

Abstract
Autoreactive T-cells clearly mediate the pancreatic β cell destruction causing Type 1 diabetes (T1D)2. However, studies in NOD mice indicate that B-cells also contribute to pathogenesis since their ablation by introduction of an Igμnull mutation elicits T1D resistance. T1D susceptibility is restored in NOD.Igμnull mice that are irradiated and reconstituted with syngeneic bone marrow (SBM) plus NOD B-cells, but not SBM alone. Thus, we hypothesized some non-MHC T1D susceptibility (Idd) genes contribute to disease by allowing development of pathogenic B-cells. Supporting this hypothesis was the finding, that unlike those from NOD donors, engraftment with B-cells from H2g7 MHC matched, but T1D-resistant, NOR mice failed to restore full disease susceptibility in NOD.Igμnull recipients. T1D resistance in NOR mice is mainly encoded within the Idd13, Idd5.2 and Idd9/11 loci. B-cells from NOD congenic stocks containing Idd9/11 or Idd5.1/5.2 resistance loci respectively derived from the NOR or C57BL/10 strains were characterized by suppressed diabetogenic activity. Immature autoreactive B-cells in NOD mice have an impaired ability to be rendered anergic upon antigen engagement. Interestingly, both Idd5.1/5.2 and Idd9/11 resistance loci were found to normalize this B-cell tolerogenic process, which may represent a mechanism contributing to the inhibition of T1D.
Keywords: B-cells, Autoimmunity, Diabetes, Rodent, Tolerance/Suppression/Anergy
Introduction
Type 1 Diabetes (T1D) in humans and the NOD mouse model results from T-cell mediated autoimmune destruction of insulin-secreting pancreatic β cells. Although B-cells are also among the earliest leukocytes to infiltrate the pancreatic islets of NOD mice (1, 2) and the specific autoantibodies they secrete are excellent diagnostic markers of future risk for T1D in both humans and NOD mice (3), their presence was believed to be secondary to T-cell activation. However, an important role for B-cells in T1D development was discovered when NOD mice made deficient in this lymphocyte population, either by introducing a functionally inactivated Ig μ heavy chain gene (termed NOD.Igμnull) or treatment with IgM specific antibodies, were strongly protected from disease (4, 5). One study has described a human T1D patient that was deficient in B-cells (6). While of interest, this report does not universally exclude a role for B-cells in T1D development in humans since it is likely that different patient populations may have varying sets of contributory factors leading to disease. Hence, while their role may be by-passed in some cases, B-cells may still significantly contribute to T1D in a large proportion of human patients.
Although the mechanism is not clear, maternal transmission of autoantibodies appear to have an early diabetogenic role in NOD mice (7). However, susceptibility of NOD.Igμnull mice to T1D could only be restored by reconstitution with NOD B-cells, and not by infusion of autoantibodies, revealing another pathogenic mechanism for this lymphocyte population (8). Successive studies revealed that NOD B-cells also make an important contribution to T1D as a subset of APC with a preferential ability to expand autoreactive CD4+ T-cell responses (8-11). Such a role was attributed to their unique ability to specifically capture, through membrane-bound Ig molecules (or BCR), β-cell autoantigens for subsequent MHC class II mediated presentation (12, 13). An implication of this finding is that in addition to defects underlying the generation of autoreactive T-cells, another essential component of T1D susceptibility may include disruptions in mechanisms that normally prevent the development or activation of B-cells expressing self-reactive BCR. This was investigated by comparative analyses of transgenic stocks of NOD and non-autoimmune prone C57BL/6 (B6) mice in which virtually all B-cells express a BCR specific for hen egg lysozyme (IgHEL), and develop in an environment where this protein was absent or expressed as a nominal membrane-bound (mHEL) or soluble neo-self-antigen (sHEL) (14). Immature IgHEL B-cells of NOD and B6 origin did not differ in their ability to be deleted upon high avidity engagement of mHEL molecules. However, immature IgHEL B-cells in NOD mice were found to have a significantly lower ability than those of B6 origin to be deleted or anergized following low avidity Ig engagement of sHEL (14).
Multiple susceptibility (Idd) genes located both within and outside particular MHC haplotypes interactively contribute to T1D in both humans and mice (15, 16). Given their important role in T1D development it seemed likely that the pathogenic action of at least some Idd genes in NOD mice (and potentially humans) is to allow the development and/or functional activation of autoreactive B-cells. Indeed, the mechanism by which the non-expressed H2-Ea allele within the NOD H2g7 MHC haplotype contributes to T1D development partly entails a B-cell component (17). In this study, we investigated whether specific non-MHC Idd genes also regulate the diabetogenic potential of B-cells in NOD mice. We found genes on proximal Chromosome (Chr.) 1 (Idd5) and distal Chr. 4 (Idd9/11) both contribute to T1D by affecting B-cells in NOD mice, potentially by regulating responsiveness to activation and tolerance induction stimuli.
Materials and Methods
Mice
Mice were housed at The Jackson Laboratory (Bar Harbor, ME) under SPF conditions with free access to food (NIH31A/6% fat diet; Ralston Purina, Richmond, IN) and acidified water or under similar conditions at the Garvan Institute animal facility (Sydney, Australia). Female mice were used in all experiments. Derivation of NOD.Igμnull (currently at N10; (5)), NOR/Lt (18), NOD.Idd5B10 (Line R444 @ N13 (19) and R467 @N>10 (20)), NOD.Idd13NOR (Line D2Mit490-Mit144NOR @ N21; (21)), NOD.Idd5NOR and NOD.Idd9/11NOR mouse lines (N5; (22)) are previously described. Three additional back-crosses of NOD.Idd5B10 line R444 to NOD/Lt were performed to produce sub-congenic stocks containing B10-derived genomic regions from D1Mit74 to 303 and D1Mit249 to 132, which were then fixed to homozygosity. The R2s Idd5B10 subcongenic strain was produced by additional backcrossing of the previously described R2 line (20).
NOD/Lt, B6, and C57BL/10 (B10) mice used for flow cytometric analysis and in vitro experiments were purchased from Animal Resources Centre (Perth, Australia) and NOR/Lt mice from the Walter Eliza Hall Institute (Melbourne, Australia). NOD and B6 mice carrying the IgHEL and sHEL transgenes were previously described (14, 23, 24). IgHEL and sHEL transgenes were each introduced into NOD.Idd5B10 (R444) and NOD.Idd9/11NOR congenic mice by intercrossing each transgenic with congenic mice, followed by back-crosses to the congenic strains to fix the B10 or NOR-derived regions to homozygosity. NOD, B6 and congenic background stocks hemizygous for the IgHEL or sHEL transgenes were intercrossed to produce IgHEL/sHEL double transgenic mice. All experimental procedures utilizing mice were approved by The Jackson Laboratory’s Animal Care and Use Committee or the Garvan Institute Animal Ethics Committee.
Flow cytometric analyses
Bone marrow (BM), blood, peritoneal cavity, lymph node (LN) or splenic leukocyte suspensions from the indicated mice were assessed for proportions of B-cell subsets by multicolor flow cytometry using the FACSCalibur instrument with CellQuest acquisition software (BD Biosciences, San Jose, CA), and previously described methods and reagents (14). Analysis of flow cytometric data was performed using FlowJo software (Tree Star Inc. Ashland, OR).
