

Abstract
Near-IR MCD and variable temperature, variable field (VTVH) MCD have been applied to Naphthalene 1,2-dioxygenase (NDO) to describe the coordination geometry and electronic structure of the mononuclear non-heme ferrous catalytic site in the resting and substrate-bound forms with the Rieske 2Fe2S cluster oxidized and reduced. The structural results are correlated with the crystallographic studies of NDO and other related Rieske non-heme iron oxygenases to develop molecular level insights into the structure/function correlation for this class of enzymes. The MCD data for resting NDO with the Rieske center oxidized indicate the presence of a six-coordinate high-spin ferrous site with a weak axial ligand which becomes more tightly coordinated when the Rieske center is reduced. Binding of naphthalene to resting NDO (Rieske oxidized and reduced) converts the six-coordinate sites into five-coordinate (5c) sites with elimination of a water ligand. In the Rieske oxidized form the 5c sites are square pyramidal, but transform to a 1:2 mixture of trigonal bipyramial/square pyramidal sites when the Rieske center is reduced. Thus the geometric and electronic structure of the catalytic site in the presence of substrate can be significantly affected by the redox state of the Rieske center. The catalytic ferrous site is primed for the O2 reaction when substrate is bound in the active site in the presence of the reduced Rieske site. These structural changes insure that two electrons and the substrate are present before the binding and activation of O2, which avoids the uncontrolled formation and release of reactive oxygen species.
Introduction
Non-heme iron containing oxygenases mediate a wide range of chemical transformations where the coordination geometry and the electronic structure of the iron site are thought to be optimized for catalytic function.1,2 Much less had been known about the active site in the non-heme iron containing oxygenases relative to the heme systems as the non-heme iron centers are less spectroscopically accessible, particularly at the ferrous oxidation level. Our group has developed a methodology for the analysis of non-heme ferrous active sites emphasizing magnetic circular dichroism (MCD) spectroscopy to observe the ligand-field (LF) excited-states and variable temperature variable field (VTVH) MCD spectroscopy to obtain the ground-state sublevel splittings for these non-Kramers ions that rarely exhibit electron paramagnetic resonance (EPR) signals.3 This MCD methodology has been applied to study α-ketoglutarate-dependent dioxygenases,4-9 extradiol dioxygenases,10,11 and pterin-dependent hydroxylases,12-14 providing insight into the coordination geometry and electronic structure of the catalytic ferrous site. However, our knowledge of Rieske dioxygenases has been relatively limited due to the difficulty in obtaining spectroscopic information on the mononuclear ferrous catalytic site in the presence of the Rieske 2Fe2S cluster in this class of enzymes.
Rieske dioxygenases catalyze the stereo- and regio-specific dioxygenation of aromatic compounds to cis-dihydrodiols, as a first step in the metabolism of aromatic compounds in bacteria.15 They biodegrade toxic aromatic compounds, thus attracting interest in the development of bioremediation technology15 and are useful for synthetic applications.16
Recent x-ray crystallographic studies of the oxygenase component of Rieske non-heme iron oxygenases (ROs)17 have shown common structural features of a mononuclear non-heme iron site and a Rieske 2Fe2S cluster.18-32 The mononuclear iron site activates O2 for reaction with substrate, while the Rieske center transfers an electron to the mononuclear iron site during the catalytic cycle. The crystal structures of ROs show that each α-subunit contains a Rieske 2Fe2S cluster and a mononuclear non-heme iron center separated by ~44 Å. But the functional pair appears to involve the Rieske cluster and mononuclear non-heme iron in neighboring subunits separated by ~12 Å (Figure 1). The mononuclear non-heme iron site is coordinated by two histidine residues and one carboxylate residue, termed a 2-His-1-Carboxylate facial triad, which is a versatile platform of non-heme iron containing oxygenases.33 In most RO crystal structures, the carboxylate residue is bidentately coordinated to the iron,19-25,28,29 although a monodentate structure is also reported in a few cases.26,27,30-32 Additional water molecules are found to be coordinated to the iron, forming a five- or six-coordinate catalytic ferrous site, depending on the number water molecules. The Rieske diiron centers are bridged by two sulfides, one iron center is terminally coordinated by two histidine residues and the other iron by two cysteine residues forming four-coordinate iron sites. The catalytic site and electron transfer site can be bridged by a conserved aspartic acid residue present at the subunit interface, which may be important for electron transfer34 or regulation35,36 during the catalytic cycle. Several spectroscopic studies have been performed on this class of enzymes, including resonance Raman,37,38 electron nuclear double resonance (ENDOR),39-43 EPR,35,36,44-49 nuclear magnetic resonance (NMR),50 extended x-ray absorption fine structure (EXAFS),51,52 and x-ray absorption near edge spectroscopy (XANES),51,52 but most of those studies have focused on the nature of the Rieske center or indirectly probed the catalytic ferrous site using metal substituted or nitric-oxide (NO) bound enzymes. The mononuclear ferrous site is far less studied because it is more difficult to access spectroscopically.
Figure 1.
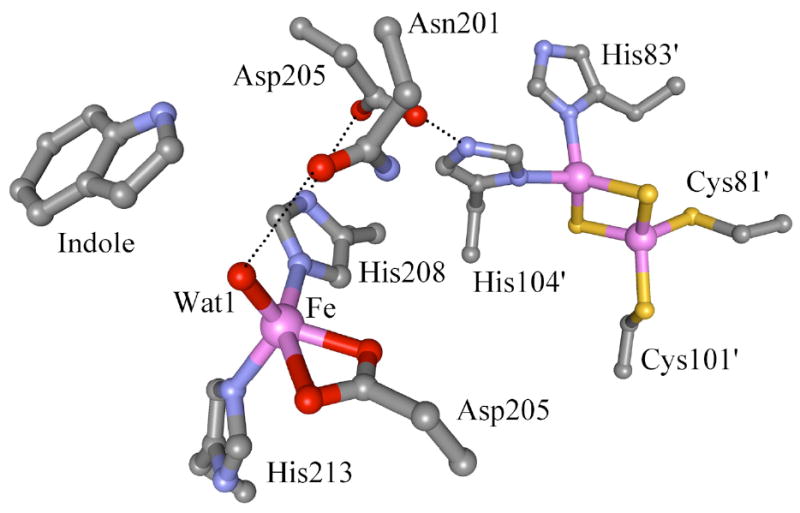
One of the specific crystal structures of Rieske dioxygenase from naphthalene dioxygenase (NDO) with bound indole (PDB: 1EG9)
One of the most thoroughly studied Rieske dioxygenases, naphthalene 1,2-dioxygenase (NDO), catalyzes the conversion of naphthalene to cis-(1R,2S)-dihydroxy-1,2-dihydrohaphthalene in the assimilation of carbon from naphthalene (Scheme 1).53,54 Spectroscopic and kinetic studies established that NDO can catalyze the cis-dihydroxylation of naphthalene in a single-turnover reaction if both the Rieske and mononuclear non-heme iron centers are reduced.45 It was further shown that the binding of the O2 surrogate nitric oxide (NO) to the ferrous catalytic site is regulated by both binding of substrate and the reduction of the Rieske 2Fe2S cluster, suggesting that structural reorganization occurs at the vicinity of the ferrous site during these steps.45 There is, in fact, evidence from crystallography for an allosteric effect of the redox state of the Rieske site in 2-oxoquinoline 8-monooxygenase (OMO).25 It was found that reduction of the Rieske 2Fe2S cluster modulates the resting ferrous catalytic site through a chain of conformational changes across the subunit interface, resulting in the displacement of the non-heme iron and its histidine ligand away from a substrate-binding site. The resting ferrous catalytic site changes its coordination number from five-to six-coordinate, which would be relevant to the inhibition of NO binding to resting ferrous sites with the Rieske center reduced as found for NDO45 and other RDOs.36,47,48 However, the crystal structure of the catalytic ferrous site in NDO with a reduced Rieske center is modeled as five-coordinate,21 inconsistent with the results found for OMO.
Scheme 1.
