since the identification of the first gene mutated in this disorder in 1997. The commitment of the patients with DBA, their families, their clinicians, and the dedicated scientists, clinical and laboratory based, with whom we have the privilege to work is largely responsible for this progress. Improvement in the quantity and quality of life for patients with DBA is one of the great successes of twenty-first century translational medicine. The authors would also like to thank, in particular, Adrianna Vlachos, M.D., Johnson Liu, M.D., Eva Atsidaftos, M.A., and Hanna Gazda, M.D for their invaluable advice and assistance. One of us (JML) would like to thank David G. Nathan, M.D. and Blanche Alter, M.D., M.P.H. for their introduction to the field of bone marrow failure. We owe a considerable debt of gratitude to Marie and Manny Arturi and the Diamond Blackfan Anemia Foundation who have supported and sustained the science and the scientists who work in this area. Indeed the marriage of laboratory and clinical science to the management of children and adults with DBA has been brokered by and large through their efforts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young NSAB. Inherited Bone Marrow Failure Syndromes: Introduction. Philadelphia: WB Saunders Company; 1994. [Google Scholar]
- 2.Gripp KW, McDonald-McGinn DM, La Rossa D, et al. Bilateral microtia and cleft palate in cousins with Diamond-Blackfan anemia. Am J Med Genet. 2001;101:268–274. doi: 10.1002/ajmg.1329. [DOI] [PubMed] [Google Scholar]
- 3.Lipton JM, Federman N, Khabbaze Y, et al. Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol. 2001;23:39–44. doi: 10.1097/00043426-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Dianzani I, Loreni F. Diamon\d-Blackfan anemia: a ribosomal puzzle. Haematologica. 2008;93:1601–1604. doi: 10.3324/haematol.2008.000513. [DOI] [PubMed] [Google Scholar]
- 5.Josephs H. Anemia of infancy and early childhood. Medicine. 1936;15:307–451. [Google Scholar]
- 6.Diamond LK, Blackfan KD. Hypoplastic Anemia. American Journal of Diseases of Children. 1938;56:464–467. [Google Scholar]
- 7.Diamond LK, Wang WC, Alter BP. Congenital hypoplastic anemia. Adv Pediatr. 1976;22:349–378. [PubMed] [Google Scholar]
- 8.Glader BE, Backer K, Diamond LK. Elevated erythrocyte adenosine deaminase activity in congenital hypoplastic anemia. N Engl J Med. 1983;309:1486–1490. doi: 10.1056/NEJM198312153092404. [DOI] [PubMed] [Google Scholar]
- 9.Vlachos A, Klein GW, Lipton JM. The Diamond Blackfan Anemia Registry: tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. J Pediatr Hematol Oncol. 2001;23:377–382. doi: 10.1097/00043426-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Gustavsson P, Garelli E, Draptchinskaia N, et al. Identification of microdeletions spanning the Diamond-Blackfan anemia locus on 19q13 and evidence for genetic heterogeneity. Am J Hum Genet. 1998;63:1388–1395. doi: 10.1086/302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 12.Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol. 2004;125:243–252. doi: 10.1111/j.1365-2141.2004.04890.x. [DOI] [PubMed] [Google Scholar]
- 13.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balaban EP, Buchanan GR, Graham M, Frenkel EP. Diamond-Blackfan syndrome in adult patients. Am J Med. 1985;78:533–538. doi: 10.1016/0002-9343(85)90352-3. [DOI] [PubMed] [Google Scholar]
- 15.Willig TN, Niemeyer CM, Leblanc T, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Societe d’Hematologie et d’Immunologie Pediatrique (SHIP), Gesellshaft fur Padiatrische Onkologie und Hamatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr Res. 1999;46:553–561. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Campagnoli MF, Garelli E, Quarello P, et al. Molecular basis of Diamond-Blackfan anemia: new findings from the Italian registry and a review of the literature. Haematologica. 2004;89:480–489. [PubMed] [Google Scholar]
- 17.Ohga S, Mugishima H, Ohara A, et al. Diamond-Blackfan anemia in Japan: clinical outcomes of prednisolone therapy and hematopoietic stem cell transplantation. Int J Hematol. 2004;79:22–30. doi: 10.1007/BF02983529. [DOI] [PubMed] [Google Scholar]