Generation of mixed BM/B-cell chimeras
Cohorts of female NOD.Igμnull mice were lethally irradiated in two 600R split doses from a 137Cs source between 4-6 weeks of age, and then reconstituted by i.v. injection with 5×106 T-cell depleted syngeneic BM (SBM) cells admixed with 5×106 purified splenic B-cells from indicated strains. Splenic B-cells were purified using the previously described (8) MACS negative depletion system (Miltenyi Biotec Inc. Auburn, CA.) which routinely achieved >90% purity. Control chimeras consisted of NOD.Igμnull female mice reconstituted with 5×106 SBM cells only. BM/B-cell recipients were then monitored weekly between 8 and 21 weeks post-reconstitution for T1D development (see below). Upon diabetes onset or at the end of incidence study (21 weeks post-reconstitution), spleens from recipient mice were assessed by flow cytometry to determine percentage reconstitution of B-cells, CD4+ and CD8+ T-cells using previously published methods and reagents (8).
Assessment of diabetes development
T1D development was assessed by measuring glycosuric values with Ames Diastix (Bayer Corporation Diagnostics Division, Elkhart, IN). Values of >500mg/dL were considered indicative of T1D onset. Statistical differences in T1D onset between different experimental groups were tested using Kaplan-Meier life table analyses.
Proliferation Assays
B-cells were purified from pooled spleens or LN of indicated strains as described above. Proliferative responses of triplicate aliquots of 1 × 105 B-cells stimulated with 1μg/ml AffiniPure goat anti-mouse IgM F(ab’)2 fragments (Jackson ImmunoResearch Laboratories, West Grove, PA) in the presence or absence of the CD40-specific mAb HM40-3 (BD Biosciences) at a concentration of 5μg/ml were assessed as previously described (14). Control wells contained no stimulatory agents. Proliferative responses are presented as mean Δcpm ± SEM for triplicate wells of each sample.
Tolerance induction assays of T1 B-cells
Transitional type-1 (T1) B-cells were enriched from pooled red blood cell (RBC) depleted spleens of indicated mice by performing a streptavidin-microbead (Miltenyi Biotec) magnetic depletion of T-cells, monocytes/granulocytes and T2/follicular B-cells stained with biotinylated monoclonal antibodies specific for CD3 (145-2C11), Mac-1 (M1/70) and CD23 (B3B4), respectively (BD Biosciences). Recovered cells were subsequently stained with mAbs specific for B220 (RA3-6B2), CD21/35 (7G6) and CD23 (B3B4) conjugated to allophycocyanin (AP), FITC, and PE (BD Biosciences) respectively, in addition to treatment with propidium iodide (PI; Sigma-Aldrich, St Louis, MO) to exclude dead cells. FACSAria flow cytometer (BD Biosciences) was used to sort viable T1 B-cells, defined as B220+, CD21/35−, CD23− and PI−, to greater than 99% purity. To perform the deletion assays, triplicate aliquots of 1 × 105 cells were seeded into 96 well plates in 200μl of complete RPMI 1640 media with varying concentrations of AffiniPure goat anti-mouse IgM F(ab’)2 fragments. Cell were incubated for 24 hours at 37°C in a 5% CO2 incubator and then harvested and stained with Annexin V-FITC (BD Biosciences) and PI in the presence of Annexin-friendly buffer (10mM HEPES, pH7.4, 140nM NaCl, 2% dialysed FBS, 0.02% NaN3). Data are presented as the percentage of T1 B-cells that remained viable (Annexin V−, PI−) in each stimulated culture compared to non-stimulated control cultures.
Results
Non-MHC Idd loci contribute to the development of diabetogenic B-cells in NOD mice
We hypothesized that some non-MHC Idd genes functionally contribute to T1D in NOD mice by allowing the development of autoreactive B-cells. The closely NOD related, but T1D resistant, NOR strain proved to be a valuable resource for testing this hypothesis. NOR shares a large proportion of its genome (~88%) with NOD mice, including the H2g7 MHC haplotype (25), with the balance derived from the C57BLKS/J (BKS) strain, which itself is of a mixed B6 and DBA/2-like origin (25). BKS-derived genomic regions on Chr. 1, 2, and 4 are mainly responsible for the strong T1D-resistance of NOR mice (21, 22, 25). We hypothesized that some subset of these NOR non-MHC Idd resistance loci may elicit T1D protection by diminishing development of pathogenic B-cells. This possibility was tested by determining if repopulation with B-cells from NOD versus NOR donors differentially abrogated the normal T1D resistance of NOD.Igμnull recipients. Lethally irradiated NOD.Igμnull mice were reconstituted with SBM admixed with purified NOD or NOR B-cells. We previously found this approach overcomes the difficulty that unmanipulated NOD.Igμnull mice are not tolerant of directly infused B-cells, as evidenced by their ability to rapidly reject them through a CD8+ T-cell response (8). Over a 21-week observation period, T1D developed at a significantly lower frequency in NOD.Igμnull mice reconstituted with SBM plus B-cells from NOR than NOD donors (30% vs. 69.6%, respectively; Figure 1a). As discussed later, this was not due to differential repopulation by NOD versus NOR B-cells. These results indicated certain non-MHC Idd genes in NOD mice are involved in facilitating the function or development of diabetogenic B-cells.
Figure 1. Non-MHC Idd alleles from the T1D resistant NOR strain reduces the pathogenic capacity of B-cells.
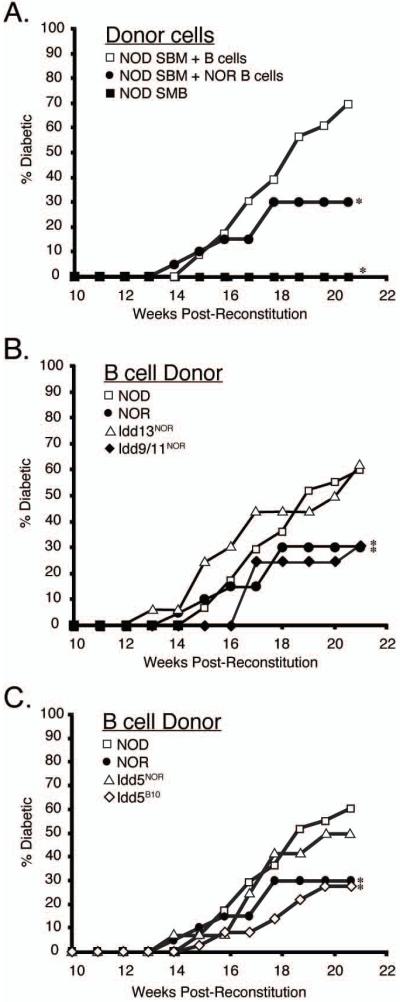
A. T1D incidence was compared in cohorts of female NOD.Igμnull mice that were lethally irradiated and reconstituted with SBM alone (closed squares, n=12) or admixed with purified splenic NOD (open squares, n=23) or NOR (closed circles, n=20) B-cells. (B) T1D incidence of female NOD.Igμnull mice reconstituted with SBM and purified splenic B-cells from either NOD.Idd13NOR (line D2Mit490-144NOR; open triangles, n=16) or NOD.Idd9/11NOR (closed diamonds, n = 16) mice and in (C) NOD.Idd5NOR (open triangles, n=12) or NOD.Idd5B10 (line R444; open diamonds, n=36) mice. For comparative purposes, disease development in NOD.Igμnull mice reconstituted with SBM and NOD (open squares, n=58) or NOR (closed circles, n=20) B-cells are also shown in graphs (B) and (C). All B-cell donor mice were 6 week old females. T1D development was assessed over 21 weeks post-reconstitution. As mice did not develop disease prior to 10 weeks after reconstitution, only weeks 10-21 are shown on graphs. Stars indicate a significant difference (p<0.05) in T1D development compared to NOD B-cell reconstituted controls using Kaplan-Meier Life-Table Analysis.
