

Abstract
Motivation: PSORTb has remained the most precise bacterial protein subcellular localization (SCL) predictor since it was first made available in 2003. However, the recall needs to be improved and no accurate SCL predictors yet make predictions for archaea, nor differentiate important localization subcategories, such as proteins targeted to a host cell or bacterial hyperstructures/organelles. Such improvements should preferably be encompassed in a freely available web-based predictor that can also be used as a standalone program.
Results: We developed PSORTb version 3.0 with improved recall, higher proteome-scale prediction coverage, and new refined localization subcategories. It is the first SCL predictor specifically geared for all prokaryotes, including archaea and bacteria with atypical membrane/cell wall topologies. It features an improved standalone program, with a new batch results delivery system complementing its web interface. We evaluated the most accurate SCL predictors using 5-fold cross validation plus we performed an independent proteomics analysis, showing that PSORTb 3.0 is the most accurate but can benefit from being complemented by Proteome Analyst predictions.
Availability: http://www.psort.org/psortb (download open source software or use the web interface).
Contact: psort-mail@sfu.ca
Supplementary Information: Supplementary data are availableat Bioinformatics online.
1 INTRODUCTION
Computational prediction of bacterial protein subcellular localization (SCL) provides a quick and inexpensive means for gaining insight into protein function, verifying experimental results, annotating newly sequenced bacterial genomes, detecting potential cell surface/secreted drug targets, as well as identifying biomarkers for microbes. In recent years, this area of computational research has achieved an impressive level of precision (Gardy and Brinkman, 2006), allowing SCL prediction tools to be reliably integrated into automated proteome annotation pipelines and to complement analyses of high-throughput proteomics experiments.
PSORTb version 2.0 (Gardy et al., 2005), the most precise bacterial SCL prediction software (Gardy and Brinkman, 2006), was introduced in 2005, and has been widely used for the SCL prediction of individual proteins as well as for whole proteomes. It generates prediction results for five major localizations for Gram-negative bacteria (cytoplasmic, inner membrane, periplasmic, outer membrane and extracellular) and four localizations for Gram-positive bacteria (cytoplasmic, cytoplasmic membrane, cell wall and extracellular). Since then, numerous SCL prediction tools have been created for bacteria using a variety of machine learning algorithms: CELLO version 2.0 (Yu et al., 2006) uses multi-layered support vector machines (SVMs); SLP-Local predicts SCLs based on local composition and distance frequencies of amino acid groups (Matsuda et al., 2005); PSL101 makes predictions based on amino acid compositions coupled with structural feature conservations (Su et al., 2007); and PSLDoc bases its SVM features on gapped di-peptides (Chang et al., 2008). Other tools such as Gpos-PLoc (Shen and Chou, 2007) and Gneg-PLoc (Chou and Shen, 2006) make predictions for bacterial proteins by clustering Swiss-Prot proteins with annotated SCLs based on their GO terms and amino acid properties using the K-nearest neighbor algorithm. Some methods, such as SubcellPredict and HensBC, combine multiple classifying algorithms in order to boost the prediction performance (Bulashevska and Eils, 2006; Niu et al., 2008). LocateP (Zhou et al., 2008) and Augur (Billion et al., 2006) differentiate between different types of membrane-anchored, cell wall-anchored and secreted proteins for Gram-positive bacterial proteomes. Based on the principle that training datasets could benefit from being genus specific, TBPred (Rashid et al., 2007) was developed specifically for the genus of Mycobacterium spp.
Even though many bacterial SCL prediction methods have been published, most of them focus on optimizing prediction accuracy—maximizing the number of positive predictions on the training dataset, at the expense of producing more false positive results. Furthermore, none of the current bacterial SCL predictors provide standalone versions of software for users. Most web servers also do not provide convenient means for analyzing whole bacterial proteomes. PSORTb remains one of the most user-friendly bacterial SCL prediction tools, providing both a web server and a standalone version, and allowing for both single and batch sequence processing. Its accompanying database, PSORTdb (Rey et al., 2005b), provides a dataset of experimentally verified protein localizations, as well as pre-computed prediction results for more than 1000 sequenced bacterial genomes available from NCBI. Because of its focus on maintaining high precision, it does not return a forced prediction if the localization score does not reach a minimum cut-off. As a result, only ∼50% of proteins encoded in Gram-negative bacterial genomes and ∼75% of proteins encoded in Gram-positive bacterial genomes receive a prediction from PSORTb. Thus, there is a need to produce an updated version with better genome coverage.
