

Summary
Porphyromonas gingivalis can inhibit chemically induced apoptosis in primary cultures of gingival epithelial cells through blocking activation of the effector caspase-3. The anti-apoptotic phenotype of P. gingivalis is conserved across strains and does not depend on the presence of fimbriae, as fimbriae-deficient mutants and a naturally occurring non-fimbriated strain were able to impede apoptosis. To dissect the survival pathways modulated by P. gingivalis, protein and gene expression of a number of components of apoptotic death pathways were investigated. P. gingivalis infection of epithelial cells resulted in the phosphorylation of JAK1 and Stat3. Quantitative real-time reverse transcription polymerase chain reaction showed that expression of Survivin and Stat3 itself, targets of activated Stat3, were elevated in P. gingivalis-infected cells. siRNA knockdown of JAK1, in combination with knockdown of Akt, abrogated the ability of P. gingivalis to block apoptosis. In contrast, cIAP-1 and cIAP-2 were not differentially regulated at either the protein or mRNA levels by P. gingivalis. One mechanism by which P. gingivalis can block apoptotic pathways in gingival epithelial cells therefore is through manipulation of the JAK/Stat pathway that controls the intrinsic mitochondrial cell death pathways. Induction of a pro-survival phenotype may prevent programmed host cell death and aid survival of P. gingivalis within gingival epithelial cells.
Introduction
Porphyromonas gingivalis, a Gram-negative anaerobe, is a major aetiological agent in severe manifestations of periodontal diseases that involve destruction of the tissues supporting the tooth (Holt and Ebersole, 2005; Feng and Weinberg, 2006). Periodontal diseases are among the most common infections of humans, and indeed reports indicate that around half of adults in developed countries have some form of chronic periodontitis (Albandar, 2005). The primary ecological niche of P. gingivalis is in the subgingival crevice, and the organism possess a array of virulence determinants including multiple colonization factors, proteolytic enzymes and immune modulins (Lamont and Jenkinson, 1998; Holt and Ebersole, 2005). However, P. gingivalis frequently colonizes the subgingival area in the absence of overt disease, and can be considered an opportunistic pathogen, causing disease only when the host-microbe balance is perturbed (Haffajee et al., 1998; Ximenez-Fyvie et al., 2000; Marsh, 2003). A dynamic and interactive association develops between P. gingivalis and the epithelial cells that line the gingival crevice and comprise a barrier to oral microbial intrusion. The long fimbriae of P. gingivalis engage an integrin receptor on the gingival epithelial cells (GECs) resulting in bacterial manipulation of downstream signalling events and culminating in bacterial internalization (Belton et al., 1999; Watanabe et al., 2001; Yilmaz et al., 2002; 2003). Intracellular P. gingivalis reside in the cytoplasm and accumulate in the perinuclear area (Belton et al., 1999). Epithelial cell association induces major changes in the expressed proteome and transcriptome of P. gingivalis (Hosogi and Duncan, 2005; Zhang et al., 2005) and the host epithelial cells react with a tailored transcriptional response (Handfield et al., 2005). In this manner both P. gingivalis and GEC adapt to their conjoint status to facilitate long-term cohabitation. Indeed, intracellular P. gingivalis cells remain viable and capable of spreading between host cells (Yilmaz et al., 2006), and epithelial cells can survive for up to 8 days after infection with P. gingivalis (Madianos et al., 1996). Moreover, epithelial cells recovered from the oral cavity show high levels of intracellular P. gingivalis (Noiri et al., 1997; Rudney et al., 2001; Vitkov et al., 2005).
Apoptosis is a highly regulated process of cell deletion that is involved in development, differentiation, homoeostasis and regulation of immune function. Dysfunction or dysregulation of the apoptotic program is involved in a variety of pathological conditions including autoimmune disease and cancer (Fadeel and Orrenius, 2005). There are two major apoptotic pathways that operate through activation of a family of cysteine proteases called caspases: the mitochondrial-mediated or intrinsic pathway; and the death receptor-mediated [e.g. tumour necrosis factor (TNF) receptor and Fas] or extrinsic pathway (Hail et al., 2006). In addition, apoptosis can be mediated through non-caspase proteases such as cathepsins, calpains and granzymes, and by ex-mitochondrial proteins such as apoptosis-inducing factor (AIF) (Hail et al., 2006). A number of intracellular bacteria evoke elevated apoptosis in host epithelial cells, a response thought to facilitate clearance of infected cells (Gao and Kwaik, 2000; Hacker et al., 2006). In contrast, several bacterial pathogens including Chlamydia, Neisseria gonorrhoeae and Salmonella can impinge on apoptotic pathways to extend host cell survival (Gao and Kwaik, 2000; Hacker et al., 2006; Simons et al., 2006). Prevention of host cell death by intracellular bacterial pathogens will prolong the integrity of their intracellular environment and thus favour bacterial persistence.
Porphyromonas gingivalis has been shown to modulate apoptotic cell death in many kinds of cells, including immune cells, fibroblasts, epithelial cells and endothelial cells, resulting in both pro- and anti-apoptotic outcomes (Hiroi et al., 1998; Gemmell et al., 1999; Graves et al., 2001; Ozaki and Hanazawa, 2001; Harris et al., 2002; Murray and Wilton, 2003; Brozovic et al., 2006; Inaba et al., 2006; Sheets et al., 2006; Urnowey et al., 2006). In primary and transformed epithelial cells derived from the gingiva, metabolically active P. gingivalis do not induce apoptosis and, moreover, inhibit chemically induced apoptotic cell death (Nakhjiri et al., 2001; Yilmaz et al., 2004; Handfield et al., 2005). This survival phenotype is associated with upregulation of anti-apoptosis genes Bcl-2 and Bfl-1 (Handfield et al., 2005), and down-regulation of the pro-apoptosis gene Bax (Nakhjiri et al., 2001). All of these genes are involved in control of mitochondrial permeability. Consistent with these transcriptional responses, P. gingivalis activates the pro-survival phosphatidylinositol 3-kinase (PI3K)/Akt pathway and blocks depolarization of the mitochondrial membrane and cytochrome c release (Yilmaz et al., 2004). Hence, the primary anti-apoptotic activity of P. gingivalis in GECs is through modulation of the intrinsic cell death pathways.