Identification of non-MHC Idd loci regulating diabetogenic B-cells
The strongest T1D protective effect in NOR mice is encoded by genes located within two closely linked loci on distal Chr. 2, designated Idd13a and Idd13b (21, 25). We therefore compared T1D development in NOD.Igμnull mice reconstituted with SBM and B-cells from standard NOD donors or a stock congenic for a segment of NOR Chr. 2 encompassing both the Idd13a and Idd13b regions (NOD.Idd13NOR) (21). Over 21 weeks both groups of recipients developed a similar high incidence of T1D (Figure 1B). Thus, while Idd13 region genes strongly contribute to T1D resistance in NOR mice, they do not function at the B-cell level.
Genes on Chr. 1 and 4, which respectively reside within portions of the previously mapped Idd5 and Idd9/11 loci, also make significant contributions to T1D resistance in NOR mice (22, 25). Thus, we tested if either of these loci dampen the diabetogenic capacity of NOR B-cells. Lethally irradiated NOD.Igμnull mice were reconstituted with SBM admixed with B-cells from NOD stocks congenic for NOR-derived Idd5 or Idd9/11 resistance regions (termed NOD.Idd5NOR and NOD.Idd9/11NOR, respectively). Over a 21 week follow-up period, T1D developed at a significantly lower frequency in NOD.Igμnull mice repopulated with B-cells from NOD.Idd9/11NOR (31%) than NOD (60%) donors (Figure 1B). It should be noted that compared to those from NOD donors, the ability of B-cells from NOD.Idd9/11NOR and NOR mice to support T1D development were equally diminished. These results indicated allelic variants of a gene(s) within the Idd9/11 region on Chr. 4 contributes to T1D susceptibility or resistance by affecting the pathogenicity of B-cells. In contrast, NOD.Idd5NOR B-cells elicited a rate of T1D development in NOD.Igμnull recipients that was slightly lower, but not significantly different than those from NOD donors (50% vs. 60%; Figure 1C).
Studies utilizing NOD mice harboring various overlapping B10-derived congenic regions have revealed Idd5-mediated T1D susceptibility results from a combination of at least three separate genes, designated Idd5.1, Idd5.2 and Idd5.3 (Figure 2) (20) (L.Wicker, unpublished observations). As the NOR Idd5 region, which is of B6 origin, only overlaps with the previously described Idd5.2 region (Figure 2), we wanted to determine whether the full complement of resistance alleles at Idd5 locus could affect the diabetogenic activity of B-cells. Irradiated NOD.Igμnull mice were reconstituted with SBM and B-cells from the NOD.Idd5B10 R444 congenic line, which contains B10-derived alleles at Idd5.1, Idd5.2 and Idd5.3 (20). Unlike those from NOD.Idd5NOR donors, NOD.Idd5B10 B-cells elicited a significantly lower frequency of T1D in NOD.Igμnull recipients than those of standard NOD origin (27.7% vs. 60.3%, respectively, Figure 1C). NOD.Igμnull mice were also reconstituted with SBM and B-cells from one of the four NOD.Idd5B10 sub-congenic lines illustrated in Figure 2. B-cells from none of the sub-congenic lines, containing different combinations of B10 derived Idd5.1, Idd5.2 and Idd5.3 variants, could completely replicate the protective effect of the originally tested NOD.Idd5B10 R444 line. B-cells derived from the sub-congenic line containing B10-derived genome between D1Mit249 and D1Mit132, which encompasses the Idd5.1, Idd5.2 and Idd5.3 genes according to recently described boundaries, came closest to the protection offered by R444 B-cells (46.4% vs. 27.7%, respectively; Figure 2). The discrepancy in protection offered by B-cells from these two Idd5 congenic lines suggests this effect could result from the presence of an additional polymorphic gene in the region of Chr. 1 where the two lines differ (i.e. between D1Mit74 and D1Mit249). However, the major conclusion from these collective results is that multiple Idd5 region genes interactively determine the extent of diabetogenic B-cell development.
Figure 2. Sub-congenic analysis of the Idd5 locus reveals epistatic contributions to the diabetogenic activity of NOD B-cells.

Schematic illustrations of Chr. 1 showing polymorphic microsatellite markers (on left) used to define congenic intervals in NOD.Idd5NOR (grey box) and various NOD.Idd5B10 (diagonally lined boxes) lines. Location of markers (in mega base pairs (Mb) from centromere) were determined using the ENSEMBL mouse genome database. On the right are shown boundaries of Idd5.1, 5.2 and 5.3 T1D susceptibility genes as defined by Wicker and colleagues. Numbers of female NOD.Igμnull mice reconstituted with SBM and various B-cells, and subsequent frequency of T1D over a 21 week follow up period are displayed above the Chr. maps. Stars are indicative of a significant difference (p<0.05) in T1D compared to the NOD B-cell reconstituted controls using Kaplan Meier Life-Table Analysis.
B-cell reconstitution levels in NOD.Igμnull recipients do not correlate with their diabetogenic capacity
It was possible quantitative differences in ability to reconstitute NOD.Igμnull recipients correlated with the capacity of the various tested B-cell populations to induce T1D. This was assessed by comparing B-cell levels in diabetic and non-diabetic mice within each group (i.e. those reconstituted with one type of B-cell) and also comparing reconstitution between different groups that had varying overall susceptibility to disease (Table I). In the majority of cases, we found no significant differences in B-cells reconstitution between diabetic versus non-diabetic mice within a particular group. In the two cases a difference was noted (NOD.Idd13NOR and NOD.Idd5B10 donor groups), B-cell reconstitution levels were lower in diabetic than non-diabetic mice. Between groups, B-cell reconstitution levels varied, but did not correlate with T1D incidence (r2= 0.0048).
Table I. Repopulation of B-cells, CD4 and CD8 T-cells in NOD.Igμnull recipientsa.
| Type of B-cells used in co- transfer with SBM (total recipients) |
% T1D |
Non-diabetic (%±SEM) |
Diabetic (%±SEM) |
Total (%±SEM) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B | CD4 | CD8 | B | CD4 | CD8 | B | CD4 | CD8 | ||
| None (n=12) | 0% | 0.4±0.2 | 29.6±7.5 | 9.8±2.6 | - | - | - | 0.4±0.2 | 29.6±7.5 | 9.8±2.6 |
| NOD (n=23) | 69.6% | 37.7±3.1 | 29.9±2.0 | 11.1±0.8 | 35.5±2.2 | 32.8±1.5 | 11.4±0.5 | 36.3±1.8 | 31.7±1.2 | 11.3±0.4 |
| NOR (n=20) | 30.0% | 17.7±2.4 | 46.2±1.6 | 18.6±0.8 | 18.2±2.6 | 49.3±1.4 | 16.0±0.8 | 17.9±1.8 | 47.1±1.2 | 17.8±0.6 |
| NOD.Idd13NOR (n=16) |
62.5% | 37.7±4.3 | 38.4±1.8 | 14.4±1.2 | 29.3±1.3 | 42.2±1.9 | 14.9±0.6 | 32.4±2.0 | 40.8±1.4 | 14.7±0.6 |
| NOD.Idd5NOR (n=12) |
50.0% | 14.7±1.6 | 53.9±1.7 | 18.6±1.3 | 20.0±3.1 | 50.5±2.8 | 16.9±1.1 | 17.3±1.8 | 52.2±1.6 | 17.8±0.8 |
| NOD.Idd9/11NOR (n=16) |
31.0% | 14.4±1.7 | 48.5±2.5 | 19.5±0.9 | 16.7±3.7 | 50.7±2.0 | 19.2±0.9 | 15.1±1.5 | 49.2±1.8 | 19.4±0.6 |
| NOD.Idd5B10 (R444) (n=33) |
27.7% | 47.8±2.1 | 33.1±1.5 | 11.0±0.6 | 40.8±1.7 | 37.0±1.5 | 12.1±0.6 | 45.6±1.7 | 34.0±1.2 | 11.3±0.5 |
Female NOD.Igμnull mice were lethally irradiated at 4-6 wks of age and reconstituted with 1:1 proportions of SBM and indicated B-cells. Proportions of splenic B-cells, CD4 and CD8 T-cells in each recipient mouse were determined either after the development of diabetes or at the end of study (i.e. 21 weeks post reconstitution; non-diabetic) by flow cytometry.