The current localization classifications for PSORTb and most existing SCL prediction software do not provide any information on proteins targeted to specialized bacterial hyperstructures/organelles such as the flagellum, the fimbrium/pilus or proteins destined to the host cell. Gneg-PLoc (Chou and Shen, 2006) attempts to address this by providing prediction categories for the nucleosome (DNA-binding proteins) and the flagellum. Gpos-PLoc (Shen and Chou, 2007) provides predictions for Gram-positive periplasmic proteins. Some studies have attempted to predict effector proteins secreted by the Type III secretion system based on N-terminal signal sequence of proteins (Arnold et al., 2009; Samudrala et al., 2009). Ideally, comprehensive SCL prediction software should incorporate predictions for these more specialized compartments in addition to reporting major SCLs.
Typically, bacterial organisms that stain Gram-positive consist of one cytoplasmic membrane and a thick cell wall, whereas a Gram-negative organism is enclosed by a thin cell wall within a periplasm and an outer membrane that surrounds the entire cell. However, some bacteria have cell structures that do not fit with the classical Gram-negative or Gram-positive cell model. For example, Mycoplasma spp. and other members of the phylum Tenericutes stain Gram-negative, yet they have no outer membrane or cell wall (Miyata and Ogaki, 2006). Deinococcus spp. has a thick cell wall and is considered as a Gram-positive organism, but they also have an outer membrane (Thompson and Murray, 1981). Therefore, to make protein SCL predictions for all prokaryotes, not only does an archaeal predictor need to be created, but we also need to be able to make a predictor that can handle the four possible bacterial cell structures that we now know are possible: Gram-positive without an outer membrane (i.e. traditional Gram-positives), Gram-negative with an outer membrane (i.e. traditional Gram-negatives), Gram-positive with an outer membrane and Gram-negative without an outer membrane. Only then is a predictor able to cover the true diversity of prokaryotic life, which will become more important as increased sampling of prokaryotes occurs through metagenomics and other projects (Wu et al., 2009).
In addition to bacterial SCL prediction algorithms, several software packages for predicting SCL of eukaryotic proteins have been developed, despite the fact that they are much harder to predict due to the greater complexity of eukaryotic cells (see http://www.psort.org for a list of available eukaryotic protein SCL predictors). However, there are no dedicated SCL prediction tools for archaea, the third domain of life whose basic cellular compartments are similar to that of a Gram-positive bacterium. Not only do they represent an entire domain of abundant organisms that inhabit the earth, they produce many thermotolerant and halotolerant enzymes that have wide industrial applications (de Champdore et al., 2007). Furthermore, identification of novel cell surface and secreted proteins can also be very helpful for designing new methods for the detection of specific archaeal species in the environment.
To address these issues, we have created PSORTb version 3.0, with a significant increase in recall of predictions as well as proteome prediction coverage while maintaining high precision (see 2.3 Software evaluations—using literature and Swiss-Prot-based datasets for definitions of precision and recall). In addition, we recognize that the current localization classification scheme does not adequately cover all bacterial proteins' detailed localization sites. Therefore, we have added new localization subcategories commonly found in many groups of bacteria—the first subcategory localization system for an SCL predictor. Options specifically for predicting archaeal proteins and proteins in organisms with membrane structures not reflecting Gram stains have also been implemented. We further improved usability by adding an online batch submission system with formatted results returned by email. For the standalone version, we have simplified the installation procedure. Finally, we examined the results of combining complementary SCL predictions in order to produce accurate predictions for the majority of prokaryotic proteomes, using an independent, proteomics-derived laboratory test dataset to aid the analysis.