Signal transduction pathways that control programmed cell death are complex and multithreaded. Indeed, in a recent systems model of apoptosis a network of 7980 intracellular signalling events directly linked to 1440 response outputs were found to accurately predict cytokine induced apoptosis (Janes et al., 2005). To gain greater insight into the modulation of the apoptotic signalling network in GECs by P. gingivalis, we investigated the signal transduction events that funnel into, and propagates from, the mitochondrial hub of the intrinsic cell death pathway. The results indicate that P. gingivalis upregulates the JAK/Stat pathway, ultimately blocking caspase-3 activation.
Results
Caspase-3 activation is inhibited by P. gingivalis
Apoptosis can occur through both caspase-dependent and caspase-independent pathways downstream of the mitochondria (Hail et al., 2006). Hence, to distinguish between these pathways in primary GECs, the ability of P. gingivalis to inhibit activation of the effector caspase-3 was investigated. We first determined the range of moi through which P. gingivalis inhibits apoptosis in GECs by assaying the level of cytoplasmic histone associated DNA, a marker of late events in the process of apoptotic cell death. As shown in Fig. 1A, P. gingivalis 33277 significantly reduced camptothecin-induced DNA fragmentation at moi of 100, 50 and 10. At an moi of 1 the anti-apoptotic effect was lost. Thus, the anti-apoptotic activity of P. gingivalis in GEC is dose-dependent and requires few infecting bacteria. Infection with P. gingivalis over the same moi inhibited camptothecin-induced activation of caspase-3 in a similar dose-dependent manner (Fig. 1B). Furthermore, P. gingivalis did not modulate expression of the caspase-independent apoptosis effector, AIF, in the GECs (not shown). These data indicate that the anti-apoptotic pathways activated by P. gingivalis in GECs ultimately converge to limit activity of the executioner caspase-3. Although caspase-3 levels did not return to baseline, visual inspection of the GECs containing P. gingivalis and treated with camptothecin did not reveal cell rounding or detachment, and the GECs could be successfully passaged (laboratory observations), demonstrating the absence of significant cell death.
Fig. 1.

Porphyromonas gingivalis infection protects gingival epithelial cells against camptothecin-induced apoptosis in a dose-dependent manner. Primary GECs were infected with P. gingivalis 33277 for 20 h at moi of 1, 10, 50 and 100 or were mock-infected (Pg-). Camptothecin (CAM) at 1 μgml−1 was added to the cells for an additional 4 h. Control represents GEC without CAM treatment.
A. ELISA for histone-associated DNA fragments in the cytoplasm.
B. Caspase-3 activity assessed by cleavage of the fluorescent substrate Ac-DEVD-AMC (arbitrary units).
Error bars represent SD, n = 3. An asterisk denotes statistical difference of P. gingivalis-infected from mock-infected, P < 0.01, t-test.
Anti-apoptotic activity is not strain specific and does not require the presence of fimbriae
To ensure that the anti-apoptotic phenotype of P. gingivalis is not unique to strain 33277, a variety of strains of P. gingivalis were tested. All of the P. gingivalis wild-type strains, comprising A7A1-28, ATCC 49417 and W83, were capable of preventing chemically induced caspase-3 activation (Fig. 2). Thus, the anti-apoptotic properties of P. gingivalis are widely distributed among strains. As these strains have differing virulence properties in animal models of disease (van Winkelhoff et al., 1993; Katz et al., 1996), this would tend to suggest that inhibition of apoptosis is important for long-term survival of the organism in the host, rather than for overt pathogenicity. Interestingly, W83 is naturally deficient in the production of both the long (FimA) and short (Mfa) fimbriae (Watanabe et al., 1992; Noiri et al., 2004). Hence, inhibition of apoptosis by W83 argues against a role for the P. gingivalis fimbriae in the process. To test this hypothesis, we examined two strains of P. gingivalis, YPF1 and SMF1, containing an insertional inactivation of the fimA and mfa genes respectively. Both YPF1 and SMF1 retained the capability to inhibit chemically induced caspase-3 activation in GEC, corroborating the lack of involvement of either fimbrial type in the process (Fig. 2). This contrasts with the situation in monocytes where the long FimA-containing fimbriae of P. gingivalis inhibited apoptosis induced by growth factor deprivation (Ozaki and Hanazawa, 2001). Thus, P. gingivalis has more than one anti-apoptotic effector molecule that can be used according to the context of the type of host cell being encountered.
Fig. 2.
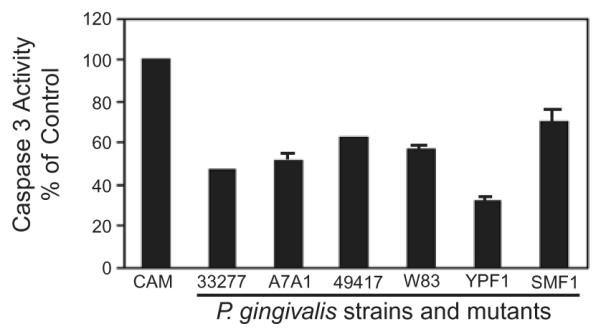
The anti-apoptotic properties of P. gingivalis are conserved across strains and are not dependent on the presence of fimbriae. GECs were infected with P. gingivalis wild type and mutant strains at moi 50 for 20 h, or were mock-infected (Pg-). YPF1 is a FimA (long fimbriae) mutant, and SMF1 is a Mfa (short fimbriae) mutant. Camptothecin at 1 μgml−1 was added to the cells for an additional 4 h. Cells were lysed, and caspase-3 activity assessed by cleavage of the fluorescent substrate Ac-DEVD-AMC. Data are expressed as per cent fluorescent intensity (arbitrary units) of camptothecin alone control (CAM). Inhibition of caspase-3 was significant (P < 0.01, t-test) for all P. gingivalis strains.