Resistance genes within Idd5B10 and Idd9/11NOR congenic regions do not normalize B-cell maturation
B-cell development in NOD mice has been reported to differ from that in non-autoimmune prone strains (14, 26, 27). Differences include a notable deficiency in immature T1 B-cells and enlarged populations of transitional type-2 (T2)/pre-marginal zone and marginal zone (MZ) subpopulations. This altered state of B-cell development may have important implications for T1D development since similar differences are observed in other autoimmune prone strains (28-30). Hence, we investigated whether differences in the levels of certain splenic B-cell subpopulations of the various mouse strains utilized as donors (all 6 week old females) may underlie their variable diabetogenic capacity. Consistent with past reports, NOD mice had lower numbers of T1 B-cells and higher numbers of pre-MZ/T2 and MZ subpopulations compared to non-autoimmune prone C57BL/10 mice (Figure 3 and Table II). We were unable to differentiate between the T2 and T3 B-cell subpopulations described by Allman et. al. (31) since NOD mice appear to be deficient in expression of the AA4.1 marker (P.A.S, unpublished observation and (32)). In spite of their decreased diabetogenic activity, NOR, NOD.Idd9/11NOR and NOD.Idd5B10 splenic B-cells demonstrated similar T1, T2/pre-MZ, follicular, and MZ profiles as those from NOD mice (Figure 3 and Table 2). We also examined various other lymphoid organs and compartments, including BM, blood, peritoneal cavity and pancreatic LN to determine whether genes within the Idd5B10 and Idd9/11NOR congenic regions could affect the development or migration of B-cells at these sites. No such changes were found (data not shown). Together, these results indicate T1D resistance alleles within the Idd5B10 and Idd9/11NOR congenic regions do not decrease B-cell pathogenicity by altering their subset distribution patterns from that seen in non-autoimmune prone strains.
Figure 3. Idd5B10 and Idd9NOR congenic regions do not normalize subset distribution of splenic B-cells in NOD mice.

Representative FACS profiles of CD21 and CD23 antibody staining on B220+ gated splenocytes from 6 week old NOD, C57BL/10, NOR, NOD.Idd5B10 and NOD.Idd9NOR female mice. Mean proportions and absolute cell numbers of total and gated B-cell subpopulations (including T1, T2/pre-MZ, FO and MZ) in spleens of four female mice of each strain are shown on Table II.
Table II. Comparison of B-cells populations in spleens of 6 week old female micea.
| Spleen cell populations | C57BL/10 | NOR | NOD | NOD.Idd5B10 | NOD.Idd9/11NOR | |
|---|---|---|---|---|---|---|
| Total Splenocytes | x106 | 46.0 ± 5.0 | 42.0 ± 1.0 | 53.5 ± 8.2 | 67.5 ± 11.2 | 61.5 ± 17.6 |
| All B220+ B-cells | % x106 |
51.7 ± 5.8 23.7 ± 1.1 |
41.2 ± 2.1 17.4 ± 2.6 |
42.9 ± 2.3 22.9 ± 1.2 |
45.5 ± 1.3 30.7 ± 2.1 |
41.6 ± 3.2 25.7 ± 3.7 |
| T1 B-cells | % x105 |
8.0 ± 1.1 36.6 ± 4.0 |
4.5 ± 0.6 19.0 ± 3.7 |
3.2 ± 0.3 16.9 ± 1.7 |
3.1 ± 0.3 20.5 ± 1.2 |
3.4 ± 0.3 21.6 ± 5.0 |
| T2/pre-MZ B-cells | % x105 |
0.9 ± 0.2 4.2 ± 0.8 |
1.7 ± 0.2 7.2 ± 0.6 |
1.6 ± 0.1 8.6 ± 0.7 |
2.5 ± 0.1 17.0 ± 1.4 |
2.4 ± 0.2 14.5 ± 1.9 |
| MZ B-cells | % x105 |
1.4 ± 0.2 6.6 ± 1.0 |
3.4 ± 0.5 13.7 ± 0.9 |
3.6 ± 0.1 19.0 ± 1.3 |
3.8 ± 0.2 25.8 ± 2.6 |
5.2 ± 0.5 31.6 ± 2.7 |
| Mature FO B-cells | % x106 |
36.3 ± 1.7 16.7 ± 1.0 |
27.2 ± 1.3 11.6 ± 1.7 |
31.6 ± 0.6 16.9 ± 0.8 |
33.5 ± 0.6 22.6 ± 1.7 |
27.1 ± 1.4 16.8 ± 2.6 |
| FO:MZ B-cell Ratio | 26.5 ± 3.6 | 8.6 ± 1.6 | 8.9 ± 0.3 | 8.9 ± 0.6 | 5.4 ± 0.8 |
Mean percentages and absolute numbers of splenic B-cell populations (±SEM) in four 6-week-old C57BL/10, NOR, NOD, NOD.Idd5B10 and NOD.Idd9/11NOR and B6 female mice were determined by FACS analysis as described in the Materials and Methods.
Idd9/11NOR increases the responsiveness of stimulated B-cells
Mature B-cells in NOD mice reportedly respond more robustly to several types of stimuli compared to those from non-autoimmune prone strains such as B6 and BALB/c (33, 34). This hyper-responsiveness may constitute a mechanism that enhances the ability of NOD B-cells to contribute to T1D. Hence, we assessed whether Idd9/11 or Idd5 loci regulate B-cell responsiveness. The ability of B-cells from NOD, NOD.Idd9/11NOR, NOD.Idd5B10 and B10 mice to proliferate upon IgM cross-linking with or without CD40 co-stimulation (mimicking antigen encounter with or without T-cell help) was compared (Figure 4A). To ensure any differential responses were not be due to subset variations, we used B-cells from pooled inguinal, axillary, cervical and mesenteric LN which consist primarily of the follicular subset. We deliberately did not take pancreatic LN as this is believed to be the primary site of lymphoid activation preceding T1D (35, 36), and thus may differ in levels of activated B-cells between each strain depending on their disease susceptibility. As previously observed, NOD B-cells proliferated significantly more than those from B10 mice under both stimulation conditions. NOD.Idd5B10 B-cells showed a similar degree of proliferation as those from standard NOD mice after anti-IgM treatment with or without CD40 stimulation, indicating genes in the congenic region do not affect this phenotype. Surprisingly, NOD.Idd9/11NOR B-cells proliferated significantly more than those from NOD mice in response to both sets of stimuli. Thus, we tested if allelic variants within the Idd9/11NOR region allowed similar super-responsiveness by NOR B-cells. To our surprise, the responsiveness of NOR B-cells to both forms of stimulation was not only greater than those from NOD, but also the NOD.Idd9/11NOR strain (Figure 4B). Therefore, while a Idd9/11 region gene(s) contributes to the ability of NOR B-cells to respond more vigorously to proliferation stimuli than those from NOD mice, other factors also control this phenotype.