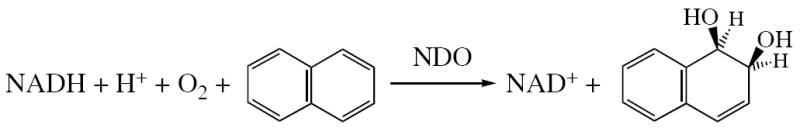
Binding of a substrate to the active site of RDOs is a key step in regulation of the reactivity toward O2. Using near infrared (NIR) MCD spectroscopy, we and others have previously studied the Rieske dioxygenase, phthalate dioxygenase (PDO),55,56 and found that the resting PDO enzyme with the Rieske center oxidized showed features indicative of a six-coordinate ferrous site; substrate binding converted the site to two different five-coordinate species. This six- to five-coordinate conversion upon substrate binding to the resting enzyme is in line with a general mechanistic strategy observed for other non-heme iron containing oxygenases where substrate binding leads to a coordinatively unsaturated ferrous site for the O2 reaction.1,2,57 However, the crystal structures of the catalytic ferrous sites in the resting and substrate-bound forms of NDO (with the Rieske center reduced) did not show a significant change in coordination environment maintaining five-coordinate structures.21 In addition, the five-coordinate ferrous catalytic site in OMO with the Rieske center oxidized also does not show a change in coordination number when a substrate is bound to the active site.25 Thus spectroscopy and currently available crystal structure data have provided different descriptions of the structural mechanism for the catalytic ferrous site in ROs.
Thus it is important to study a Rieske dioxygenase such as NDO with NIR MCD and VTVH MCD where crystallographic information is available for correlation with spectroscopy to determine whether the coordination geometry and electronic structure of the catalytic ferrous site are synergistically modulated with binding of substrate and reduction of the Rieske center. It should be noted that temperature-dependent MCD intensity requires a paramagnetic ground-state. Since the oxidized Rieske center has two ferric S = 5/2 Fe(III) ions antiferromagnetically coupled to generate an S = 0 ground-state, it will not contribute to the temperature-dependent MCD signal. The reduced Rieske site has an S = 5/2 Fe(III) ion antiferromagnetically coupled to an S = 2 Fe(II) ion to generate an S = 1/2 ground-state. While this is paramagnetic and will contribute to the temperature dependent MCD, our previous studies have shown that in the ferrous d-d transition energy region, the contributions of the reduced Rieske center are comparable in magnitude to that of the ferrous catalytic site.55 This signal can be subtracted out, using an apoenzyme which lacks the catalytic ferrous center but contains the Rieske site.
Herein we present NIR MCD and VTVH MCD analyses of the NDO enzyme addressing the coordination geometry and the electronic structure of the catalytic ferrous site in the resting and substrate-bound forms with both oxidized and reduced Rieske sites. These studies allow correlation with the crystal structures of NDO and other ROs, and provide molecular level mechanistic insights for this class of enzymes.
Experimental Section
Chemicals
All commercial reagents were used without further purification: MES (Sigma), naphthalene (99+ %; ACROS), EDTA (99+ %; Aldrich), 2,2’-dipyridyl (99 > %; Fluka), D2O (99.9 % atom % D; Cambridge Isotope Laboratories), glycerol-d3 (98 % atom % D; Cambridge Isotope Laboratories), NaOD (Sigma), sodium dithionite (Sigma).
Protein and Sample Preparation
All experiments were performed using NDO purified from Pseudomonas sp. NCIB 9816-4, as described previously.45 Mononuclear iron remaining in the isolated and purified NDO was removed by dialysis using a 100 mM MES buffer (pH 6.8) containing 10 mM EDTA and 5 mM 2,2’-dipyridyl for 15-20 h, followed by dialysis using a 100 mM MES buffer (pH 6.8) for 15-20 h in a cold room. The apo NDO enzyme with the Rieske center oxidized (apoNDOox) was further run through a Chelex-100 (Sigma) column in 100 mM MES (pH 6.8) to remove any remaining iron. The apoNDO samples were then exchanged into a deutrated 100mM MES buffer (pD 6.4), and diluted 50 % (v/v) by anaerobic addition of glycerol-d3. The concentrations of apoNDOox samples were determined via their absorbance at 280 nm, using ε = 129 mM (αβ)−1cm−1, as reported previously. The holo NDO enzyme, FeNDOox, was made by the addition of 0.9 equivalent of a degassed stock solution of Fe(NH4)2(SO4)2•6H2O in D2O to the apoNDOox-glycerol-d3 solution. For preparation of naphthalene-bound NDO samples, both apoNDOox and FeNDOox samples were exchanged into a naphthalene-saturated buffer (50 % (v/v) degassed glycerol/100 mM MES buffer at pD 6.4) by centrifugation in microcon concentrators (Microcon (Millipore)) in a refrigerated microfuge in a grove box. The concentration of naphthalene in the MES buffer/glycerol (50:50) was determined as ~400 μM (using ε = 6000 M-1cm-1 at 275 nm58).59 Reduced Rieske samples (apoNDOred and FeNDOred) were prepared by the anaerobic addition of excess sodium dithionite to the oxidized Riske samples of apoNDOox and FeNDOox.
NIR MCD Spectroscopy
NDO samples for NIR MCD spectroscopy were in the form of a glass with 50 % (v/v) degassed glycerol- d3/100 mM MES buffer at pD 6.4, and were typically 0.6 to 0.9 mM in active sites. Samples were injected anaerobically into a MCD cell made by compressing a 0.3 cm thick neoprene gasket between two quartz plates. Low-temperature NIR MCD spectra were obtained using either a JASCO 200D or JASCO 730 spectropolarimeter with a liquid N2 cooled InSb detector equipped with an Oxford Instruments Spectromag 4000 superconducting magnet/cryostat (Oxford SM4000-7 Tesla (T)). The MCD spectra shown were recorded at 5 K and 7 T with the natural CD (0 T) subtracted unless otherwise stated. To obtain the MCD spectrum of the mononuclear ferrous site of NDO, the MCD spectrum of a matched apoNDO sample was subtracted from that of FeNDO. The matched apoNDO sample came from the same degassed protein stock solution of FeNDO. VTVH MCD spectra were collected at a range of magnetic fields at each chosen temperature, measured using a calibrated Cernox resistor (Lakeshore Cryogenics, calibrated 1.5-300K) inserted into the sample cell to accurately measure the temperature. VTVH MCD data were collected at an energy where there is no contribution of the apoNDO, thus the VTVH behavior of the signal intensity can be associated with the mononuclear ferrous site. The VTVH MCD data were normalized to the maximum observed intensity over all isotherms for a given wavelength and the ground-state parameters were extracted by fitting in accordance with published procedures.60
Results and Analyses
NDOox
The 5 K 7 T MCD spectrum of resting NDO with the oxidized Rieske site is shown in Figure 2A. NIR MCD spectroscopy is a powerful method for observing the 5Eg excited-state LF splitting (Δ5Eg) of non-heme ferrous active sites, which allows assignment of ferrous coordination number (six-coordinate: two LF transitions centered at 10000 cm−1 split by ~2000 cm−1; five-coordinate: one LF transition at ~10000 cm−1 and second LF transition at ~5000 cm−1; four-coordinate: two LF transitions at ~5000-6000 cm−1).3,60 The low-temperature MCD spectrum of FeNDOox indicated by the gray line exhibits two peaks at ~8000 and 9700 cm−1, which includes spectroscopic contributions from both the oxidized Rieske site and the catalytic ferrous site. The apo spectrum shown by the gray dashed line is the temperature-independent MCD signal of the S = 0 diamagnetic oxidized Rieske site. The difference spectrum indicated with the dark green line obtained by subtracting the MCD spectrum of apoNDOox from that of FeNDOox revealed two d-d transitions centered at ~9300 cm−1 with the 5Eg split by 2750 cm−1 (Δ5Eg) indicative of a distorted six-coordinate ferrous site, in agreement with the previous MCD study of the resting PDO enzyme. The Δ5Eg splittings of resting NDO is relatively large compared to the splittings observed for PDO55,56 and other non-heme iron containing oxygenases,4-14 indicative of the presence of a relatively weak axial ligand at the ferrous site of resting NDO.
Figure 2.