- 18.Freedman MH, Amato D, Saunders EF. Erythroid colony growth in congenital hypoplastic anemia. J Clin Invest. 1976;57:673–677. doi: 10.1172/JCI108323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nathan DG, Clarke BJ, Hillman DG, Alter BP, Housman DE. Erythroid precursors in congenital hypoplastic (Diamond-Blackfan) anemia. J Clin Invest. 1978;61:489–498. doi: 10.1172/JCI108960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipton JM, Kudisch M, Gross R, Nathan DG. Defective erythroid progenitor differentiation system in congenital hypoplastic (Diamond-Blackfan) anemia. Blood. 1986;67:962–968. [PubMed] [Google Scholar]
- 21.Chan HS, Saunders EF, Freedman MH. Diamond-Blackfan syndrome. II. In vitro corticosteroid effect on erythropoiesis. Pediatr Res. 1982;16:477–478. doi: 10.1203/00006450-198206000-00015. [DOI] [PubMed] [Google Scholar]
- 22.Matsson H, Davey EJ, Frojmark AS, et al. Erythropoiesis in the Rps19 disrupted mouse: Analysis of erythropoietin response and biochemical markers for Diamond-Blackfan anemia. Blood Cells Mol Dis. 2006;36:259–264. doi: 10.1016/j.bcmd.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Alter BP, Gaston T, Lipton JM. Lack of effect of corticosteroids in W/Wv and S1/S1d mice: these strains are not a model for steroid-responsive Diamond-Blackfan anemia. Eur J Haematol. 1993;50:275–278. doi: 10.1111/j.1600-0609.1993.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 24.Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008 doi: 10.1182/blood-2008-01-132290. [DOI] [PubMed] [Google Scholar]
- 25.Uechi T, Nakajima Y, Chakraborty A, Torihara H, Higa S, Kenmochi N. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum Mol Genet. 2008;17:3204–3211. doi: 10.1093/hmg/ddn216. [DOI] [PubMed] [Google Scholar]
- 26.Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGowan KA, Li JZ, Park CY, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohene-Abuakwa Y, Orfali KA, Marius C, Ball SE. Two-phase culture in Diamond Blackfan anemia: localization of erythroid defect. Blood. 2005;105:838–846. doi: 10.1182/blood-2004-03-1016. [DOI] [PubMed] [Google Scholar]
- 29.Perdahl EB, Naprstek BL, Wallace WC, Lipton JM. Erythroid failure in Diamond-Blackfan anemia is characterized by apoptosis. Blood. 1994;83:645–650. [PubMed] [Google Scholar]
- 30.Tsai PH, Arkin S, Lipton JM. An intrinsic progenitor defect in Diamond-Blackfan anaemia. Br J Haematol. 1989;73:112–120. doi: 10.1111/j.1365-2141.1989.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 31.Ellis SR, Lipton JM. Diamond Blackfan anemia: a disorder of red blood cell development. Curr Top Dev Biol. 2008;82:217–241. doi: 10.1016/S0070-2153(07)00008-7. [DOI] [PubMed] [Google Scholar]
- 32.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 33.Gazda HT, Grabowska A, Merida-Long LB, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79:1110–1118. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue M-F, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, Ball SE, Niewiadomska E, Matysiak M, Zaucha JM, Glader B, Niemeyer C, Meerpohl JJ, Atsidaftos E, Lipton JM, Gleizes PE, Beggs AH. Ribosomal Protein L5 and L11 Mutations Are Associated with Cleft Palate and Abnormal Thumbs in Diamond-Blackfan Anemia Patients. Am J Hum Genet. 2008 doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Idol RA, Robledo S, Du HY, et al. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis. 2007;39:35–43. doi: 10.1016/j.bcmd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Flygare J, Aspesi A, Bailey JC, et al. Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood. 2007;109:980–986. doi: 10.1182/blood-2006-07-038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choesmel V, Bacqueville D, Rouquette J, et al. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109:1275–1283. doi: 10.1182/blood-2006-07-038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. doi: 10.1128/mcb.14.11.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Wolf GW, Bhat K, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Held WA, Ballou B, Mizushima S, Nomura M. Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J Biol Chem. 1974;249:3103–3111. [PubMed] [Google Scholar]
- 44.Hage AE, Tollervey D. A surfeit of factors: why is ribosome assembly so much more complicated in eukaryotes than bacteria? RNA Biol. 2004;1:10–15. [PubMed] [Google Scholar]
- 45.Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007;17:749–760. doi: 10.1016/j.cub.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D, Zhang Z, Li M, et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26:5029–5037. doi: 10.1038/sj.onc.1210327. [DOI] [PubMed] [Google Scholar]