Figure 4. A gene(s) in the Idd9/11, but not the Idd5 region affects the proliferative responsiveness of NOD B-cells.

B-cells were purified from pooled LN (cervical, axillary, mesenteric and inguinal) of three 6-8 week old NOD (black bars), NOD.Idd5B10 (white bars), NOD.Idd9/11NOR (grey bars) and C57BL/10 female mice (striped bars) in experiment (A) or four 6-8 week old female NOD (black bars), NOD.Idd9/11NOR (grey bars) and NOR (dotted bars) mice in a separate Experiment (B). Triplicate aliquots of 1×105 B-cells were stimulated with 1 μg/ml goat anti-mouse IgM F(ab’)2 fragments with or without 5μg/ml CD40 specific mAb. Proliferation over the final 24 hours of a 72 hour incubation period was measured by [3H] thymidine incorporation and is shown as mean Δ cpm (stimulated – non-stimulated) ± SEM for triplicate wells of each strain.
Neither Idd5 or Idd9/11 genes contribute to deletion resistance of immature B-cells in NOD mice
In non-autoimmune prone mice immature B-cells in the BM or at the T1 developmental stage in the spleen are susceptible to apoptosis induced by antigen engagement of the BCR. We previously showed both of these subpopulations in NOD mice have a higher threshold for deletion upon BCR cross-linking than those from non-autoimmune prone B6 mice (14). This aberrant phenotype could contribute to the increased numbers of mature self-reactive diabetogenic B-cells in NOD mice. We therefore investigated whether genes within Idd5 or Idd9/11 regions contribute to the higher deletion resistance of immature B-cells in NOD mice. Given they are the easier population to obtain at levels sufficient to allow experimental evaluation, we compared the sensitivity of purified splenic T1 B-cells from wild-type NOD, NOD.Idd5B10, NOD.Idd9/11NOR and B6 mice to apoptotic death by culturing them for 20 hours with various concentrations of anti-IgM F(ab’)2 fragments (Figure 5). Consistent with our previous observation (14), more NOD than B6 T1 B-cells remained viable after BCR stimulation. After BCR stimulation, both NOD.Idd5B10 and NOD.Idd9/11NOR T1 B-cells survived to a similar extent as those from standard NOD mice. This finding indicated that genes within the Idd5 or Idd9/11 region do not contribute to the decreased diabetogenic capacity of B-cells by restoring their ability to be deleted at an immature stage of development upon BCR engagement.
Figure 5. Neither the Idd5B10 or Idd9/11NOR regions can correct the deletion resistant phenotype of immature B-cells in NOD mice.

T1 B-cells (B220+ CD21− CD23−) were purified from the pooled spleens of four 6-8-week-old NOD (open diamonds, solid line), NOD.Idd5B10 (closed triangles, dashed line), NOD.Idd9/11NOR (open squares, solid line) or B6 (closed circles, dashed line) female mice as described in Material and Methods. Triplicate aliquots of 1×105 T1 B-cells from each pool were cultured in with the indicated concentrations of anti-IgM F(ab’)2 fragments for 20 hours. Cells were subsequently harvested and analyzed by flow cytometry for viability characterized by negative staining for Annexin V and PI. Results are displayed as a percentage of viable T1 B-cells remaining in culture after 20 hours of IgM stimulation compared to non-stimulated controls ± SD. Two separate experiments are shown.
Idd5 and Idd9/11 loci both play roles in regulating B-cell anergy
Immature IgHEL expressing B-cells in B6 mice that do not undergo deletion following low avidity engagement with the neo-self-antigen sHEL are instead rendered functionally anergic (37). Such anergic B-cells are normally short-lived and remain unresponsive (37), even when provided with BCR and CD40 activating signals (14). In contrast, identical transgenes on the NOD background resulted in B-cells being induced into only a weak anergic state, that was readily reversed by BCR and CD40 stimulation (14). Hence, we investigated whether genes within the Idd5 or Idd9/11 regions contribute to impaired B-cell anergy in NOD mice. B6, NOD, NOD.Idd5B10 and NOD.Idd9/11NOR genetic background mice hemizygous for the IgHEL and sHEL transgenes were intercrossed. Our previous study had shown that in B6 background mice, total numbers of HEL-specific B-cells in the spleen were decreased by ~30% when they developed in the presence versus the absence of their cognate antigen expressed as a soluble self-protein (14). In contrast, the presence of sHEL as a soluble self-antigen did not influence the number of HEL-specific B-cells found in the spleens of NOD background mice (14). Congenic introduction of homozygous Idd5B10 or Idd9/11NOR resistance loci to NOD background mice did not result in an enhanced ability of sHEL expressed as a soluble self-antigen to numerically limit the development of IgHEL expressing B-cells (data not shown). Nevertheless, compared to those maturing in the absence of soluble self-antigen, IgHEL expressing B-cells exposed to sHEL during their development in each of the B6, NOD, NOD.Idd5B10 and NOD.Idd9/11NOR genetic background stocks down-regulated IgM on their surface, were restricted from entering the marginal zone and secreted little specific antibody spontaneously (data not shown).
Our previous work (14), as well as the results described above, indicated that in NOD genetic background mice a state of anergy is induced in IgHEL expressing B-cells maturing in the presence of soluble self-antigen. However, unlike the case in B6 background controls, the anergic state of IgHEL expressing B-cells in NOD mice that had matured in the presence of soluble self-antigen is readily reversed by stimulation through the Ig and CD40 receptors (14). Thus, we determined if congenic introduction of the Idd5B10 or Idd9/11NOR resistance loci to NOD background mice resulted in a restored ability of IgHEL expressing B-cells maturing in the presence of soluble self-antigen to be anergized in a non-reversible fashion. B-cells were purified from the spleens of IgHEL single transgenic and IgHEL/sHEL double transgenic progeny from NOD, B6, NOD.Idd5B10 and NOD.Idd9NOR genetic background mice, and stimulated with IgM and CD40 cross-linking antibodies (Figure 6A, B and C). As expected, IgHEL B-cells from B6 control mice maturing in the presence sHEL were properly anergized as evidenced by lower levels of anti-IgM and CD40 stimulated proliferation than those that developed in the absence of cognate soluble self-antigen (Figure 6A). Also as previously observed, IgHEL expressing B-cells from NOD mice maturing in the presence of sHEL were not properly anergized since they proliferated equivalently in response to anti-IgM and CD40 stimulation as those that did not encounter soluble self-antigen during their development (Figure 6 A, B, C). Significantly, in contrast to NOD, but similar to B6 background mice, IgHEL expressing B-cells from both the NOD.Idd5B10 and NOD.Idd9/11NOR stocks did anergize properly when maturing in the presence of soluble self-antigen (Figure 6B and C). Restoration of proper anergy induction was irrespective of the baseline level of B-cell responsiveness in the NOD.Idd5B10 and NOD.Idd9/11NOR stocks. This was demonstrated by the finding that while IgHEL expressing B-cells from NOD.Idd9/11NOR mice that matured in the absence of sHEL were more responsive to anti-IgM and CD40 stimulation than those of standard NOD origin, when developing in the presence of cognate soluble self-antigen they were still functionally repressed. These collective results indicate one mechanism whereby genes within the Idd5 and Idd9/11 regions contribute to T1D development in NOD mice is by inducing defects in the ability of B-cells that recognize soluble self-antigens to be properly anergized.