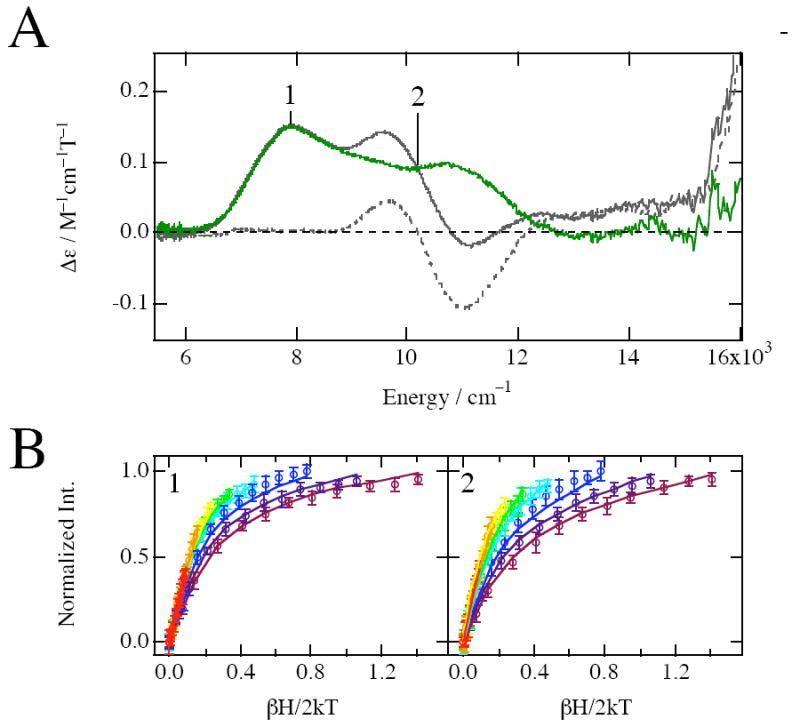
Ligand field MCD and VTVH MCD data for resting NDO with oxidized Rieske. (A) MCD spectra at 5K and 7T of resting NDO; FeNDOox (gray line), apoNDOox (gray dashed line), and the difference (FeNDOox – apoNDOox) (dark green line) spectra. (B) VTVH saturation magnetization behavior of resting NDO with oxidized Rieske measured at 7810 cm−1 (1) and 10200 cm−1 (2). The normalized data are plotted vs βH/2kT for a series of fixed temperatures (1.7, 2, 3, 5, 7, 10, 15, 25K). The temperature of isotherm curves increases as the color of curves changes from purple to red in the spectral color order. Errors in the intensities are also shown by error bars. The best fit (solid lines) to the data was generated by the parameters described in Table 1.
To obtain the ground-state ZFS of the S = 2 ferrous site, VTVH MCD data were taken at 7810 (position 1 in Figure 2A) and 10200 cm−1 (position 2 in Figure 2A), because the oxidized Rieske site does not exhibit MCD signal intensity at these energies. Figure 2B shows these data plotted vs βH/2kT (where β is the Bohr magneton and k is Boltzmann’s constant), along with the best fit to the data. The inverse relationship between MCD intensity and temperature is characteristic of C-term behavior arising from a degenerate electronic ground-state. The saturation magnetization behavior is well described by a negative ZFS non-Kramers doublet model (see ref 3 for details), and the ground-state spin Hamiltonian parameters obtained from the fit to the data are g∥ = 9.3 and δ = 3.7 cm−1 for both bands, as listed in Table 1 (δ is the energy splitting of the Ms = ±2 non-Kramers doublet ground state and g∥ is its Zeeman splitting). The difference in saturation behavior between the 7810 and 10200 cm−1 transition intensities reflects different contributions from B-terms and the polarization ratios, Mz/Mxy (Table 1). On the basis of these parameters, the 5T2g ground state energy splitting parameters (Δ = dxy − dxz,dyz; V = dxz − dyz) are determined (see ref 3) to be −Δ ~ 450 cm−1 and V ~ 160 cm−1 which are in the range found for distorted six-coordinate ferrous sites. Thus, from the MCD and VTVH MCD data, the resting NDO enzyme with an oxidized Rieske site contains a high-spin ferrous active site with a distorted six-coordinate geometry. The |V/2Δ| of 0.18 is far from the rhombic limit of 0.33, implying weak axial coordination to the catalytic ferrous site consistent with the relatively large Δ5Eg.
Table 1.
Summary of VTVH MCD analyses
| NDOox | NDOox-Naph | NDOred | NDOred-Naph | ||||
|---|---|---|---|---|---|---|---|
| transitions | 7950 | 10700 | 10290 | 8060 | 10350 | 8165 | 10575 |
| Δ5Eg | 2750 | > 5290 | 2290 | > 3165 | > 5575 | ||
| δ / cm-1 | 3.7 ± 0.1 | 3.7 ± 0.1 | 3.3 ± 0.1 | 3.0 ± 0.1 | 3.0 ± 0.1 | 1.2 ± 0.1b | 3.8 ± 0.1 |
| g | 9.3 ± 0.1 | 9.3 ± 0.1 | 8.9 ± 0.1 | 9.2 ± 0.1 | 9.3 ± 0.1 | 8.5 ± 0.1b | 9.0 ± 0.1 |
| −Δ / cm-1 | 450 ± 125 | 450 ± 125 | 800 ± 150 | 310 ± 50 | 350 ± 50 | -a | 700 ± 200 |
| |V/2Δ| | 0.18 ± 0.01 | 0.18 ± 0.01 | 0.30 ± 0.01 | 0.14 ± 0.01 | 0.14 ± 0.01 | -a | 0.30 ± 0.01 |
| V | 160 ± 50 | 160 ± 50 | 500 ± 110 | 90 ± 20 | 100 ± 20 | -a | 425 ± 135 |
| Mz/Mxy | −0.6 | −0.5 | −0.2 | 0.04 | −0.2 | −1.0b | −0.2 |
| B-term | 1.1 | 4.3 | 1.0 | 0.5 | 1.9 | 2.2b | 1.2 |
5T2g Hamiltonian not applicable to the trigonal bipyramidal 5E’’
VTVH MCD data analyses obtained at 8890 cm-1 provided δ of 1.4 cm-1 ± 0.1, g∥ of 8.5 ± 0.1, Mz/Mxy of -1.0 %, and B-term of 2.3 %
NDOox-Naph
Figure 3A shows the 5 K 7 T MCD spectra of naphthalene-bound holo- and apo-NDO with the Rieske site oxidized. The gray line is the spectrum of FeNDOox-Naph, the gray dashed line is of the apoNDOox-Naph, and the navy line is the difference spectrum (FeNDOox-Naph – apoNDOox-Naph). Although the apoNDOox-Naph spectrum is essentially the same as apoNDOox, the spectrum of FeNDOox-Naph exhibits a large increase in signal intensity at ~10000 cm−1 compared to the spectrum of FeNDOox (Figure 2A). The difference spectrum (navy line) clearly demonstrates the presence of a single intense MCD band at 10290 cm−1, which suggests formation of a five-coordinate ferrous site. A five-coordinate site also has a second ligand transition in the energy range of ~5000 cm−1, which is not observed in FeNDOox-Naph but can be below our instrument cutoff.
Figure 3.
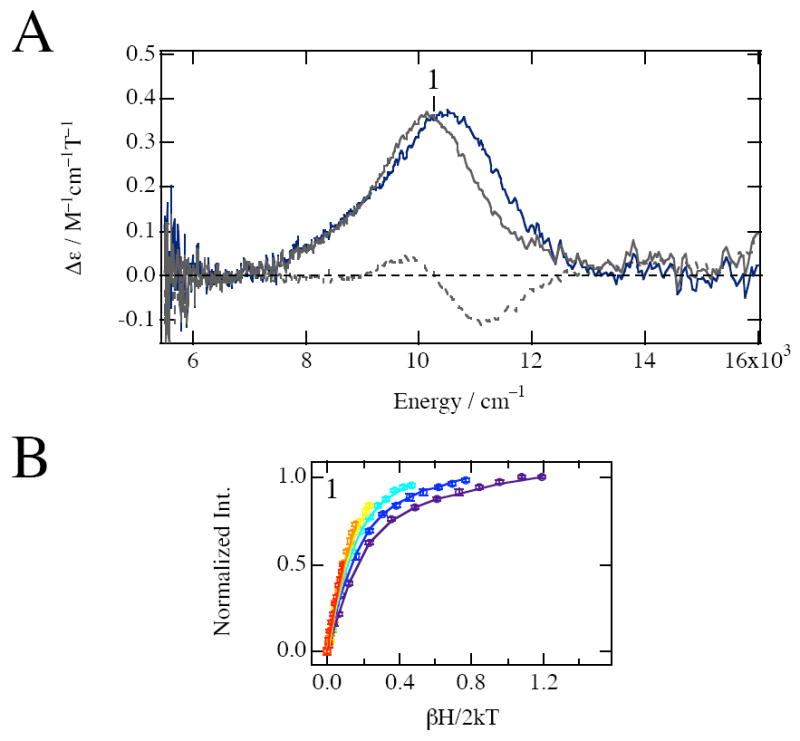
Ligand field MCD and VTVH MCD data for naphthalene-bound NDO with oxidized Rieske. (A) MCD spectra at 5K and 7T of naphthalene-bound NDO with oxidized Rieske; FeNDOox-Naph (gray line), apoNDOox-Naph (gray dashed line), and the difference (FeNDOox-Naph − apoNDOox-Naph) (dark blue line) spectra. (B) VTVH saturation magnetization behavior of naphthalene-bound NDO with oxidized Rieske measured at 10340 cm−1 (1). The normalized data are plotted vs βH/2kT for a series of fixed temperatures (2, 3, 5, 10, 15, 25K). The temperature of isotherm curves increases as the color of curves changes from purple to red in the spectral color order. Errors in the intensities are also shown by error bars. The best fit (solid lines) to the data was generated by the parameters described in Table 1.