Elevation of Stat3 is associated with P. gingivalis infection
Inhibition of caspase-3, along with elevation of Bcl2 and block of cytochrome c release demonstrated previously (Nakhjiri et al., 2001; Yilmaz et al., 2004), is indicative of an anti-apoptotic pathway involving the transcription factor Stat3 (Zhong et al., 1994; Calo et al., 2003). To examine this possibility, the levels of phosphorylated Stat3 in GECs following challenge with P. gingivalis were determined by immunoblotting with specific Stat3, and phospho (tyrosine 705)-Stat3 antibodies (Fig. 3). Quantitative densitometry showed that the ratio of phosphorylated Stat3/total Stat3 was increased after 30 min at moi of 1, 10 and 50, with up to a sixfold induction observed at moi 50. After 2 h, the level of phosphorylation decreased, however, at moi 50, activated Stat3 levels were twofold higher than in the control uninfected cells. At 16 h of infection with P. gingivalis, phosphorylated Stat3 levels returned to baseline. Furthermore, the blots revealed that the total amount of Stat3, as expressed by the ratio of Stat3/actin, was transiently increased in a dose-dependent manner after 30 min of P. gingivalis infection (Fig. 3A). In addition to phosphorylation on tyrosine 705, activation of Stat3 usually requires other post-translational modifications, including phosphorylation on serine 727 (O’Shea et al., 2005; Liu et al., 2006), that were not tested in this study. However, as the stat3 gene is autoregulated (Narimatsu et al., 2001), the increased levels of Stat3 protein are consistent with functional activation of this transcription factor by P. gingivalis.
Fig. 3.

Porphyromonas gingivalis infection transiently activates Stat3. GEC were infected with P. gingivalis at moi of 1, 10 and 50, or were mock-infected (control) for 30 min (A), 2 h (B) or 16 h (C). Cell lysates were analysed by immunoblotting with antibodies to Stat3, or phosphorylated (P-) Stat3. Blots were then stripped and probed with β-actin antibodies as a loading control. Blots were analysed by scanning densitometry and ratios of P-Stat3/Stat3 and Stat3/μ-actin determined relative to ratios in control cells. Data are representative of three independent experiments.
JAK is activated by P. gingivalis
Upstream of the Stat transcription factors in eukaryotic signal transduction pathways are the JAK kinases. JAK mediated phosphorylation of Stat enables Stat dimerization, through reciprocal interaction of each Stat SH2 domain, followed by nuclear translocation and DNA binding (Aaronson and Horvath, 2002). JAKs in turn are activated by transient phosphorylation of a number of tyrosine residues (Khwaja, 2006). Therefore, to further define the signalling events impacted by P. gingivalis, an immunoblot analysis with JAK1 and phospho-JAK1 antibodies was used to monitor activation of JAK. As shown in Fig. 4, 30 min infection with P. gingivalis resulted in phosphorylation of JAK1, an effect that was dose-dependent. At moi 50 a greater than twofold induction of JAK phosphorylation was evident. After 2 h, activation was less pronounced, and phosphorylated JAK/total JAK ratios returned to baseline levels after 16 h. Together with the Stat activation data, these observations provide compelling evidence for the involvement of the JAK/Stat signalling pathway in the P. gingivalis-mediated protection of GECs from chemically induced apoptosis.
Fig. 4.

Porphyromonas gingivalis infection transiently activates Jak1. GEC were infected with P. gingivalis at moi of 1, 10 and 50, or were mock-infected (control) for 30 min (A), 2 h (B) or 16 h (C). Cell lysates were analysed by immunoblotting with antibodies to Jak1, or phosphorylated (P-) Jak1. Blots were analysed by scanning densitometry and ratios of P-Jak1/Jak1 determined relative to ratios in control cells. Data are representative of three independent experiments.
Knockdown of JAK and Akt abrogates the anti-apoptotic activity of P. gingivalis
Having shown that P. gingivalis rapidly induces phosphorylation of JAK we investigated the functionality of this activity through siRNA knockdown of JAK. Moreover, as P. gingivalis also activates Akt (Yilmaz et al., 2004), that can stimulate distinct anti-apoptotic pathways through GSK-3 (Sen et al., 2003), both JAK1 and Akt were transcriptionally silenced. As shown in Fig. 5, whereas down-regulation of JAK or Akt alone was ineffective, down-regulation of both JAK and Akt resulted in abrogation of the ability of P. gingivalis to block camptothecin induced activation of caspase-3. These results corroborate the role of JAK/Stat and suggest a process whereby P. gingivalis induces a pro-survival phenotype in epithelial cells through activation of at least two functionally distinct pathways including JAK/Stat and Akt.
Fig. 5.

Simultaneous knockdown of Jak1 and Akt abrogates the anti-apoptotic activity of P. gingivalis.
A. GECs were transfected with siRNA to Akt, Jak1, or both Akt and JAK. Controls were no target siRNA (SiNT) and transfection agent alone (control). GECs were infected with P. gingivalis at moi 50 for 20 h and then treated with camptothecin at 1 μgml−1 for 4 h. Caspase-3 activity was determined by a fluorescent substrate assay and data are expressed as per cent of mock-infected camptothecin treated cells (CAM). An asterisk denotes statistically significant (P < 0.01, t-test) from control and siNT.
B. Scanning densiotometry of JAK1 (left panel) or Akt (right panel) expression in GECs after transfection with siRNA to Jak1, Akt or AKt and Jak1. Protein levels were determined by Western blotting and are expressed as per cent of the NT-siRNA control. Data are representative of three independent experiments.
Survivin levels are elevated by P. gingivalis
The JAK/Stat pathway regulates expression of a number of genes including the apoptotic inhibitor Survivin (Hsieh et al., 2005; Wheatley and McNeish, 2005). Thus, we postulated that elevated levels of Survivin would be present in GEC infected with P. gingivalis. Figure 6 shows that P. gingivalis at moi 10 and 50 induced over twofold higher levels of Survivin in GEC at 30 min and 2 h. By 16 h post infection, Survivin levels were equivalent between infected and uninfected cells. These data are consistent with P. gingivalis induction of the JAK/Stat signalling pathway, and implicate Survivin as one of the mediators of apoptosis inhibition.