2 METHODS
2.1 Training dataset
The training dataset contains data from ePSORTdb 2.0 (Rey et al., 2005b), which was used to build PSORTb 2.0, Swiss-Prot version 49 (Wu et al., 2006), plus protein localization data obtained from manual literature search (the latter comprises 30% of the dataset). From Swiss-Prot, protein localizations were based on the ‘Comments—Subcellular location’ field with review. A natural language processing predictive model, TeGRR (Melli et al., 2007), was used as a text mining technique on literature abstracts to confirm the validity of the Swissprot SCL annotation. Organisms were separated into Gram-positive and Gram-negative groups based on their phylum/class and literature review. Bacteria belonging to the phyla of Actinobacteria, Chloroflexi and Deinococcus-Thermus of the order Thermales, Firmicutes of class Bacilli and most Clostridia were categorized as Gram-positive bacteria. Bacteria in phylum groups not mentioned above were categorized as Gram-negative. For proteins from the Swissprot library with annotated subcellular locations, those labeled as ‘fragment’, ‘by similarity’, ‘probable’ and ‘potential’ were removed. Those that were annotated with very specialized localizations such as ‘chlorosome’ and ‘chromatophore’ were not used for this dataset. Proteins that were labeled with ambiguous terms such as ‘cell envelope’ were manually confirmed for their specific localization if possible, or discarded if the precise localization could not be determined. Some protein entries were manually retrieved from the literature as well as the EcoSal database (http://www.ecosal.org) and the Pseudomonas Genome Database (Winsor et al., 2008). The archaeal testing dataset was obtained in a similar fashion as the bacterial dataset. The training dataset for building the archaeal predictor was created by combining archaeal proteins with Gram-positive/Gram-negative cytoplasmic and cytoplasmic membrane proteins, as well as Gram-positive cell wall and extracellular proteins, as this was found to notably increase accuracy when evaluated using archaeal proteins. In total, the Gram-negative training dataset has expanded from 1572 to 8230 proteins; the Gram-positive dataset has increased from 576 to 2652 proteins, and 810 archaeal proteins have been added to the training dataset. The full training dataset is available at http://www.psort.org/dataset/datasetv3.html.
2.2 Software implementation and updates
2.2.1 New localization subcategories
To account for proteins targeted to some of the common bacterial hyperstructures and host-destined SCLs, new subcategory localizations have been introduced in PSORTb 3.0, as listed in Table 1. This represents, to our knowledge, the first implementation of subcategories for primary SCL localizations, for an SCL predictor. These subcategory localizations for a protein were identified using the SCL-BLAST module, which infers localization by homology using criteria that are of measured high precision (Nair and Rost, 2002). Proteins detected to have a secondary localization are also predicted as one of the four main categories for Gram-positive bacteria or one of five main compartments for Gram-negative bacteria (or similarly for those bacteria with atypical cell structures). Any protein exported past the outer-most layer of the bacterial cell is considered as extracellular, whereas proteins localized to one of the membranes that are part of a hyperstructure (such as the flagellum) are identified both as an inner or outer membrane protein as well as a protein of that hyperstructure. The basal components of the flagellum are not annotated as such, since they are often homologous to proteins that are not part of the flagellar apparatus (for example, a general ATPase).
Table 1.
New subcategory SCLs predicted by PSORTb 3.0
| SCL subcategories | Description |
|---|---|
| Host-associated | Any proteins destined to the host cell cytoplasm, cell membrane or nucleus by any of the bacterial secretion systems |
| Type III secretion | Components of the Type III secretion apparatus |
| Fimbrial | Components of a bacterial or archaeal fimbrium or pilus |
| Flagellar | Components of a bacterial or archaeal flagellum |
| Spore | Components of a spore |
2.2.2 Implementation changes to software
The implementation of the new version of PSORTb is similar to version 2.0 (Gardy et al., 2005), with the following changes: motifs that provided false prediction results were either updated or removed. SCL-BLASTdbs for both Gram-positive and Gram-negative options were updated with the newly expanded dataset. The transmembrane α-helix predictor module HMMTOP (Tusnady and Simon, 2001) was replaced with S-TMHMM, an open source transmembrane α-helix predictor (Viklund and Elofsson, 2004). The program was modified such that the software reports the number of helices predicted. As with the PSORTb 2.0 set-up, this module first examined if an alpha helix was predicted in the first 70 amino acid residues; if so, this helix would be subtracted. It then examined the rest of the protein sequence, returning a positive prediction if more than two helices were found, to ensure high precision. Although this leads to membrane-associated proteins being under-predicted by this module, such proteins are instead predicted by the SCL-BLAST module and SVMs (mentioned below).
All SVMs, except for the Gram-negative outer membrane SVM module and Gram-positive cytoplasmic SVM module, were retrained with the new dataset following the protocols of PSORTb 2.0 paper (Gardy et al., 2005). The aforementioned two SVMs were not updated because the new SVMs did not improve significantly in performance when retrained. For PSORTb 2.0, we made use of an implementation of generalized suffix tree (Wang et al., 1994) to extract frequent subsequences, which occur in more than a predefined fraction of total number of proteins of interest. These frequent subsequences were used as features to discriminate localizations of related proteins. The implementation first sampled a subset of related proteins, then extracted frequent subsequences from this subset and finally checked whether these frequent subsequences were frequent in all related proteins. This method may miss some frequent subsequences or produce false positives. To overcome this issue, we used another augmentation of generalized suffix tree (Matias et al., 1998). The algorithm guarantees returning all the frequent subsequences and its running time is in the order of the total length of the related protein sequences.