- 47.Horn HF, Vousden KH. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene. 2008;27:5774–5784. doi: 10.1038/onc.2008.189. [DOI] [PubMed] [Google Scholar]
- 48.Lindstrom MS, Deisenroth C, Zhang Y. Putting a finger on growth surveillance: insight into MDM2 zinc finger-ribosomal protein interactions. Cell Cycle. 2007;6:434–437. doi: 10.4161/cc.6.4.3861. [DOI] [PubMed] [Google Scholar]
- 49.Maggi LB, Jr, Kuchenruether M, Dadey DY, et al. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol Cell Biol. 2008;28:7050–7065. doi: 10.1128/MCB.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brady SN, Yu Y, Maggi LB, Jr, Weber JD. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol Cell Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cathie IAB. Erythrogenesis imperfecta. Arch Dis. 1950;25:313–324. doi: 10.1136/adc.25.124.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aase JMSD. Congenital anemia and triphalangeal thumbs: A new syndrome. J Pediatr. 1969;74:471–472. doi: 10.1016/s0022-3476(69)80208-8. [DOI] [PubMed] [Google Scholar]
- 53.Alter BP. Thumbs and anemia. Pediatrics. 1978;62:613–614. [PubMed] [Google Scholar]
- 54.Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006;46:558–564. doi: 10.1002/pbc.20642. [DOI] [PubMed] [Google Scholar]
- 55.Teber OA, Gillessen-Kaesbach G, Fischer S, et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004;12:879–890. doi: 10.1038/sj.ejhg.5201260. [DOI] [PubMed] [Google Scholar]
- 56.Fazen LE, Elmore J, Nadler HL. Mandibulo-facial dysostosis. (Treacher-Collins syndrome) Am J Dis Child. 1967;113:405–410. doi: 10.1001/archpedi.1967.02090190051001. [DOI] [PubMed] [Google Scholar]
- 57.Lipton JMAB. Diamond Blackfan anemia. Boca Raton, FL: CRC Press; 1993. [Google Scholar]
- 58.Wise CA, Chiang LC, Paznekas WA, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc Natl Acad Sci U S A. 1997;94:3110–3115. doi: 10.1073/pnas.94.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.So RB, Gonzales B, Henning D, Dixon J, Dixon MJ, Valdez BC. Another face of the Treacher Collins syndrome (TCOF1) gene: identification of additional exons. Gene. 2004;328:49–57. doi: 10.1016/j.gene.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 60.Dixon J, Edwards SJ, Anderson I, Brass A, Scambler PJ, Dixon MJ. Identification of the complete coding sequence and genomic organization of the Treacher Collins syndrome gene. Genome Res. 1997;7:223–234. doi: 10.1101/gr.7.3.223. [DOI] [PubMed] [Google Scholar]
- 61.Dixon MJ. Treacher Collins syndrome. Hum Mol Genet. 1996;5(1391–1396) doi: 10.1093/hmg/5.supplement_1.1391. [DOI] [PubMed] [Google Scholar]
- 62.Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci U S A. 2004;101:10709–10714. doi: 10.1073/pnas.0402492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzales B, Henning D, So RB, Dixon J, Dixon MJ, Valdez BC. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum Mol Genet. 2005;14:2035–2043. doi: 10.1093/hmg/ddi208. [DOI] [PubMed] [Google Scholar]
- 64.Dixon J, Brakebusch C, Fassler R, Dixon MJ. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum Mol Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
- 65.Jones NC, Lynn ML, Gaudenz K, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alter BP. Inherited bone marrow failure syndromes. Philadelphia, PA: WB Saunders; 2003. [Google Scholar]
- 67.Yaris N, Erduran E, Cobanoglu U. Hodgkin lymphoma in a child with Diamond Blackfan anemia. J Pediatr Hematol Oncol. 2006;28:234–236. doi: 10.1097/01.mph.0000210413.63478.91. [DOI] [PubMed] [Google Scholar]
- 68.Janov AJ, Leong T, Nathan DG, Guinan EC. Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine (Baltimore) 1996;75:77–78. doi: 10.1097/00005792-199603000-00004. [DOI] [PubMed] [Google Scholar]
- 69.Alter BP, Greene MH, Velazquez I, Rosenberg PS. Cancer in Fanconi anemia. Blood. 2003;101:2072. doi: 10.1182/blood-2002-11-3597. [DOI] [PubMed] [Google Scholar]