Fig. 6.

Porphyromonas gingivalis infection transiently increases levels of survivin. GEC were infected with P. gingivalis at moi of 1, 10 and 50, or were mock-infected (control) for 30 min (A), 2 h (B) or 16 h (C). Cell lysates were analysed by immunoblotting with antibodies to survivin. Blots were then stripped and probed with β-actin antibodies as a loading control (not shown). Blots were analysed by scanning densitometry and ratios of survivin/β-actin determined relative to ratios in control cells. Data are representative of three independent experiments.
The anti-apoptotic properties of P. gingivalis do not involve cIAP1/2
Inhibition of caspase-3 can also occur through the Inhibitor of Apoptosis proteins cIAP-1 and cIAP-2, components that can be regulated by Akt (Johnson et al., 2004). Therefore, we next considered the possibility that P. gingivalis can also protect GEC from apoptosis through upregulation of cIAP-1 and cIAP-2. However, immunoblots of GEC infected with P. gingivalis did not show elevation of cIAP/actin ratios, as compared with uninfected cells (Fig. 7). Thus, modulation of apoptotic pathways by P. gingivalis is limited to a discrete set of anti-apoptotic signal transduction events.
Fig. 7.

Porphyromonas gingivalis infection does not elevate levels of cIAP-1 or cIAP-2. GEC were infected with P. gingivalis at moi of 1, 10 and 50, or were mock-infected (control) for 30 min (A), 2 h (B) or 16 h (C). Cell lysates were analysed by immunoblotting with antibodies cIAP-1 or cIAP-2. Blots were then stripped and probed with β-actin antibodies as a loading control (not shown). Blots were analysed by scanning densitometry and ratios of cIAP/β-actin determined relative to ratios in control cells. Data are representative of three independent experiments.
Regulation of protein expression occurs at the mRNA level
To corroborate that the increase in expression of Stat3 is the result of increased transcriptional activity and to confirm the lack of cIAP regulation, quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed on mRNA extracted from GEC infected with P. gingivalis (Fig. 8). mRNA levels for Stat3 were increased over twofold compared with control at 30 min and 2 h, consistent with the immunoblot data. In contrast, levels of cIAP-1 or cIAP-2 message were not altered between infected and uninfected conditions. At the transcriptional level cIAP-1 and cIAP-2 are controlled by the transcription factor NF-κb (Wang et al., 1998) and our previous results demonstrated that P. gingivalis does not activate NF-κb in GEC (Watanabe et al., 2001). Collectively, these data would tend to exclude a role for the NF-κB/cIAP pathway in protection against apoptosis by P. gingivalis in GEC.
Fig. 8.
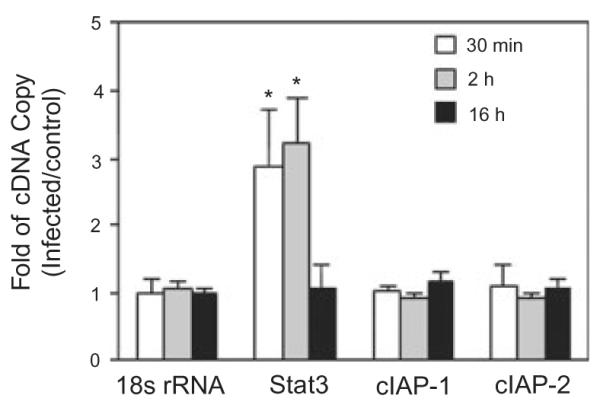
Stat3, but not cIAP-1/2, gene expression is transiently increased by P. gingivalis infection. Gene expression was measured by quantitative RT-PCR on mRNA from GEC infected with P. gingivalis at moi 50 for the times indicated, or mock-infected (control). 18sRNA was included as an internal control. Fold change was calculated by dividing the copy number of the gene transcript in P. gingivalis infected cells by the copy number in control cells. Significant differences infected/control (P < 0.01, t-test) are labelled with an asterisk.
Discussion
Epithelial cells are among the first host cell types encountered by organisms that colonize mucosal surfaces, and many mucosal pathogens are capable of entry and survival within epithelial cells. Bacteria that have an intracellular component to their lifestyle will thus benefit from the maintenance of epithelial cell integrity. This selective pressure appears to have driven the development of bacterial strategies to manipulate the apoptotic pathways to prolong the lifespan of host cells. N. gonorrhoeae, for example, can protect epithelial cells from chemically induced apoptotic cell death (Binnicker et al., 2003). However, often the situation is less definitive with pathogens exhibiting both pro- and anti-apoptotic phenotypes, depending on contextual and temporal cues (Byrne and Ojcius, 2004; Knodler et al., 2005). P. gingivalis is one such organism. Apoptosis can be induced in some cell types by P. gingivalis components such as proteases (Sheets et al., 2006). Other cellular constituents such as fimbriae and LPS can either suppress or induce apoptosis depending on the host cell type (Isogai et al., 1996; Hiroi et al., 1998; Ozaki and Hanazawa, 2001; Murray and Wilton, 2003). In primary and transformed GECs, metabolically active P. gingivalis do not induce apoptosis and indeed protect the cells from chemically induced apoptosis (Nakhjiri et al., 2001; Yilmaz et al., 2004; Handfield et al., 2005). In contrast, heat-killed P. gingivalis stimulate apoptotic death in these cells (Brozovic et al., 2006). A broad framework that would accommodate much of these disparate data involves extracellular P. gingivalis elaborating toxic factors such as proteases that kill some types of host cells. Once attached to, or inside the host epithelial cell, P. gingivalis limits protease production (Zhang et al., 2005) and manipulates host cell signalling pathways to prevent apoptosis. The balance of these activities will determine the ultimate fate of the host cell.