VTVH MCD spectroscopy was applied to probe the ground-state splitting by monitoring the signal intensity at 10340 cm−1 (position 1 in Figure 3A) for a series of different fields at fixed temperatures. At this transition energy, only the mononuclear ferrous site contributes to the MCD signal intensity because the MCD spectrum of apoNDOox-Naph has a crossing point. Figure 3B shows these data plotted vs βH/2kT, along with the best fit to the data. The saturation magnetization behavior is well described by the negative ZFS non-Kramers model, and the ground-state spin Hamiltonian parameters are listed in Table 1. The decrease in the nesting of the isotherm curves in Figure 3B compared to the curves observed for a six-coordinate ferrous site in FeNDOox in Figure 2B reflects a smaller δ value of 3.3.61 Using the δ and g∥ from the VTVH MCD fit, the 5T2g energy splittings are −Δ ~ 800 and V ~ 500 cm−1, which are typical for a five-coordinate site. Thus, from the MCD spectrum and VTVH MCD data, FeNDOox-Naph contains a high-spin ferrous catalytic site with a five-coordinate geometry. The formation of a five-coordinate site is consistent with the high intensity of the MCD signal compared with the signal from a six-coordinate site in FeNDOox; A five-coordinate site would have increased absorption intensity relative to a six-coordinate site, because its low symmetry allows higher energy intense transitions to mix into parity forbidden d-d transitions. MCD intensity is proportional to absorption intensity.62
NDOred
The reduced Rieske site with a paramagnetic S = 1/2 ground-state shows C-term MCD intensity. Therefore, to access the spectroscopic data on the ferrous catalytic site in resting NDO with the Rieske site reduced, we recorded the MCD spectra of both FeNDOred and apoNDOred from the same batch of stock protein under identical conditions, and the apo contribution was subtracted from the FeNDOred spectrum. Figure 4A shows the 5 K 7 T MCD spectrum of resting NDO with the Rieske site reduced. The gray line is the spectrum of FeNDOred, the gray dashed line is of the apoNDOred, and the green line is their difference spectrum. The MCD spectrum of apoNDOred reveals that the signal due to the Rieske center has changed considerably, providing direct spectroscopic evidence that the two sets of samples are indeed in different oxidation states; conditions that are difficult to ascertain and maintain in crystallographic experiments. The difference spectrum shows two peaks centered around 9200 cm−1, implying the presence of a six-coordinate ferrous catalytic site. The smaller Δ5Eg splitting of 2290 cm−1 compared to the 2750 cm−1 splitting observed for the ferrous site in FeNDOox (in Figure 2A) indicates that the weak axial ligand in the non-heme ferrous site becomes stronger upon reduction of the Rieske site.
Figure 4.
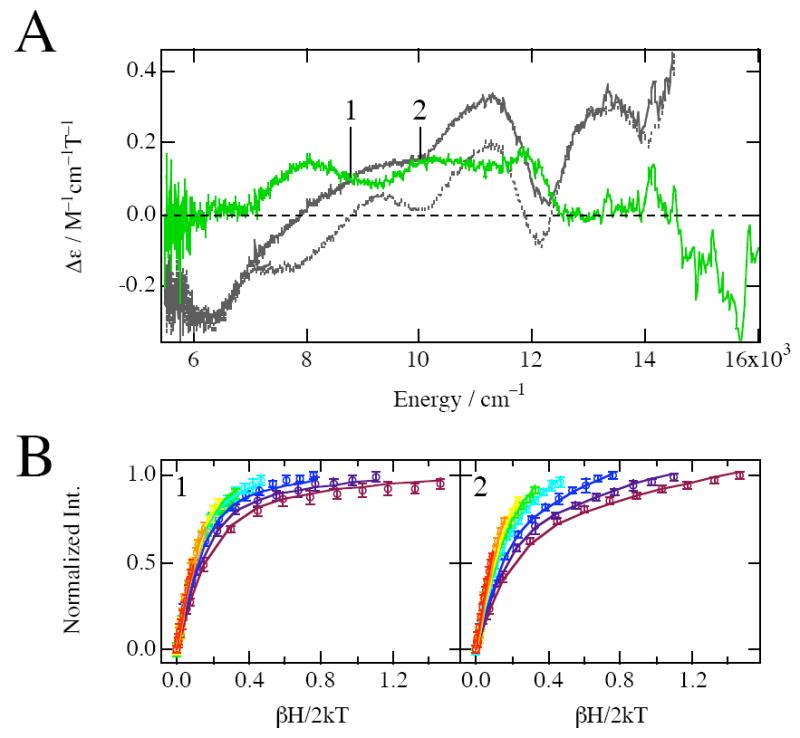
Ligand field MCD and VTVH MCD data for resting NDO with reduced Rieske. (A) MCD spectra at 5K and 7T of resting NDO with reduced Rieske; FeNDOred (gray line), apoNDOred (gray dashed line), and the difference (FeNDOred – apoNDOred) (light green line) spectra. (B) VTVH saturation magnetization behavior of resting NDO with reduced Rieske measured at 8810 cm−1 (1) and 10010 cm−1 (2). The normalized data are plotted vs βH/2kT for a series of fixed temperatures (1,7, 2, 3, 5, 7, 10, 15, 25K). The temperature of isotherm curves increases as the color of curves changes from purple to red in the spectral color order. Errors in the intensities are also shown by error bars. The best fit (solid lines) to the data was generated by the parameters described in Table 1.
VTVH MCD data on the non-heme ferrous site were obtained at 8810 cm−1 (position 1 in Figure 4A) and 10010 cm−1 (position 2 in Figure 4A) for a series of different fields at fixed temperatures. At these transition energies the apoNDOred spectrum of the S = 1/2 Rieske center (dashed line in Figure 4A) has no MCD signal intensity. Figure 4B shows these data plotted vs βH/2kT, along with the best fit to the data. The saturation magnetization behaviors at these transition energies are well described by the negative ZFS non-Kramers model. The ground-state spin Hamiltonian parameters obtained from the fits to the data are g∥ = 9.3 and δ = 3.0 cm−1 for both bands as listed in Table 1. The 5T2g splittings are determined from these to be −Δ ~ 330 cm−1 and V ~ 95 cm−1, which are in a range for distorted octahedral six-coordinate ferrous sites. The difference in saturation behavior between the 8810 and 10010 cm−1 transition intensities again reflects the different contributions from the B-terms and polarization ratios (Table 1). Thus, from the MCD and VTVH MCD data, resting FeNDOred contains a high-spin ferrous active site with a six-coordinate distorted octahedral geometry. The rhombicity |V/2Δ| of 0.14 is a little smaller than that of NDOox (0.18), which would seem inconsistent with the lower 5Eg excited-state splitting. However, the excited-state splitting pattern depends on σ d-orbital interactions with the ligands, while the ground-state splittings reflects π d-orbital interactions with the ligands, in particular the carboxylate.1,3 Importantly, -Δ and V both decrease in FeNDOred compared to FeNDOox consistent with a stronger 6th ligand. Thus, upon reduction of the Rieske site, a structural change is transmitted across the subunit interface to the ferrous site, which results in stronger axially coordinated water, while the π-donor carboxylate ligation becomes perturbed. Importantly, the MCD spectral features of the ferrous catalytic site in FeNDOox and FeNDOred are generally the same, and thus alteration of the redox state of the Rieske site does not significantly affect the coordination number of the mononuclear ferrous site in the absence of a substrate.