Figure 6. B-cell anergy induction is controlled by Idd5 and Idd9/11 region genes.

B-cells were purified from pooled spleens of three 6-8 week old IgHEL single transgenic or IgHEL/sHEL double transgenic female mice. In each experiment, proliferation of purified B-cells from single (black bars) and double transgenic (grey bars) mice on the NOD genetic background were compared to single (white bars) and double transgenic mice (striped bars) on the (A) B6, (B) NOD.Idd5B10 or (C) NOD.Idd9/11NOR genetic background. B-cells were cultured with or without 1μg/ml anti-IgM F(ab’)2 fragments in combination with 5μg/ml anti-CD40 mAb. Proliferation over the final 24 hours of a 72 hour culture period was measured by [3H] thymidine incorporation and is presented as mean Δ cpm (stimulated – non-stimulated) ± SEM for triplicate wells of each strain. Each graph is representative of one of two experiments with similar results.
Discussion
This study demonstrated the development of autoreactive B-cells which serve as a critical subset of APC in T-cell mediated autoimmune T1D in NOD mice, is at least partly a consequence of pathogenic functions mediated by genes within the Idd5 and Idd9/11 disease susceptibility loci on Chr. 1 and 4, respectively. Specifically, mechanisms normally enabling B-cells to be rendered functionally anergic following low avidity Ig engagement of soluble self-antigen are impaired in NOD mice by the activity of genes within the Idd5 and Idd9/11 loci. Our experiments show clear differences in these tolerance induction mechanisms using highly purified B-cells from standard NOD mice and stocks congenic for Idd5 or Idd9/11 resistance variants. Hence, we are confident at least some polymorphic genes within the Idd5 and Idd9/11 loci act intrinsically through B-cells to allow or prevent the development of functional autoreactive clonotypes. The association of these Idd susceptibility loci with the development of pathogenic B-cells in NOD mice provides further affirmation of the important role this lymphocyte population plays in T1D development. It was also noted that while B-cells of NOR, NOD.Idd5B10, and NOD.Idd9/11NOR origin supported an overall significantly lower rate of T1D development in NOD.Igμnull recipients than those from NOD donors, in all these groups the initial kinetics of disease onset were quite similar. We currently do not have an explanation for this phenomenon.
Enlarged numbers of splenic MZ B-cells have been reported in NOD mice (14, 27, 38), which is of interest since this population is characterized by an ability to mount rapid responses to blood borne antigens and an enhanced capacity to act as APC for naïve CD4 T-cells (39). Furthermore, a link between MZ B-cells and T1D development has been proposed due to evidence showing that: [1] insulin specific B-cells have an enhanced capacity to enter the MZ compartment (40); [2] depletion of MZ B-cells protects NOD mice from disease (27); [3] linkage analysis of an F2 cross between NOD and B6 mice indicated a significant association between the Idd11 susceptibility locus and the enlarged MZ compartment (38). In spite of this evidence, we found resistance alleles within Idd5B10 or Idd9/11NOR congenic regions did not decrease the diabetogenic capacity of NOD B-cells by diminishing their entry into MZ compartment. Furthermore, our findings are consistent with a recent report showing NOD mice carrying a B6-derived Idd11 congenic region continue to have an enlarged MZ B-cell compartment (41). The discrepancy in results between linkage analyses and congenic mice casts some doubt over the ability of Idd11 to regulate MZ B-cell development. Nevertheless, our congenic stock data cannot rule out the possibility that the regulation of MZ B-cells may require interactions between genes in the Idd9/11 region and elsewhere.
NOD B-cells respond more robustly to several types of stimuli compared to those from the non-autoimmune prone B6 and BALB/c strains (34). Underlying NOD B-cell hyper-responsiveness may be dysregulations of signaling pathways such as the heightened activation potential of the cardinal pro-inflammatory transcription factor NF-κB, and also increased expression of costimulatory molecules such as CD40, CD72, CD80 and CD86 (11, 33, 42). These molecular differences may lower the activation threshold for NOD B-cells and enhance their capacity to interpret exposure to self-antigen in a pro-inflammatory context, thereby contributing further to the breakdown of tolerance in this strain. While our results are consistent with the hyper-responsiveness of NOD B-cells compared to those from non-autoimmune prone B10 and B6 mice, resistance alleles within the Idd5B10 or Idd9/11NOR regions did not dampen this phenotype. In contrast, NOR and NOD.Idd9/11NOR B-cells both proliferated more vigorously in response to activation stimuli than those of NOD origin. This indicates the super-responsiveness of NOR B-cells is controlled by a gene(s) within the Idd9/11 locus. Strong responsiveness following Ig engagement is essential for activation-induced cell death (AICD), a tolerance mechanism that normally leads to the elimination of autoimmune B-cells receiving chronic stimulation from self-antigens (43). Despite being hyper-responsive, NOD B-cells have been previously reported to be more resistant to AICD compared to those from non-autoimmune prone B6 and BALB/c mice (34), perhaps contributing to enhanced development of self-reactive clones. Interestingly, based on analyses of an otherwise NOD genetic background stock, the ability of a congenically introduced NOR derived gene(s) in the Idd9/11 region to enhance Ig mediated signaling responses did not also result in an increased capacity of immature B cells to undergo AICD upon engagement of a self antigen. The shared resistance of NOD and NOR B-cells to AICD was also previously observed by Hussain et. al. (34). This may be due to the fact that while differing at the Idd9/11 locus, NOD and NOR mice share many other T1D susceptibility genes which could contribute to the impaired ability of B-cells from both strains to undergo AICD. However, while not affecting susceptibility to AICD, our data indicate that the ability of the NOR derived Idd9/11 resistance locus to allow for enhanced Ig receptor mediated responsiveness is likely to increase the ability of autoreactive B-cells to be anergized following engagement of self-antigen.
The impaired ability of immature NOD B-cells to undergo deletion upon Ig cross-linking is likely to result in increased numbers of self-reactive clonotypes that mature and serve as APC for diabetogenic CD4 T-cells. However, our studies showed that allelic variants causing diabetes resistance within the Idd5 or Idd9/11 regions were both unable to correct the deletion resistance of immature B-cells in NOD mice. The inability of Idd5 genes to control deletion of immature B-cells was particularly surprising given previous reports have mapped a locus to this region in NOD mice that causes apoptosis resistance in total lymphocytes, mature T-cells and thymocytes (44-46). Although this difference may be indicative of distinct genetic control of apoptosis resistance in T and B-cells of NOD mice, we cannot discount that genes within Idd5 may still be able to regulate the deletion of B-cells upon interaction with other Idd genes. This may also be the case for T-cells since the impaired deletion of thymocytes in NOD mice has been demonstrated to be the product of several Idd genes in addition to Idd5 (47, 48). Nevertheless, our data indicate that both the Idd5B10 or Idd9/11NOR regions independently decrease the development of diabetogenic B-cells through the regulation of some mechanism other than the induction of deletion.