Intracellular P. gingivalis have a number of anti-apoptotic effects on GECs including: upregulation of Bcl-2, Bfl-1, cMYC, SGK, TNFAIP3 and CFLAR; down-regulation of Bax and CDC2L2; activation of phosphati-dylinositol 3-kinase (PI3K)/Akt pathway; and blockage of depolarization of the mitochondrial membrane and cytochrome c release (Yilmaz et al., 2004; Handfield et al., 2005). The diverse nature of these properties indicates that P. gingivalis can impinge on apoptotic pathways on multiple levels. Many of these regulated genes and pathways revolve around the mitochondria, key components of the intrinsic apoptotic death mechanism, although this does not exclude a role for P. gingivalis in regulation of the extrinsic pathway. In the current study we have defined in finer molecular detail the major intrinsic anti-apoptotic pathways controlled by P. gingivalis.
The intrinsic pathway of apoptosis is dependent on mitochondrial outer membrane permeabilization. Mitochondrial proteins, such as cytochrome c, are then released, which leads to the formation of the apoptosome (Spierings et al., 2005). The apoptosome binds and activates initiator caspases such as caspase-9, which in turn cleave and activate effector caspases such as caspase-3 (Shen and White, 2001). Activated caspase-3 cleaves a variety of substrates including the inhibitory subunit of DNA ladder nuclease, β-catenin, the cytoskeletal proteins fodrin and gelsolin, and PAK2, a member of the p21-activated kinase family, all of which contribute to the process and appearance of apoptotic cell death (Earnshaw et al., 1999). However, cell death can also proceed downstream of mitochondrial outer membrane permeabilization independently of caspase activation through the action of lethal factors such as AIF (Hail et al., 2006). Inhibition of apoptosis in primary GECs by P. gingivalis was mediated through the caspase-dependent apoptotic pathway and did not involve AIF. Upstream of the executioner caspase-3 and of the mitochondria, in terms of pro-survival pathways, are the Stat transcription factors and the JAK family of cytoplasmic tyrosine kinases. The JAK-Stat pathway transmits information from extracellular polypeptide signals, often cytokines, directly to target gene promoters (Aaronson and Horvath, 2002). The JAKs are activated by auto-phosphorylation and subsequently activate Stats by tyrosine phosphorylation. Targets of Stat3 include anti-apoptotic genes such as Bcl-2, Bcl-XL and Survivin, along with genes involved in cell cycle progression such as Fos, Cyclin-D and c-Myc (Dauer et al., 2005; Hsieh et al., 2005). Herein we demonstrated that both Stat3 and JAK1 were transiently phosphorylated by P. gingivalis and were associated with the pro-survival phenotype in GECs. Furthermore, Survivin, a bifunctional protein under the control of Stat3, that regulates cell division and suppresses apoptosis, was upregulated at the mRNA and protein levels following P. gingivalis infection. Survivin functions to suppress apoptosis by inactivating caspase-3 both through direct binding and through inhibition of caspase-9 (Johnson and Howerth, 2004; Sah et al., 2006). Thus, elevated levels of Survivin are consistent with the result that P. gingivalis prevents activation of caspase-3 by camptothecin. Coupled with our previous results that P. gingivalis upregulates expression of Bcl-2 and cMyc (Handfield et al., 2005), these data indicate that the JAK-Stat pathway is a major component of the anti-apoptotic activity of P. gingivalis. As JAKs are cytoplasmic, this may partially explain why metabolically active P. gingivalis cells that reside in the epithelial cell cytoplasm, are anti-apoptotic; whereas heat killed P. gingivalis cells, that are non-invasive, are pro-apoptotic. The mechanism by which P. gingivalis activates JAK remains to be determined.
The findings of the current study, along with earlier reports (Yilmaz et al., 2004), indicate that P. gingivalis can modulate at least two major anti-apoptotic pathways, namely the PI3K/Akt and JAK/Stat pathways. The importance of both pathways is indicated by the inability of either JAK or Akt knockdown alone to antagonize the anti-apoptotic property of P. gingivalis. In contrast, knock-down of both Akt and JAK prevented inhibition of apoptosis by P. gingivalis. A third major anti-apoptotic pathway involves NF-κb-dependent transcriptional activation of pro-survival factors such as cIAP-1 and cIAP-2 (Wang et al., 1998). However, neither cIAP-1 nor cIAP-2 was upregulated at the protein or transcriptional levels by P. gingivalis. Furthermore, our previous studies found that P. gingivalis did not activate NF-κb in GECs (Watanabe et al., 2001). NF-κb is also a major component of the extrinsic apoptotic pathway involving TNF and FasL (Beyaert et al., 2002), which would indicate that this pathway is not impacted by P. gingivalis. Furthermore, as Akt can be involved in activation of NF-κb (Amiri and Richmond, 2005), these findings demonstrate a degree of precision on the part of P. gingivalis as it accomplishes apoptotic inhibition. The ability of P. gingivalis to promote a pro-survival phenotype in GECs may contribute to its success as a colonizer of the oral epithelium.
Experimental procedures
Bacterial strains and culture conditions
Porphyromonas gingivalis strains ATCC 33277, ATCC 49417, W83 and A7A1-28 (low passage clinical isolate), YPF1 (fimA−), SMF1 (mfa−) were grown anaerobically at 37°C in Trypticase Soy Broth supplemented with yeast extract (1 mg ml−1), haemin (5 μg ml−1) and menadione (1 μg ml−1).
Eukaryotic cell lines
Primary cultures of GECs were generated as described previously (Oda and Watson, 1990; Lamont et al., 1995). Briefly, healthy gingival tissue was collected from patients undergoing surgery for removal of impacted third molars and following Institutional Review Board Guidelines. Basal epithelial cells were separated and cultured in keratinocyte growth medium (KGM; Cambrix, East Rutherford, NJ), at 37°C in 5% CO2. GECs were used at passage 3–5 and at 80% confluence.