NDOred-Naph
Figure 5A shows the 5 K 7 T MCD spectra of naphthalene-bound NDO with the reduced Rieske site. Although the spectral features of apoNDOred-Naph shown by the gray dash line are essentially the same as that of apoNDOred, there is a difference in the spectral pattern between FeNDOred-Naph (shown by the gray line) and the corresponding substrate free form, in particular an increase of the MCD signal at ~11000 cm−1 in the substrate-bound form. The MCD difference spectrum (FeNDOred-Naph − apoNDOred-Naph) indicated by the aqua line exhibits two bands at 8165 cm-1 and 10575 cm-1. VTVH MCD data were used to assign the species associated with the new bands by monitoring the MCD intensity at 8890 (position 1 in Figure 5A) and 10010 cm−1 (position 2 in Figure 5A) for a series of different fields at fixed temperatures. Again, at these energies the reduced Rieske site in apoNDOred-Naph does not exhibit signal intensity, which enables VTVH MCD analysis on the ferrous catalytic site. Figure 5B shows these data plotted vs βH/2kT, along with the best fits. However, a band fitting analysis of the two MCD signals of NDOred-Naph indicates that the MCD signal intensity at 8890 cm-1 has a 65% contribution from a band centered at 8165 cm-1 and 35% from a 10575 cm-1 band (at 7T, 5K; Figure S2 in Supporting Information). The VTVH MCD data obtained at 10010 cm-1 has no contribution from the lower energy band. To access to the VTVH MCD data for the 8165 cm-1 band, normalized VTVH MCD intensities experimentally obtained from the VTVH MCD analysis at 8890 cm-1 were corrected for the 35% contribution from the 10575 cm-1 band (at 7T, 5K) and renormalized; This was accomplished by subtraction of 35% of the intensity of normalized VTVH MCD data collected at 10010 cm-1 from the normalized VTVH MCD intensities experimentally obtained at 8890 cm-1. The resulting curves were then renormalized. Figure 5C shows these data plotted vs βH/2kT, along with the best fit. The saturation magnetization behaviors of both bands are well described by the negative ZFS non-Kramers model, but in contrast to FeNDOox-Naph, fits to FeNDOred-Naph can only be achieved by using different ground-state parameters for the two transitions as listed in Table 1. The energy of the LF transitions observed for each species and the VTVH MCD data indicate the presence of two different five-coordinate ferrous sites. The second, low energy, LF transition would be below the 5000 cm−1 cutoff of our NIR MCD instruments. The species associated with 10000 cm−1 MCD transition is characterized by a δ of 3.8 cm−1 and g∥ of 9.0. The 5T2g splittings calculated from those spin Hamiltonian parameters are almost the same as those obtained for the oxidized substrate bound form indicative of a square pyramidal ferrous site, thus the species associated with the 10000 cm−1 band in FeNDOred-Naph is equivalent to the five-coordinate ferrous site found for FeNDOox-Naph. The decreased MCD signal intensity of NDOred-Naph compared to FeNDOox-Naph at 10000 cm-1 indicates that ~1/3 of the square pyramidal sites is converted to a second 5c species in FeNDOred-Naph (Figure S1). This species is associated with 8165 cm−1 band and is characterized by a small δ value of 1.2 cm−1 which is consistent with a five-coordinate trigonal bipyramidal ferrous catalytic site. The small value of δ for the trigonal bipyramidal site reflects the out-of-state interaction of the spin-orbit coupling of the 5E’’ ground-state with both the 5E’ and 5A1 excited-states (vide infra).3,60 For the square pyramidal ferrous site the spin-orbit coupling is in-state (5T2g) which leads to a larger ZFS.
Figure 5.
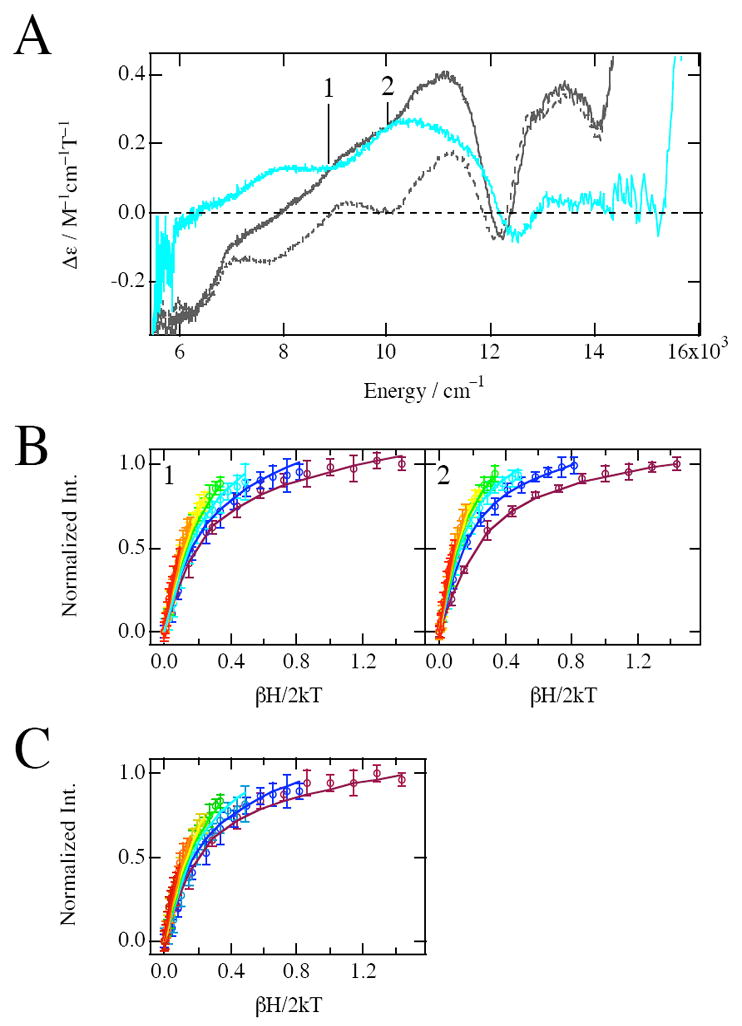
Ligand field MCD and VTVH MCD data for naphthalene-bound NDO with reduced Rieske. (A) MCD spectra at 5K and 7T of naphthalene-bound NDO with reduced Rieske; FeNDOred-Naph (gray line), apoNDOred-Naph (gray dashed line), and the difference (FeNDOred-Naph − apoNDOred-Naph) (aqua line) spectra. (B) VTVH saturation magnetization behavior of naphthalene-bound NDO with reduced Rieske measured at 8890 cm−1 (1) and 10010 cm−1 (2). The normalized data are plotted vs βH/2kT for a series of fixed temperatures (1,7, 3, 5, 7, 10, 15, 25K). The temperature of isotherm curves increases as the color of curves changes from purple to red in the spectral color order. Errors in the intensities are also shown by error bars. The best fit (solid lines) to the data was generated by the parameters described in Table 1. (C) Corrected VTVH saturation magnetization behavior of naphthalene-bound NDO with reduced Rieske obtained by subtraction of 35% of the intensities of normalized VTVH MCD data calculated at 10010 cm-1 from the normalized VTVH MCD intensities experimentally obtained at 8890cm-1. The normalized data are plotted vs βH/2kT for a series of fixed temperatures (1,7, 3, 5, 7, 10, 15, 25K). The temperature of isotherm curves increases as the color of curves changes from purple to red in the spectral color order. Errors in the intensities are also shown by error bars. The best fit (solid lines) to the data was generated by the parameters described in Table 1.
d-orbital energy levels
From the experimental excited-state splittings and ground-state VTVH MCD analyses, experimental d-orbital energy level diagrams have been constructed for the ferrous catalytic sites in FeNDOox, FeNDOox-Naph, FeNDOred, and FeNDOred-Naph (Figure 6). For the resting NDO enzymes (FeNDOox and FeNDOred in the first and third columns), the observed 5Eg excited-state d-orbital energies at ~8000 and 10000 cm−1 are consistent with a distorted six-coordinate ferrous center. The 5T2g ground-state dπ-orbitals lie at 0, 160, and 530 cm−1 and at 0, 95, 385 cm−1 in FeNDOox and FeNDOred, respectively, which are typical splittings observed for six-coordinate complexes with oxygen and nitrogen ligand sets.1,3,60 The energies of these five d-orbitals are similar, implying that in the absence of substrate, the geometric and electronic structure of the catalytic ferrous site are not significantly perturbed with change in the redox state of the Rieske site. This is in contrast to the x-ray crystal study of OMO, but consistent with the observation that O2 does not bind to the ferrous mononuclear iron in the absence of substrate for either redox state of the Rieske cluster.45
Figure 6.
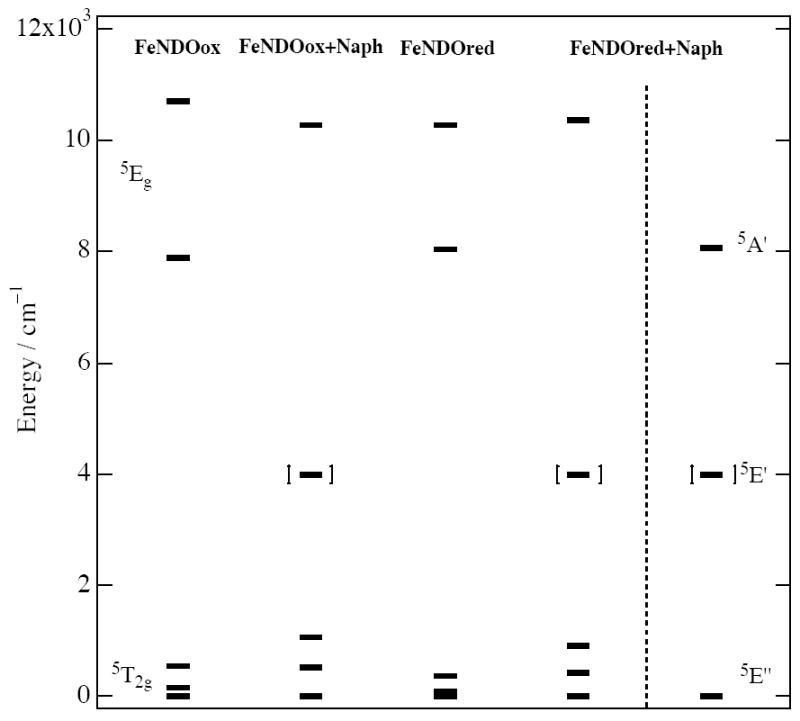
Experimentally determined d-orbital energy levels. Ligand field parameters are listed in Table 1.