- 70.Rosenberg PS, Alter BP, Link DC, et al. Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol. 2008;140:210–213. doi: 10.1111/j.1365-2141.2007.06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lensch MW, Rathbun RK, Olson SB, Jones GR, Bagby GC., Jr Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: a view from the window of Fanconi anemia. Leukemia. 1999;13:1784–1789. doi: 10.1038/sj.leu.2401586. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Sejas DP, Zhang X, et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest. 2007;117:3283–3295. doi: 10.1172/JCI31772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol. 2007;27:1056–1068. doi: 10.1128/MCB.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21:4246–4255. doi: 10.1128/MCB.21.13.4246-4255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boon K, Caron HN, van Asperen R, et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20:1383–1393. doi: 10.1093/emboj/20.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oskarsson T, Trumpp A. The Myc trilogy: lord of RNA polymerases. Nat Cell Biol. 2005;7:215–217. doi: 10.1038/ncb0305-215. [DOI] [PubMed] [Google Scholar]
- 77.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 78.Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu JM, Ellis SR. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107:4583–4588. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- 80.Morimoto K, Lin S, Sakamoto K. The functions of RPS19 and their relationship to Diamond-Blackfan anemia: a review. Mol Genet Metab. 2007;90:358–362. doi: 10.1016/j.ymgme.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 81.Palis J, Segel GB. Developmental biology of erythropoiesis. Blood Rev. 1998;12:106–114. doi: 10.1016/s0268-960x(98)90022-4. [DOI] [PubMed] [Google Scholar]
- 82.Weinberg RS, He LY, Alter BP. Erythropoiesis is distinct at each stage of ontogeny. Pediatr Res. 1992;31:170–175. doi: 10.1203/00006450-199202000-00016. [DOI] [PubMed] [Google Scholar]
- 83.Dessypris EN, Krantz SB, Roloff JS, Lukens JN. Mode of action of the IgG inhibitor of erythropoiesis in transient erythroblastopenia of children. Blood. 1982;59:114–123. [PubMed] [Google Scholar]
- 84.Gasser C. Aplastische anämie (cronische erythroblastophthise) und cortison. Schhhweiz Med Wochenschr. 1951;81:1241–1242. [PubMed] [Google Scholar]
- 85.Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997;89:739–761. [PubMed] [Google Scholar]
- 86.Nisbet-Brown E, Olivieri NF, Giardina PJ, et al. Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet. 2003;361:1597–1602. doi: 10.1016/S0140-6736(03)13309-0. [DOI] [PubMed] [Google Scholar]
- 87.Alter BP, Kumar M, Lockhart LL, Sprinz PG, Rowe TF. Pregnancy in bone marrow failure syndromes: Diamond-Blackfan anaemia and Shwachman-Diamond syndrome. Br J Haematol. 1999;107:49–54. doi: 10.1046/j.1365-2141.1999.01668.x. [DOI] [PubMed] [Google Scholar]
- 88.Vlachos A, Federman N, Reyes-Haley C, Abramson J, Lipton JM. Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant. 2001;27:381–386. doi: 10.1038/sj.bmt.1702784. [DOI] [PubMed] [Google Scholar]
- 89.Roy V, Perez WS, Eapen M, et al. Bone marrow transplantation for diamond-blackfan anemia. Biol Blood Marrow Transplant. 2005;11:600–608. doi: 10.1016/j.bbmt.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 90.Kuliev A, Rechitsky S, Tur-Kaspa I, Verlinsky Y. Preimplantation genetics: Improving access to stem cell therapy. Ann N Y Acad Sci. 2005;1054:223–227. doi: 10.1196/annals.1345.028. [DOI] [PubMed] [Google Scholar]
- 91.Wagner JE, Kahn JP, Wolf SM, Lipton JM. Preimplantation testing to produce an HLA-matched donor infant. JAMA. 2004;292:803–804. doi: 10.1001/jama.292.7.803-b. author reply 804. [DOI] [PubMed] [Google Scholar]
- 92.Wynn RF, Grainger JD, Carr TF, Eden OB, Stevens RF, Will AM. Failure of allogeneic bone marrow transplantation to correct Diamond-Blackfan anaemia despite haemopoietic stem cell engraftment. Bone Marrow Transplant. 1999;24:803–805. doi: 10.1038/sj.bmt.1701982. [DOI] [PubMed] [Google Scholar]
- 93.Flygare J, Olsson K, Richter J, Karlsson S. Gene therapy of Diamond Blackfan anemia CD34(+) cells leads to improved erythroid development and engraftment following transplantation. Exp Hematol. 2008;36:1428–1435. doi: 10.1016/j.exphem.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 94.Pospisilova D, Cmejlova J, Hak J, Adam T, Cmejla R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica. 2007;92:e66–67. doi: 10.3324/haematol.11498. [DOI] [PubMed] [Google Scholar]
- 95.Anthony JC, Yoshizawa F, Anthony TG, Vary TC, Jefferson LS, Kimball SR. Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J Nutr. 2000;130:2413–2419. doi: 10.1093/jn/130.10.2413. [DOI] [PubMed] [Google Scholar]
- 96.Sakai D, Trainor PA. Treacher Collins syndrome: Unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 2008 doi: 10.1016/j.biocel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451:335–339. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]