Epithelial cell apoptosis
Apoptotic cell death in GECs was assayed by ELISA-based detection of histone associated DNA fragments (Roche Mannhein, Germany). Camptothecin 1 μg ml−1 was used to induce apoptosis in epithelial cells at 20 h post-P. gingivalis or mock infection. After 4 h of camptothecin incubation, camptothecin-treated or untreated control cells were lysed and cytoplasmic extracts were added to wells of ELISA plates coated with monoclonal antibodies against histone. The presence of histone-associated DNA fragments was then detected in a sandwich ELISA using anti-DNA peroxidase with 2, 2′-azino-di-[3-ethylbenzthiazoline-sulphonate] diammonium salt substrate.Absorbance was measured at 405 nm and background at 490 nm.
Caspase-3 (DEVDase) activity assay
A caspase-3 activity assay was performed on epithelial cells under the same conditions as the DNA fragmentation assessment. Caspase-3 activity was evaluated fluorimetrically with the caspase-3 fluorescent substrate acetyl-Asp-Gly-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC; BD Biosciences, San Jose, CA) according manufacturer’s protocol. Cells were washed with cold PBS, lysed and centrifuged to remove cell debris. Reactions in 96-well plates contained 20 μl of cell lysate with 180 μl of protease assay buffer and 20 μM (final concentration) Ac-DEVD-AMC. Fluorescence was recorded in a Victor microplate fluorescence reader (Perkin Elmer, Wellesley, MA) at excitation and emission wavelengths of 355 and 460 nm respectively. DEVDase activity was calculated as the average arbitrary fluorescence intensity of duplicate samples after incubation for 1 h.
Western immunoblotting
Lysates of control (uninfected) and P. gingivalis-infected epithelial cells were separated by SDS-PAGE. Electrophoretically separated proteins were then transferred polyvinylidene difluoride membranes by electroblotting and blocked with 5% non-fat dry milk in TBS (PBS containing 0.1% Tween 20) for 1 h at room temperature. Blots were probed with the following primary anti-bodies: anti-Phospho-Stat3 (Tyr705), 1:1000; anti-Stat3 1:1000; anti-Phospho-Jak1 (Tyr1022/1023), 1:1000; anti-cIAP1, 1:1000 (Cell Signaling, Beverly, MA); anti-JAK1, 1:200; anti-Survivin, 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA); and anti-cIAP2, 1:100 (Abcam, Cambridge, MA), at 4°C overnight. Horse-radish peroxidase-conjugated species-specific secondary antibodies along with ECL Western Blotting Detection Reagents (Amersham Biosciences, Piscataway, NJ) were used to visualize antigen-antibody binding. The blot was then stripped and probed with anti-β-actin antibodies 1:10 000 (Abcam) as a loading control. Densiometric analysis was performed on bands using Kodak 1D 3.6 software.
RNA interference
Gingival epithelial cells at 50% confluence were transfected in Opti-MEM medium using 40 nM of siRNA duplexes in Oligo-fectamine (Invitrogen, Carlsbad, CA). siRNA was targeted to Akt1/2, and/or Jak1 (Santa Cruz). No-target siRNA (Dharmacon, Lafayette, CO) was used as a negative control. Cells were used in caspase-3 assays at 48 h incubation post transfection. Gene knockdown was confirmed by Western blotting with antibody to Jak1, or Akt1/2 (Santa Cruz).
Real-time quantitative PCR
Total RNA was isolated from triplicate independent control- and P. gingivalis-infected epithelial cell cultures using Trizol LS Reagent (Invitrogen). Genomic DNA contamination was removed by DNase I (Ambion, Austin TX) digestion and samples were further purified using the RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA (1 μg) from each sample was reverse transcribed using the iScript™ cDNA Synthesis kit (Bio-Rad, Hercules, CA). Real-time quantitative PCR was conducted in duplicate for each cDNA sample with the iCycler iQ Real-time PCR detection system using iQ™ SYBR Green Supermix (Bio-Rad). Two microlitre template cDNA was added to final volume of 25 μl with 1 × SYBR Green Supermix and 4 μM of the following primer pairs:
| STAT3: | Forward: 5’-TGGCACTTGTAATGGCGTCTTC-3’, Reverse: 5’-CAGCAGGGAGGAGTCACCAG-3’ |
| cIAP-1 | Forward: 5’-TGTTCCAGTTCAGCCTGAGCAG-3’ Reverse: 5’-CACCTCAAGCCACCATCACAAC-3’ |
| cIAP-2 | Forward: 5’-CCTCAAGCCACCATCACAAC-3’ Reverse: 5’-GCCATCTAGTGTTCCAGTTCAG-3’ |
| 18s rRNA | Forward: 5’-CGCCGCTAGAGGTGAAATTC-3’ Reverse: 5’-TCTTGGCAAATGCTTTCGCT-3’ |
18s rRNA was used as an endogenous control. Real-time results were analysed using iCycler™ iQ Optical System software version 3.0a (Bio-Rad). The melt curve profile was analysed to verify a single peak for each sample, indicating primer specificity.
Preparation of standards
Specific DNA products for each gene under investigation were synthesized from chromosomal DNA using standard PCR methods and visualized by gel electrophoresis to verify that a single specific product had been generated. Each product was purified using the QIAquick PCR Purification Kit (Qiagen), and quantified using the GeneQuant spectrophotometer. DNA product copy number was calculated using the formula (Yin et al., 2001):
A 10-fold dilution series of each DNA standard was prepared for SQ of 108 to 104 copies μl−1. These were used in duplicate in each real-time PCR assay to allow the real-time PCR software to estimate SQ of that gene in cDNA samples.
Acknowledgements
This study was supported by NIDCR DE11111, DE16593 and DE16715.