The observed excited-state energy for the substrate-bound NDO with the oxidized Rieske site (FeNDOox-Naph) occurs at 10290 cm−1. The ground-state LF splittings of 0, 500, and 1050 cm−1 are in the range found for five-coordinate square pyramidal geometries.1,3,60 Therefore, a six- to five-coordinate conversion of the non-heme ferrous catalytic site occurs upon substrate binding to the resting NDO enzyme. Again this result is not consistent with the crystallography on ROs, but in line with solution studies which showed rapid oxygen binding following addition of substrate when the Rieske center is reduced.45
Upon reduction of the Rieske site in the substrate bound form, two different five-coordinate species are generated in a 2:1 mixture (fourth and fifth columns in Figure 6). The observed excited-state energy of the five-coordinate square pyramidal species is 10575 cm−1, and the 5T2g ground-state dπ-orbital splittings are 0, 425, and 910 cm−1. The trigonal bipyramidal five-coordinate species exhibits a LF transition at 8165 cm−1. A trigonal ferrous LF produces a 5E’’ ground-state with (dxy, dyz) lowest in energy, a 5E’ (dx2-y2, dxy) as the first excited-state, and a 5A1’ (dz2) as the highest LF excited-state. Thus, there is large change in the geometric and electronic structure of the catalytic ferrous site upon reduction of the Rieske site in the presence of substrate, which could contribute to its O2 reactivity.
Discussion
Correlation to crystallography
Recent structural information on the Rieske non-heme iron oxygenases provides a starting point for mechanistic studies. More than 34 crystal structures of 11 different enzymes are available from the Protein Data Bank as of June 2007. However, in most of the crystallographic studies the coordination environment and redox state of the metal centers are not well defined. An important role of spectroscopy is to provide complementary information on the geometric and electronic structures of the metal centers providing molecular level insight into the catalytic mechanism.
The MCD data demonstrate the presence of a six-coordinate catalytic ferrous site with a weak axial ligand in resting NDO when the Rieske site is oxidized, but the x-ray crystal structures of the corresponding NDO species show a five-coordinate ferrous site.21,63 A water would likely be the missing ligand in the crystal structures of resting NDO because the structure of the ferrous site of resting (OMO) (with a reduced Rieske site) shows two water molecules coordinated to a six-coordinate iron (other ligands: two histidines and one bidentate carboxylate).25 The catalytic ferrous site of the resting NDO enzyme with the Rieske cluster reduced was also characterized by the MCD data as being six-coordinate. Again, the crystal structure of resting NDO with a reduced Rieske center assigned the ferrous site as five-coordinate.21
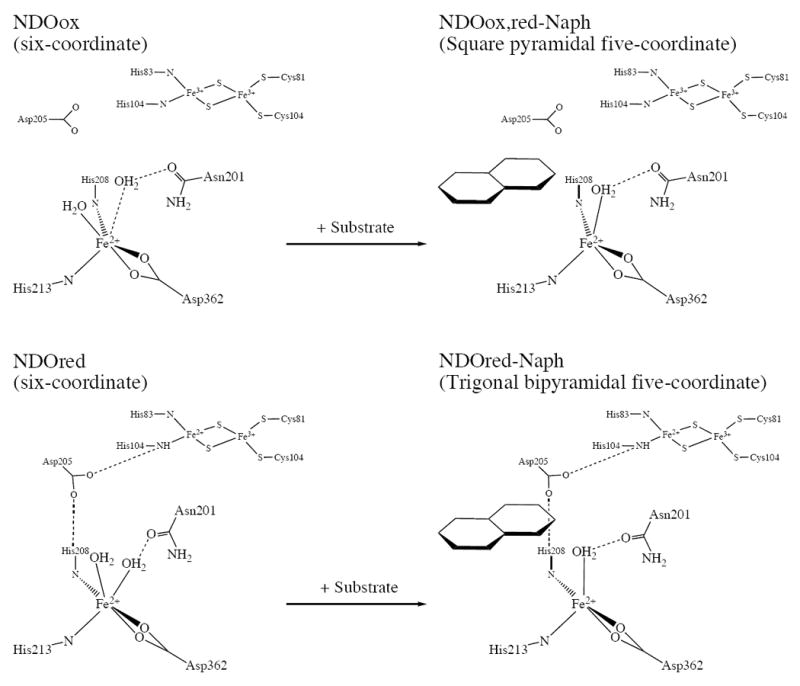
Although the x-ray structure of naphthalene-bound NDO with the Rieske site oxidized is not available, our MCD data indicate that the catalytic ferrous site now has a five-coordinate square pyramidal geometry. The crystal structure of the corresponding substrate-bound ferrous OMO enzyme was in fact modeled as having a distorted five-coordinate square pyramidal site. Reduction of the Rieske site in the substrate-bound form is found to convert 1/3 of the five-coordinate catalytic ferrous sites to trigonal bipyramidal sites. This distortion to a trigonal bipyramidal structure is consistent with the crystal structure of the ferrous site in substrate-bound NDO with the Rieske center reduced which can be viewed as a distorted five-coordinate trigonal bipyramidal site with a water, His213, and one of the oxygen atoms of Asp362 defining the trigonal plane.21 From the crystal structures of OMO, reduction of the Rieske site is believed to protonate the histidine ligand of the Rieske center that now H-bonds to an Asp, which in turn bridges to a histidine ligand coordinated to the ferrous site resulting in a conformational change that alters the active site geometry (Figure 1). From the MCD data, reduction of the Rieske center also changes the five-coordinate catalytic ferrous site structure in the presence of substrate. This structural reorganization in the Rieske dioxygenase could play a role in the electron transfer from the Rieske cluster or regulation of oxygen activation chemistry at the mononuclear iron. A summary of the coordination environments of the catalytic ferrous site in NDO constructed with the spectroscopy and crystallography is presented in Scheme 2.
Scheme 2.
Mechanistic considerations
The six- to five-coordinate conversion upon substrate binding observed for the ferrous site of NDO is in line with the general mechanistic strategy observed for other non-heme iron containing oxygenases in creating a coordinatively unsaturated site for the binding and activation of O2 only in the presence of substrate(s).1,2,57 The allosteric effect on the ferrous site upon reduction of the Rieske site in the absence and presence of the substrate would further provide a mechanism for regulation of the reactivity toward O2.
The formation of a relatively strong axial six-coordinate ferrous site in resting NDO when the Rieske site is reduced has relevance to NO binding studies of NDO45 and other RDOs;36,47,48 NO can bind to the resting ferrous site with the Rieske site oxidized, but not when the Rieske is reduced. The weak axial six-coordinate ferrous catalytic site with the oxidized Rieske site would allow binding of NO to eliminate a weakly coordinated water, but when the Rieske center is reduced the tighter six-coordinate site would inhibit NO binding. This structural change suggests a regulation mechanism limiting O2 binding to the resting ferrous site when the Rieske center is reduced. From our previous studies, O2 binding to a coordinatively unsaturated ferrous site with one-electron reduction to superoxide is unfavorable.64 However, the two-electron reduction of O2 would be favorable which is possible with the Rieske center reduced.65 Thus this allosteric effect in resting NDO would avoid uncontrolled formation and release of activated O2 species. This additional regulation of the non-heme ferrous catalytic site parallels the behavior of other non-heme iron containing oxygenases which require both cofactor and substrate for activation of O2. It was found from previous MCD studies of the α-KG and pterin dependent non-heme iron oxygenases that when only the redox active cofactor is present the site is six-coordinate preventing the O2 reaction until the substrate binds to the active site.1,2
From the crystal structures of resting OMO, reduction of the Rieske site displaces the non-heme iron ~0.8 Å away from a substrate binding site, and apparently increases the distance between the iron and a proximal residue, Asn215 (corresponding to Asn201 in NDO), allowing binding of an exogeneous 6th water ligand.25 In addition, crystal structures of resting OMO exhibit large conformational changes in His221 (corresponding to His208 in NDO) upon reduction of the Rieske center (a 1.5 Å shift in its Cα atom and 20° twist of its side chain).25,66 Thus in resting NDO reduction of the Rieske site might displace the iron due to perturbation on His208, creating a more open site. allowing a tightly bound 6th ligand (H2O). The conversion of the square pyramidal to the trigonal bipyramidal structure in naphthalene-bound NDO upon reduction of the Rieske center would also appear to be due to perturbation of the histidine ligand (His208) coordinated to the catalytic iron and involved in the ET pathway. The structural alteration might only occur in one α subunit of the α3β3 structure based on the partial conversion.