Anergy induction serves as another important mechanism for the maintenance of B-cell tolerance to soluble antigens (49), such as the T1D-relevant autoantigen insulin (50). Using the IgHEL/sHEL transgenic model, we previously showed that unlike those from B6 controls, NOD B-cells developing in the presence of cognate soluble self-antigen are maintained in only a very weak anergic state that can be readily reversed by BCR and CD40 stimulation (14). The aberrant ability of self-reactive B-cells in NOD mice to overcome anergy after BCR and CD40 stimulation would seem relevant to their increased capacity to act as diabetogenic APC, since this defect should allow them to rapidly expand after initially encountering their antigen in the presence of CD40 ligand positive β-cell autoreactive CD4 T-cells. A recent study by Acaveda-Suarez et. al. (40) addressed whether similar anergy defects give rise to B-cells specific for pancreatic β-cell antigens in NOD mice. This was achieved by introducing an insulin-specific Ig transgene into NOD and B6 mice. In contrast to results in IgHEL/sHEL transgenic mice, anergy induction was equivalent in NOD and B6 B-cells expressing this particular insulin-reactive Ig transgene. However, this result was inconsistent with the fact that insulin-specific autoantibodies are detected in standard NOD but not B6 mice (51), implying non-anergic B-cells specific for this β-cell autoantigen are generated in the former strain. One possible reason for this contradiction might be the insulin-reactive B-cells which remain functional in standard NOD mice bypass tolerance induction because the Ig they express have a lower functional avidity for antigen than the transgenic variant studied by Acaveda-Suarez et. al. Our current study strongly implicates defective induction of B-cell anergy as an important contributory factor to T1D development in NOD mice. This conclusion is supported by the finding that compared to those from standard NOD mice, B-cells from NOD stocks congenic for the Idd9/11NOR or Idd5B10 resistance regions had a decreased ability to support T1D development in NOD.Igμnull recipients, and also showed an enhanced ability to be anergized when maturing in the presence of cognate soluble self-antigen.
In future studies, B-cell phenotypes controlled by the Idd5 and Idd9/11 loci will be used to aid in susceptibility gene discovery by both a candidate approach and microarray analyses. Understanding the underlying molecular and genetic aspects leading to the development of autoreactive B-cells contributing to T1D may not only advance our understanding of disease pathogenesis, but also illuminate potential new targets for therapeutic intervention in humans.
Footnotes
P.S. is supported by Peter Doherty Fellowship from the National Health and Medical Research Council (NHMRC) and grants from the Cecilia Kilkeary Foundation, Clive and Vera Ramaciotti Foundation, NHMRC (402727) and the Juvenile Diabetes Research Foundation (JDRF). D.V.S. is supported by grants from the National Institutes of Health (DK46266 and DK51090) and the Juvenile Diabetes Research Foundation.
- AICD
- Activation induced cell death
- B6
- C57BL/6
- B10
- C57BL/10
- BM
- bone marrow
- HEL
- hen egg lysozyme
- NOD
- non-obese diabetic
- NOR
- non-obese resistant
- SBM
- syngeneic bone marrow
- T1D
- Type 1 Diabetes
- PI
- propidium iodide
References
- 1.Fox CJ, Danska JS. Independent genetic regulation of T-cell and antigen-presenting cell participation in autoimmune islet inflammation. Diabetes. 1998;47:331–338. doi: 10.2337/diabetes.47.3.331. [DOI] [PubMed] [Google Scholar]
- 2.Jarpe AJ, Hickman MR, Anderson JT, Winter WE, Peck AB. Flow cytometric enumeration of mononuclear cell populations infiltrating the islets of Langer-hans in prediabetic NOD mice: development of a model of autoimmune insulitis for type I diabetes. Reg Immunol. 1990;3:305–317. [PubMed] [Google Scholar]
- 3.Gottlieb PA, Eisenbarth GS. Diagnosis and treatment of pre-insulin dependent diabetes. Annu Rev Med. 1998;49:391–405. doi: 10.1146/annurev.med.49.1.391. [DOI] [PubMed] [Google Scholar]
- 4.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–946. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 5.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T-cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, Roep BO. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345:1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 7.Greeley SA, Katsumata M, Yu L, Eisenbarth GS, Moore DJ, Goodarzi H, Barker CF, Naji A, Noorchashm H. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 8.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T-cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–3918. [PubMed] [Google Scholar]
- 9.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol. 1998;161:1163–1168. [PubMed] [Google Scholar]
- 10.Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, Song HK, Noto LE, Jevnikar AM, Barker CF, Naji A. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T-cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163:743–750. [PubMed] [Google Scholar]
- 11.Wheat W, Kupfer R, Gutches DG, Rayat GR, Beilke J, Scheinman RI, Wegmann DR. Increased NF-kappa B activity in B-cells and bone marrow-derived dendritic cells from NOD mice. Eur J Immunol. 2004;34:1395–1404. doi: 10.1002/eji.200324490. [DOI] [PubMed] [Google Scholar]
- 12.Silveira PA, Baxter AG. The NOD mouse as a model of SLE. Autoimmunity. 2001;34:53–64. doi: 10.3109/08916930108994126. [DOI] [PubMed] [Google Scholar]
- 13.Hulbert C, Riseili B, Rojas M, Thomas JW. B-cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol. 2001;167:5535–5538. doi: 10.4049/jimmunol.167.10.5535. [DOI] [PubMed] [Google Scholar]
- 14.Silveira PA, Dombrowsky J, Johnson E, Chapman HD, Nemazee D, Serreze DV. B-cell selection defects underlie the development of diabetogenic APCs in nonobese diabetic mice. J Immunol. 2004;172:5086–5094. doi: 10.4049/jimmunol.172.8.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Santamaria P. Dissecting autoimmune diabetes through genetic manipulation of non-obese diabetic mice. Diabetologia. 2003;46:1447–1464. doi: 10.1007/s00125-003-1218-1. [DOI] [PubMed] [Google Scholar]
- 16.Serreze DV, Leiter EH. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr Dir Autoimmun. 2001;4:31–67. doi: 10.1159/000060527. [DOI] [PubMed] [Google Scholar]
- 17.Johnson EA, Silveira P, Chapman HD, Leiter EH, Serreze DV. Inhibition of autoimmune diabetes in nonobese diabetic mice by transgenic restoration of H2-E MHC class II expression: additive, but unequal, involvement of multiple APC subtypes. J Immunol. 2001;167:2404–2410. doi: 10.4049/jimmunol.167.4.2404. [DOI] [PubMed] [Google Scholar]
- 18.Prochazka M, Serreze DV, Frankel WN, Leiter EH. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes. 1992;41:98–106. doi: 10.2337/diab.41.1.98. [DOI] [PubMed] [Google Scholar]
- 19.Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes. 2000;49:1744–1747. doi: 10.2337/diabetes.49.10.1744. [DOI] [PubMed] [Google Scholar]
- 20.Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL, Howson JM, Smink LJ, Kingsnorth A, Lyons PA, Gregory S, Rogers J, Todd JA, Peterson LB. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol. 2004;173:164–173. doi: 10.4049/jimmunol.173.1.164. [DOI] [PubMed] [Google Scholar]