References
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Albandar JM. Epidemiology and risk factors of periodontal diseases. Dent Clin North Am. 2005;49:517–532. doi: 10.1016/j.cden.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005;24:301–313. doi: 10.1007/s10555-005-1579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton CM, Izutsu KT, Goodwin PC, Park Y, Lamont RJ. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223. doi: 10.1046/j.1462-5822.1999.00022.x. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Van Loo G, Heyninck K, Vandenabeele P. Signaling to gene activation and cell death by tumor necrosis factor receptors and Fas. Int Rev Cytol. 2002;214:225–272. doi: 10.1016/s0074-7696(02)14007-1. [DOI] [PubMed] [Google Scholar]
- Binnicker MJ, Williams RD, Apicella MA. Infection of human urethral epithelium with Neisseria gonorrhoeae elicits an upregulation of host anti-apoptotic factors and protects cells from staurosporine-induced apoptosis. Cell Microbiol. 2003;5:549–560. doi: 10.1046/j.1462-5822.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- Brozovic S, Sahoo R, Barve S, Shiba H, Uriarte S, Blumberg RS, Kinane DF. Porphyromonas gingivalis enhances FasL expression via up-regulation of NFkappaB-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology. 2006;152:797–806. doi: 10.1099/mic.0.28472-0. [DOI] [PubMed] [Google Scholar]
- Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat Rev Microbiol. 2004;2:802–808. doi: 10.1038/nrmicro1007. [DOI] [PubMed] [Google Scholar]
- Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157–168. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24:3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- Feng Z, Weinberg A. Role of bacteria in health and disease of periodontal tissues. Periodontol. 2006;40:50–76. doi: 10.1111/j.1600-0757.2005.00148.x. 2000. [DOI] [PubMed] [Google Scholar]
- Gao LY, Kwaik YA. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. 2000;8:306–313. doi: 10.1016/s0966-842x(00)01784-4. [DOI] [PubMed] [Google Scholar]
- Gemmell E, Prajaneh S, Grieco DA, Taylor JJ, Seymour GJ. Apoptosis in Porphyromonas gingivalis-specific T-cell lines. Oral Microbiol Immunol. 1999;14:331–338. doi: 10.1034/j.1399-302x.1999.140601.x. [DOI] [PubMed] [Google Scholar]
- Graves DT, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, et al. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. 2001;80:1875–1879. doi: 10.1177/00220345010800100301. [DOI] [PubMed] [Google Scholar]
- Hacker G, Kirschnek S, Fischer SF. Apoptosis in infectious disease: how bacteria interfere with the apoptotic apparatus. Med Microbiol Immunol. 2006;195:11–19. doi: 10.1007/s00430-005-0239-4. [DOI] [PubMed] [Google Scholar]
- Haffajee AD, Cugini MA, Tanner A, Pollack RP, Smith C, Kent RL, Jr, Socransky SS. Subgingival microbiota in healthy, well-maintained elder and periodontitis subjects. J Clin Periodontol. 1998;25:346–353. doi: 10.1111/j.1600-051x.1998.tb02454.x. [DOI] [PubMed] [Google Scholar]
- Hail N, Jr, Carter BZ, Konopleva M, Andreeff M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis. 2006;11:889–904. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- Handfield M, Mans JJ, Zheng G, Lopez MC, Mao S, Progulske-Fox A, et al. Distinct transcriptional profiles characterize oral epithelium–microbiota interactions. Cell Microbiol. 2005;7:811–823. doi: 10.1111/j.1462-5822.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- Harris JI, Russell RR, Curtis MA, Aduse-Opoku J, Taylor JJ. Molecular mediators of Porphyromonas gingivalis-induced T-cell apoptosis. Oral Microbiol Immunol. 2002;17:224–230. doi: 10.1034/j.1399-302x.2002.170404.x. [DOI] [PubMed] [Google Scholar]
- Hiroi M, Shimojima T, Kashimata M, Miyata T, Takano H, Takahama M, Sakagami H. Inhibition by Porphyromonas gingivalis LPS of apoptosis induction in human peripheral blood polymorphonuclear leukocytes. Anticancer Res. 1998;18:3475–3479. [PubMed] [Google Scholar]
- Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the ‘red complex’, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. 2000. [DOI] [PubMed] [Google Scholar]
- Hosogi Y, Duncan MJ. Gene expression in Porphyromonas gingivalis after contact with human epithelial cells. Infect Immun. 2005;73:2327–2335. doi: 10.1128/IAI.73.4.2327-2335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh FC, Cheng G, Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Res Commun. 2005;335:292–299. doi: 10.1016/j.bbrc.2005.07.075. [DOI] [PubMed] [Google Scholar]
- Inaba H, Kawai S, Kato T, Nakagawa I, Amano A. Association between epithelial cell death and invasion by microspheres conjugated to Porphyromonas gingivalis vesicles with different types of fimbriae. Infect Immun. 2006;74:734–739. doi: 10.1128/IAI.74.1.734-739.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai E, Isogal H, Kimura K, Fujii N, Takagi S, Hirose K, Hayashi M. In vivo induction of apoptosis and immune responses in mice by administration of lipopolysaccharide from Porphyromonas gingivalis. Infect Immun. 1996;64:1461–1466. doi: 10.1128/iai.64.4.1461-1466.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffen-burger DA, Yaffe MB. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Howerth EW. Survivin: a bifunctional inhibitor of apoptosis protein. Vet Pathol. 2004;41:599–607. doi: 10.1354/vp.41-6-599. [DOI] [PubMed] [Google Scholar]
- Johnson NC, Dan HC, Cheng JQ, Kruk PA. BRCA1 185delAG mutation inhibits Akt-dependent, IAP-mediated caspase 3 inactivation in human ovarian surface epithelial cells. Exp Cell Res. 2004;298:9–16. doi: 10.1016/j.yexcr.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Katz J, Ward DC, Michalek SM. Effect of host responses on the pathogenicity of strains of Porphyromonas gingivalis. Oral Microbiol Immunol. 1996;11:309–318. doi: 10.1111/j.1399-302x.1996.tb00187.x. [DOI] [PubMed] [Google Scholar]
- Khwaja A. The role of Janus kinases in haemopoiesis and haematological malignancy. Br J Haematol. 2006;134:366–384. doi: 10.1111/j.1365-2141.2006.06206.x. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Finlay BB, Steele-Mortimer O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem. 2005;280:9058–9064. doi: 10.1074/jbc.M412588200. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AM, Lo RK, Wong CS, Morris C, Wise H, Wong YH. Activation of STAT3 by G alpha(s) distinctively requires protein kinase A, JNK, and phosphati-dylinositol 3-kinase. J Biol Chem. 2006;281:35812–35825. doi: 10.1074/jbc.M605288200. [DOI] [PubMed] [Google Scholar]