The formation of two coordination geometries in naphthalene-bound NDO with the reduced Rieske site relates to the ENDOR results for the substrate complexes of NO-bound NDO.42,43 The ENDOR study showed that there were two substrate binding sites (A and B).42,43 When the Rieske center is reduced, the substrate is displaced 0.5 Å away from the iron (the B site) compared with the A site which is dominant with the Rieske center oxidized. The MCD data suggest that this change in the substrate position with change in the redox state of the Rieske site is correlated to a change in the coordination geometry of the catalytic ferrous site from square pyramidal to trigonal bipyramidal. The partial formation of the second species observed by ENDOR is also consistent with the MCD results on the catalytic ferrous site.
From the crystal structures of NDO-O2 adducts,21 O2 is suggested to bind to the iron in a side-on η2-O2-Fe structure. The trigonal bipyramidal site could control binding of O2 in this orientation by elimination of its apical water ligand. The observed weak axial ligand set of NDO would generate a high-spin ferric-(hydro)peroxide through two-electron transfer (one electron from the catalytic ferrous site and the second electron from the reduced Rieske center). It was found from previous model and computational studies that homolytic cleavage of a peroxide O-O bond is not favorable at a high-spin ferric site compared to its low-spin analog.67,68 However, heterolytic cleavage of the O-O bond was not evaluated in these studies. In fact, the sub-stoichimetric incorporation of 18O into product via peroxide shunt turnover in the presence of 18O labeled H2O2, H2O, or O2 may suggest that the O-O bond is cleaved prior to substrate attack.46 A recent mechanistic study of NDO using diagnostic probe molecules has suggested that their monooxygenation reaction is mediated by an Fe5+-oxo-hydroxo intermediate.69 Alternatively, this high oxidation state would be difficult to achieve in a non-heme ligand environment.70 Further study is required to elucidate the nature and reactivity of the FeO2 species reported for NDO.
In summary, the MCD spectra and VTVH MCD data and analyses presented here provide new insight into the coordination geometry and electronic structure of the catalytic ferrous site in naphthalene 1,2- and related Rieske dioxygenases. New aspects emerging from this study are: (1) resting NDO has a six-coordinate ferrous catalytic site; (2) substrate binding to resting NDO causes a six- to five-coordinate conversion at this ferrous site; (3) a relatively weakly bound water ligand at ferrous catalytic site in the resting enzyme becomes more tightly coordinated upon reduction of the Rieske site; (4) a square pyramidal five-coordinate catalytic ferrous site in the substrate-bound enzyme is partly converted to a trigonal bipyramidal site when the Rieske site is reduced; the geometric and electronic structure of the catalytic site in the presence of substrate is thus significantly affected by the redox state of the Rieske center. These molecular structural insights are relevant to protein dynamics that regulate the reactivity toward O2 during the catalytic cycle, where both substrate binding and reduction of the Rieske center are required for O2 activation.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health, Grants No. GM40392 (E.I.S.) and GM24689 (J.D.L.). T.O. acknowledges the Uehara Memorial Foundation for a research fellowship.
Footnotes
Supporting Information: Figure S1-12; This material is available via http://pubs.acs.org.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Neidig ML, Solomon EI. Chem Commun. 2005:5843–5863. doi: 10.1039/b510233m. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Pavel EG, Loeb KE, Campochiaro C. Coord Chem Rev. 1995;144:369–460. [Google Scholar]
- 4.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 1998;120:743–753. [Google Scholar]
- 5.Zhou J, Gunsior M, Bachmann BO, Townsend CA, Solomon EI. J Am Chem Soc. 1998;120:13539–13540. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 7.Neidig ML, Kavana M, Moran GR, Solomon EI. J Am Chem Soc. 2004;126:4486–4487. doi: 10.1021/ja0316521. [DOI] [PubMed] [Google Scholar]
- 8.Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI. Proc Natl Acad Sci USA. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neidig ML, Brown CD, Kavana M, Choroba OW, Spencer JB, Moran GR, Solomon EI. J Inorg Biochem. 2006;100:2108–2116. doi: 10.1016/j.jinorgbio.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Mabrouk PA, Orville AM, Lipscomb JD, Solomon EI. J Am Chem Soc. 1991;113:4053–4061. [Google Scholar]
- 11.Davis MI, Wasinger EC, Decker A, Pau MYM, Vaillancourt FH, Bolin JT, Eltis LD, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2003;125:11214–11227. doi: 10.1021/ja029746i. [DOI] [PubMed] [Google Scholar]
- 12.Loeb KE, Westre TE, Kappock TJ, Mitic N, Glasfeld E, Caradonna JP, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:1901–1915. [Google Scholar]
- 13.Kemsley JN, Mitic N, Zaleski KL, Caradonna JP, Solomon EI. J Am Chem Soc. 1999;121:1528–1536. [Google Scholar]
- 14.Kemsley JN, Wasinger EC, Datta S, Mitic N, Acharya T, Hedman B, Caradonna JP, Hodgson KO, Solomon EI. J Am Chem Soc. 2003;125:5677–5686. doi: 10.1021/ja029106f. [DOI] [PubMed] [Google Scholar]
- 15.Gibson DT, Parales RE. Curr Opin Biotechnol. 2000;11:236–243. doi: 10.1016/s0958-1669(00)00090-2. [DOI] [PubMed] [Google Scholar]
- 16.Hudlicky T, Gonzalez D, Gibson DT. Aldrichimica Acta. 1999;32:35–62. [Google Scholar]
- 17.Some Rieske cluster containing non-heme iron enzymes are known to mediate monooxygenation as a native reaction. Rieske non-heme iron oxygenases (ROs) include both dioxygenases and monooxygenases.