- 21.Serreze DV, Bridgett M, Chapman HD, Chen E, Richard SD, Leiter EH. Subcongenic analysis of the Idd13 locus in NOD/Lt mice: evidence for several susceptibility genes including a possible diabetogenic role for beta 2-microglobulin. J Immunol. 1998;160:1472–1478. [PubMed] [Google Scholar]
- 22.Reifsnyder PC, Li R, Silveira PA, Churchill G, Serreze DV, Leiter EH. Conditioning the genome identifies additional diabetes resistance loci in Type I diabetes resistant NOR/Lt mice. Genes Immun. 2005 doi: 10.1038/sj.gene.6364241. [DOI] [PubMed] [Google Scholar]
- 23.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 24.Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol. 2002;32:3657–3666. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 25.Serreze DV, Prochazka M, Reifsnyder PC, Bridgett MM, Leiter EH. Use of recombinant congenic and congenic strains of NOD mice to identify a new insulin-dependent diabetes resistance gene. J Exp Med. 1994;180:1553–1558. doi: 10.1084/jem.180.4.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B-cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol. 2004;34:2387–2395. doi: 10.1002/eji.200324744. [DOI] [PubMed] [Google Scholar]
- 27.Noorchashm H, Moore DJ, Lieu YK, Noorchashm N, Schlachterman A, Song HK, Barker CF, Naji A. Contribution of the innate immune system to autoimmune diabetes: a role for the CR1/CR2 complement receptors. Cell Immunol. 1999;195:75–79. doi: 10.1006/cimm.1999.1522. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Li H, Ni D, Weigert M. Anti-DNA B-cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196:1543–1552. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuster H, Martin T, Marcellin L, Garaud JC, Pasquali JL, Korganow AS. Expansion of marginal zone B-cells is not sufficient for the development of renal disease in NZBxNZW F1 mice. Lupus. 2002;11:277–286. doi: 10.1191/0961203302lu191oa. [DOI] [PubMed] [Google Scholar]
- 30.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B-cell subsets reveals multiple selection points during peripheral B-cell maturation. J Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 32.Langmuir PB, Bridgett MM, Bothwell AL, Crispe IN. Bone marrow abnormalities in the non-obese diabetic mouse. Int Immunol. 1993;5:169–177. doi: 10.1093/intimm/5.2.169. [DOI] [PubMed] [Google Scholar]
- 33.Hussain S, Delovitch TL. Dysregulated B7-1 and B7-2 expression on nonobese diabetic mouse B-cells is associated with increased T-cell costimulation and the development of insulitis. J Immunol. 2005;174:680–687. doi: 10.4049/jimmunol.174.2.680. [DOI] [PubMed] [Google Scholar]
- 34.Hussain S, Salojin KV, Delovitch TL. Hyperresponsiveness, resistance to B-cell receptor-dependent activation-induced cell death, and accumulation of hyperactivated B-cells in islets is associated with the onset of insulitis but not type 1 diabetes. Diabetes. 2004;53:2003–2011. doi: 10.2337/diabetes.53.8.2003. [DOI] [PubMed] [Google Scholar]
- 35.Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T-cells in NOD mice. J Exp Med. 2002;196:369–377. doi: 10.1084/jem.20011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of isleT-cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodnow CC. Transgenic mice and analysis of B-cell tolerance. Annu Rev Immunol. 1992;10:489–518. doi: 10.1146/annurev.iy.10.040192.002421. [DOI] [PubMed] [Google Scholar]
- 38.Rolf J, Motta V, Duarte N, Lundholm M, Berntman E, Bergman ML, Sorokin L, Cardell SL, Holmberg D. The enlarged population of marginal zone/CD1d(high) B lymphocytes in nonobese diabetic mice maps to diabetes susceptibility region Idd11. J Immunol. 2005;174:4821–4827. doi: 10.4049/jimmunol.174.8.4821. [DOI] [PubMed] [Google Scholar]
- 39.Pillai S, Cariappa A, Moran ST. Marginal zone B-cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 40.Acevedo-Suarez CA, Hulbert C, Woodward EJ, Thomas JW. Uncoupling of anergy from developmental arrest in anti-insulin B-cells supports the development of autoimmune diabetes. J Immunol. 2005;174:827–833. doi: 10.4049/jimmunol.174.2.827. [DOI] [PubMed] [Google Scholar]
- 41.Brodnicki TC, O’Donnell K, Quirk F, Tarlinton DM. Congenic nonobese diabetic mouse strains fail to confirm linkage of a marginal zone B lymphocyte phenotype to the Idd11 locus on chromosome 4. J Immunol. 2006;176:701–702. doi: 10.4049/jimmunol.176.2.701. author reply 702. [DOI] [PubMed] [Google Scholar]
- 42.Rojas A, Xu F, Rojas M, Thomas JW. Structure and function of CD72 in the non-obese diabetic (NOD) mouse. Autoimmunity. 2003;36:233–239. doi: 10.1080/0891693031000141059. [DOI] [PubMed] [Google Scholar]
- 43.Parry SL, Hasbold J, Holman M, Klaus GG. Hypercross-linking surface IgM or IgD receptors on mature B-cells induces apoptosis that is reversed by costimulation with IL-4 and anti-CD40. J Immunol. 1994;152:2821–2829. [PubMed] [Google Scholar]
- 44.Colucci F, Bergman ML, Penha-Goncalves C, Cilio CM, Holmberg D. Apoptosis resistance of nonobese diabetic peripheral lymphocytes linked to the Idd5 diabetes susceptibility region. Proc Natl Acad Sci U S A. 1997;94:8670–8674. doi: 10.1073/pnas.94.16.8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garchon HJ, Luan JJ, Eloy L, Bedossa P, Bach JF. Genetic analysis of immune dysfunction in non-obese diabetic (NOD) mice: mapping of a susceptibility locus close to the Bcl-2 gene correlates with increased resistance of NOD T-cells to apoptosis induction. Eur J Immunol. 1994;24:380–384. doi: 10.1002/eji.1830240217. [DOI] [PubMed] [Google Scholar]
- 46.Bergman ML, Cilio CM, Penha-Goncalves C, Lamhamedi-Cherradi SE, Lofgren A, Colucci F, Lejon K, Garchon HJ, Holmberg D. CTLA-4−/− mice display T-cell-apoptosis resistance resembling that ascribed to autoimmune-prone non-obese diabetic (NOD) mice. J Autoimmun. 2001;16:105–113. doi: 10.1006/jaut.2000.0474. [DOI] [PubMed] [Google Scholar]
- 47.Liston A, Lesage S, Gray DH, O’Reilly LA, Strasser A, Fahrer AM, Boyd RL, Wilson J, Baxter AG, Gallo EM, Crabtree GR, Peng K, Wilson SR, Goodnow CC. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity. 2004;21:817–830. doi: 10.1016/j.immuni.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 48.Bergman ML, Duarte N, Campino S, Lundholm M, Motta V, Lejon K, Penha-Goncalves C, Holmberg D. Diabetes protection and restoration of thymocyte apoptosis in NOD Idd6 congenic strains. Diabetes. 2003;52:1677–1682. doi: 10.2337/diabetes.52.7.1677. [DOI] [PubMed] [Google Scholar]
- 49.Seo SJ, Mandik-Nayak L, Erikson J. B-cell anergy and systemic lupus erythematosus. Curr Dir Autoimmun. 2003;6:1–20. doi: 10.1159/000066853. [DOI] [PubMed] [Google Scholar]
- 50.Rojas M, Hulbert C, Thomas JW. Anergy and not clonal ignorance determines the fate of B-cells that recognize a physiological autoantigen. J Immunol. 2001;166:3194–3200. doi: 10.4049/jimmunol.166.5.3194. [DOI] [PubMed] [Google Scholar]
- 51.Bonifacio E, Atkinson M, Eisenbarth G, Serreze D, Kay TW, Lee-Chan E, Singh B. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes. 2001;50:2451–2458. doi: 10.2337/diabetes.50.11.2451. [DOI] [PubMed] [Google Scholar]