- Madianos PN, Papapanou PN, Nannmark U, Dahlen G, Sandros J. Porphyromonas gingivalis FDC381 multiplies and persists within human oral epithelial cells in vitro. Infect Immun. 1996;64:660–664. doi: 10.1128/iai.64.2.660-664.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–294. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- Murray DA, Wilton JM. Lipopolysaccharide from the periodontal pathogen Porphyromonas gingivalis prevents apoptosis of HL60-derived neutrophils in vitro. Infect Immun. 2003;71:7232–7235. doi: 10.1128/IAI.71.12.7232-7235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhjiri SF, Park Y, Yilmaz O, Chung WO, Watanabe K, El-Sabaeny A, et al. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol Lett. 2001;200:145–149. doi: 10.1111/j.1574-6968.2001.tb10706.x. [DOI] [PubMed] [Google Scholar]
- Narimatsu M, Maeda H, Itoh S, Atsumi T, Ohtani T, Nishida K, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol. 2001;21:6615–6625. doi: 10.1128/MCB.21.19.6615-6625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noiri Y, Ozaki K, Nakae H, Matsuo T, Ebisu S. An immunohistochemical study on the localization of Porphyromonas gingivalis, Campylobacter rectus and Actinomyces viscosus in human periodontal pockets. J Periodontal Res. 1997;32:598–607. doi: 10.1111/j.1600-0765.1997.tb00937.x. [DOI] [PubMed] [Google Scholar]
- Noiri Y, Li L, Yoshimura F, Ebisu S. Localization of Porphyromonas gingivalis-carrying fimbriae in situ in human periodontal pockets. J Dent Res. 2004;83:941–945. doi: 10.1177/154405910408301210. [DOI] [PubMed] [Google Scholar]
- Oda D, Watson E. Human oral epithelial cell culture I. Improved conditions for reproducible culture in serum-free medium. In Vitro Cell Dev Biol. 1990;26:589–595. doi: 10.1007/BF02624208. [DOI] [PubMed] [Google Scholar]
- O‘Shea JJ, Kanno Y, Chen X, Levy DE. Cell signaling. Stat acetylation – a key facet of cytokine signaling? Science. 2005;307:217–218. doi: 10.1126/science.1108164. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Hanazawa S. Porphyromonas gingivalis fimbriae inhibit caspase-3-mediated apoptosis of monocytic THP-1 cells under growth factor deprivation via extracellular signal-regulated kinase-dependent expression of p21 Cip/WAF1. Infect Immun. 2001;69:4944–4950. doi: 10.1128/IAI.69.8.4944-4950.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infect Immun. 2001;69:2700–2707. doi: 10.1128/IAI.69.4.2700-2707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah NK, Khan Z, Khan GJ, Bisen PS. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006;244:164–171. doi: 10.1016/j.canlet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Sen P, Mukherjee S, Ray D, Raha S. Involvement of the Akt/PKB signaling pathway with disease processes. Mol Cell Biochem. 2003;253:241–246. doi: 10.1023/a:1026020101379. [DOI] [PubMed] [Google Scholar]
- Sheets SM, Potempa J, Travis J, Fletcher HM, Casiano CA. Gingipains from Porphyromonas gingivalis W83 synergistically disrupt endothelial cell adhesion and can induce caspase-independent apoptosis. Infect Immun. 2006;74:5667–5678. doi: 10.1128/IAI.01140-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001;82:55–84. doi: 10.1016/s0065-230x(01)82002-9. [DOI] [PubMed] [Google Scholar]
- Simons MP, Nauseef WM, Griffith TS, Apicella MA. Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol. 2006;8:1780–1790. doi: 10.1111/j.1462-5822.2006.00748.x. [DOI] [PubMed] [Google Scholar]
- Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, Green DR. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science. 2005;310:66–67. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- Urnowey S, Ansai T, Bitko V, Nakayama K, Takehara T, Barik S. Temporal activation of anti- and pro-apoptotic factors in human gingival fibroblasts infected with the periodontal pathogen, Porphyromonas gingivalis: potential role of bacterial proteases in host signalling. BMC Microbiol. 2006;6:26. doi: 10.1186/1471-2180-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vitkov L, Krautgartner WD, Hannig M. Bacterial internalization in periodontitis. Oral Microbiol Immunol. 2005;20:317–321. doi: 10.1111/j.1399-302X.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yamaji Y, Umemoto T. Correlation between cell-adherent activity and surface structure in Porphyromonas gingivalis. Oral Microbiol Immunol. 1992;7:357–363. doi: 10.1111/j.1399-302x.1992.tb00636.x. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yilmaz O, Nakhjiri SF, Belton CM, Lamont RJ. Association of mitogen-activated protein kinase pathways with gingival epithelial cell responses to Porphyromonas gingivalis infection. Infect Immun. 2001;69:6731–6737. doi: 10.1128/IAI.69.11.6731-6737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- van Winkelhoff AJ, Appelmelk BJ, Kippuw N, de Graaff J. K-antigens in Porphyromonas gingivalis are associated with virulence. Oral Microbiol Immunol. 1993;8:259–265. doi: 10.1111/j.1399-302x.1993.tb00571.x. [DOI] [PubMed] [Google Scholar]
- Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000;27:648–657. doi: 10.1034/j.1600-051x.2000.027009648.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz O, Young PA, Lamont RJ, Kenny GE. Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology. 2003;149:2417–2426. doi: 10.1099/mic.0.26483-0. [DOI] [PubMed] [Google Scholar]
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun. 2006;74:703–710. doi: 10.1128/IAI.74.1.703-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JL, Shackel NA, Zekry A, McGuinness PH, Richards C, Putten KV, et al. Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol Cell Biol. 2001;79:213–221. doi: 10.1046/j.1440-1711.2001.01002.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang T, Chen W, Yilmaz O, Park Y, Jung IY, et al. Differential protein expression by Porphyromonas gingivalis in response to secreted epithelial cell components. Proteomics. 2005;5:198–211. doi: 10.1002/pmic.200400922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]