- 18.Ferraro DJ, Gakhar L, Ramaswamy S. Biochem Biophys Res Commun. 2005;338:175–190. doi: 10.1016/j.bbrc.2005.08.222. [DOI] [PubMed] [Google Scholar]
- 19.Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Structure. 1998;6:571–586. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 20.Carredano E, Karlsson A, Kauppi B, Choudhury D, Parales RE, Parales JV, Lee K, Gibson DT, Eklund H, Ramaswamy S. J Mol Biol. 2000;296:701–712. doi: 10.1006/jmbi.1999.3462. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Science. 2003;299:1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 22.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. J Biol Inorg Chem. 2005;10:483–489. doi: 10.1007/s00775-005-0657-1. [DOI] [PubMed] [Google Scholar]
- 23.Gakhar L, Malik ZA, Allen CC, Lipscomb DA, Larkin MJ, Ramaswamy S. J Bacteriol. 2005;187:7222–7231. doi: 10.1128/JB.187.21.7222-7231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferraro DJ, Okerlund AL, Mowers JC, Ramaswamy S. J Bacteriol. 2006;188:6986–6994. doi: 10.1128/JB.00707-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins BM, Svetlitchnaia T, Dobbek H. Structure. 2005;13:817–824. doi: 10.1016/j.str.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. J Mol Biol. 2004;342:1041–1052. doi: 10.1016/j.jmb.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 27.Ferraro DJ, Brown EN, Yu C-L, Parales RE, Gibson DT, Ramaswamy S. BMC Struct Biol. 2007;7:10. doi: 10.1186/1472-6807-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nojiri H, Ashikawa Y, Noguchi H, Nam J-W, Urata M, Fujimoto Z, Uchimura H, Terada T, Nakamura S, Shimizu K, Yoshida T, Habe H, Omori T. J Mol Biol. 2005;351:355–370. doi: 10.1016/j.jmb.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 29.Ashikawa Y, Fujimoto Z, Noguchi H, Habe H, Omori T, Yamane H, Nojiri H. Structure. 2006;14:1779–1789. doi: 10.1016/j.str.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Friemann R, Ivkovic-Jensen MM, Lessner DJ, Yu C-L, Gibson DT, Parales RE, Eklund H, Ramaswamy S. J Mol Biol. 2005;348:1139–1151. doi: 10.1016/j.jmb.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T. J Bacteriol. 2005;187:2483–2490. doi: 10.1128/JB.187.7.2483-2490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakoncic J, Jouanneau Y, Meyer C, Stojanoff V. FEBS J. 2007;274:2470–2481. doi: 10.1111/j.1742-4658.2007.05783.x. [DOI] [PubMed] [Google Scholar]
- 33.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 34.Parales RE, Parales JV, Gibson DT. J Bacteriol. 1999;181:1831–1837. doi: 10.1128/jb.181.6.1831-1837.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beharry ZM, Eby DM, Coulter ED, Viswanathan R, Neidle EL, Phillips RS, Kurtz DM., Jr Biochemistry. 2003;42:13625–13636. doi: 10.1021/bi035385n. [DOI] [PubMed] [Google Scholar]
- 36.Tarasev M, Pinto A, Kim D, Elliott SJ, Ballou DP. Biochemistry. 2006;45:10208–10216. doi: 10.1021/bi060219b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuila D, Fee JA, Schoonover JR, Woodruff WH, Batie CJ, Ballou DP. J Am Chem Soc. 1987;109:1559–1561. [Google Scholar]
- 38.Kuila D, Schoonover JR, Dyer RB, Batie CJ, Ballou DP, Fee JA, Woodruff WH. Biochim Biophys Acta. 1992;1140:175–183. doi: 10.1016/0005-2728(92)90007-o. [DOI] [PubMed] [Google Scholar]
- 39.Cline JF, Hoffman BM, Mims WB, LaHaie E, Ballou DP, Fee JA. J Biol Chem. 1985;260:3251–3254. [PubMed] [Google Scholar]
- 40.Gurbiel RJ, Batie CJ, Sivaraja M, True AE, Fee JA, Hoffman BM, Ballou DP. Biochemistry. 1989;28:4861–4871. doi: 10.1021/bi00437a051. [DOI] [PubMed] [Google Scholar]
- 41.Gurbiel RJ, Doan PE, Gassner GT, Macke TJ, Case DA, Ohnishi T, Fee JA, Ballou DP, Hoffman BM. Biochemistry. 1996;35:7834–7845. doi: 10.1021/bi960380u. [DOI] [PubMed] [Google Scholar]
- 42.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. J Am Chem Soc. 2003;125:2034–2035. doi: 10.1021/ja028781m. [DOI] [PubMed] [Google Scholar]
- 43.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. J Am Chem Soc. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]
- 44.Coulter ED, Moon N, Batie CJ, Dunham WR, Ballou DP. Biochemistry. 1999;38:11062–11072. doi: 10.1021/bi9904499. [DOI] [PubMed] [Google Scholar]
- 45.Wolfe MD, Parales JV, Gibson DT, Lipscomb JD. J Biol Chem. 2001;276:1945–1953. doi: 10.1074/jbc.M007795200. [DOI] [PubMed] [Google Scholar]
- 46.Wolfe MD, Lipscomb JD. J Biol Chem. 2003;278:829–835. doi: 10.1074/jbc.M209604200. [DOI] [PubMed] [Google Scholar]
- 47.Wolfe MD, Altier DJ, Stubna A, Popescu CV, Munck E, Lipscomb JD. Biochemistry. 2002;41:9611–9626. doi: 10.1021/bi025912n. [DOI] [PubMed] [Google Scholar]
- 48.Tarasev M, Rhames F, Ballou DP. Biochemistry. 2004;43:12799–12808. doi: 10.1021/bi0490587. [DOI] [PubMed] [Google Scholar]
- 49.Tarasev M, Ballou DP. Biochemistry. 2005;44:6197–6207. doi: 10.1021/bi047724y. [DOI] [PubMed] [Google Scholar]
- 50.Tierney DL, Gassner GT, Luchinat C, Bertini I, Ballou DP, Penner-Hahn JE. Biochemistry. 1999;38:11051–11061. doi: 10.1021/bi990431y. [DOI] [PubMed] [Google Scholar]
- 51.Tsang HT, Batie CJ, Ballou DP, Penner-Hahn JE. Biochemistry. 1989;28:7233–40. doi: 10.1021/bi00444a015. [DOI] [PubMed] [Google Scholar]
- 52.Tsang Him-Tai, Batie CJ, Ballou DP, Penner-Hahn JE. J Biol Inorg Chem. 1996;1:24–33. [Google Scholar]
- 53.Jeffrey AM, Yeh HJ, Jerina DM, Patel TR, Davey JF, Gibson DT. Biochemistry. 1975;14:575–584. doi: 10.1021/bi00674a018. [DOI] [PubMed] [Google Scholar]
- 54.Ensley BD, Gibson DT, Laborde AL. J Bacteriol. 1982;149:948–954. doi: 10.1128/jb.149.3.948-954.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pavel EG, Martins LJ, Ellis WR, Jr, Solomon EI. Chem Biol. 1994;1:173–183. doi: 10.1016/1074-5521(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 56.Gassner GT, Ballou DP, Landrum GA, Whittaker JW. Biochemistry. 1993;32:4820–4825. doi: 10.1021/bi00069a017. [DOI] [PubMed] [Google Scholar]
- 57.Shu L, Chiou YM, Orville AM, Miller MA, Lipscomb JD, Que L., Jr Biochemistry. 1995;34:6649–6659. doi: 10.1021/bi00020a010. [DOI] [PubMed] [Google Scholar]
- 58.Berlman IB. Handbook of Fluorescence Spectra of Aromatic Molecules. Academic Press; 1971. [Google Scholar]
- 59.There are no Kd values in literature for naphthalene dioxygenases because the substrates are colorless, the binding is tight, and the Rieske cluster masks small spectroscopic changes from the Fe(II). A value for the Km from steady state kinetics is 12 μM (Lipscomb et al. unpublished results). The reaction is completely saturated by 300 μM and mostly saturated at 100 μM. We have estimated Kd by competition with ANS which is fluorescent when bound to NDO From this the Kd for naphthalene is in the range of 20-40 μM (Lipscomb et al. unpublished results). For the studies presented here the yield of NDOox-Naph (Kd ~ 30 μM) and NDOred-Naph (Kd = 12 μM) can be estimated as 88% and 96%, respectively.
- 60.Pavel EG, Kitajima N, Solomon EI. J Am Chem Soc. 1998;120:3949–3962. [Google Scholar]
- 61.As δ increases the nesting of the saturation magnetization curves increases. An increased degree of nesting is reflected in an increase in the spacing among a series of isotherm curves taken over the same temperature range.
- 62.Solomon EI, Hanson MA. In: Inorganic Electronic Structure and Spectroscopy. Solomon EI, Lever ABP, editors. II. Wiley-Interscience; New York: 1999. pp. 1–129. [Google Scholar]
- 63.Although in ref 20 the oxidation state of the Rieske cluster in the x-ray crystal structure is described as oxidized, the x-ray beam is known to reduce the Rieske cluster ( Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. J Inorg Biochem. 2000;78:83–87. doi: 10.1016/s0162-0134(99)00213-5.).
- 64.Schenk G, Pau MYM, Solomon EI. J Am Chem Soc. 2004;126:505–515. doi: 10.1021/ja036715u. [DOI] [PubMed] [Google Scholar]
- 65.Holm R, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 66.The crystal structure of the catalytic ferrous site in resting OMO with an oxidized Rieske center shows a distorted square pyramidal 5c site having a His225-Fe bond along its z-axis. The largest ligand-metal-ligand angles in this species are between trans equatorial ligands, (His221)N-Fe-O(Asp365) and (Asp365)O-Fe-O(H2O), which are 147° and 160°, respectively Upon reduction of the Rieske cluster of resting OMO, these angles change to 156° and 150°, respectively Assuming that (His221)N-Fe-O(Asp365) becomes the new z-axis of the catalytic ferrous site in the Rieske reduced resting enzyme, the equatorial ligand angles between (His225)N-Fe-O(H2O), (H2O)O-Fe-O(Asp365), and (Asp365)O-Fe-N(His225) are 112°, 150°, and 91°, respectively The corresponding angles in the catalytic ferrous site in resting OMO with the Rieske center oxidized are 98°, 160°, and 93°, respectively, thus showing a larger deviation from an idealized trigonal bipyramidal structure.
- 67.Lehnert N, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 68.Lehnert N, Neese F, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2002;124:10810–10822. doi: 10.1021/ja012621d. [DOI] [PubMed] [Google Scholar]
- 69.Chakrabarty S, Austin RN, Deng D, Groves JT, Lipscomb JD. J Am Chem Soc. 2007;129:3514–3515. doi: 10.1021/ja068188v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Solomon EI, Decker A, Lehnert N. Proc Natl Acad Sci USA. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.