A Bayesian network combines all module predictions and generates one final localization result based on the performance accuracies of each of the updated modules.
2.2.3 New prediction categories for archaea and atypical prokaryotic organisms
The SCL predictor for archaea was implemented with similar components as the Gram-positive predictor, producing predictions for four localizations and two subcategory localizations (flagellum and fimbrium), but using the archaeal training dataset mentioned above. Any motifs that reduced the precision for archaeal SCL prediction were removed.
Two other categories were implemented for bacteria with atypical cellular structures–organisms that stain Gram-positive but have an outer membrane, and organisms that stain Gram-negative but have no outer membrane. For the former category, the Gram-negative pipeline was employed, which enables outer membrane and periplasmic localizations to be predicted. For the latter category, the Gram-positive modules were used, but the cell wall localization prediction was disabled, since the intended organisms (i.e. Tenericutes) lack cell walls.
2.2.4 Software usability improvements
To improve usability of the new software version, the web interface of PSORTb 3 now allows user to upload a batch job (such as an entire proteome), and a formatted results file is returned to the user by email when computations are completed. The installation process of the standalone software has also been improved such that the process requires fewer packages and can be installed in a more automated manner. PSORTb 3.0 works with most versions of Linux as well as Mac OS X (except Snow Leopard at press time).
2.3 Software evaluations—using literature and Swiss-Prot-based datasets
Five-fold cross validation was performed on the updated Gram-positive bacteria, Gram-negative bacteria and archaeal datasets using the approach as described in the PSORTb 2.0 paper (Gardy et al., 2005). To use this new dataset to evaluate the performance of other SCL predictors, proteins from the training set of PSORTb 2.0 were subtracted from this evaluation dataset, since this particular set of proteins is included in the training dataset of most of the bacterial SCL prediction tools. To improve the robustness of the assessment of accuracy, homology reduction was performed on the testing datasets using CD-HIT (Li and Godzik, 2006) such that none of the sequences in the testing set exhibited >80% identity with other sequences in the set. Performance metrics used to evaluate different software include precision, defined as TP/(TP + FP); recall, defined as TP/(TP + FN); accuracy, defined as (TP + TN)/(TP + TN + FP + FN); and Matthew's Coefficient Constant (MCC), defined as
| (1) |
The following web servers were benchmarked for their predictive capabilities, in addition to PSORTb versions 2.0 and 3.0: CELLO version 2.0 (Yu et al., 2006), Gneg-PLoc (Chou and Shen, 2006), Gpos-PLoc (Shen and Chou, 2007), and Proteome Analyst version 2.5 (PA 2.5; Lu et al., 2004), whose performance was previously shown to be comparable to PSORTb 2.0 (Gardy and Brinkman, 2006). Proteome Analyst 3.0 (PA 3.0), an unpublished method, was also included in this benchmark analysis, though it could only be evaluated using a new proteomics-derived experimental dataset, since we could not confirm that our test data was not in the training data for this software. Methods that are specific to an organism, such as TBPred (Rashid et al., 2007), and methods that do not allow for user submission of protein sequences, such as LocateP (Zhou et al., 2008) and Augur (Billion et al., 2006), could not be included in this comparison. Two of the recently developed methods, PSL101 (Su et al., 2007) and PSLDoc (Chang et al., 2008) were not tested since the servers could not handle large testing datasets. Once the level of precision was determined for each software, those with highest precision were also evaluated for ‘proteome coverage’, i.e. the proportion of proteins predicted in a deduced proteome from a genome, at that level of precision.
2.4 Proteomics analysis
We performed a laboratory analysis to construct an experimental dataset of proteins from a Gram-negative bacterium, Pseudomonas aeruginosa PA01, which was used to assess PSORTb 2.0, PSORTb 3.0, PA 2.5 and PA 3.0. This represents an independent dataset that includes hypothetical and uncharacterized proteins with previously unknown SCLs. P.aeruginosa is a bacterium noted for its diverse metabolic capacity and large genome/proteome size, and so represents an excellent organism with which to generate such a dataset (Stover et al., 2000). To generate this experimental dataset, we extracted protein samples from the cytoplasmic, periplasmic and secreted fractions of P.aeruginosa PA01. The resulting proteins in each fraction were digested to peptides and differentially labeled using formaldehyde isotopologues (Chan and Foster, 2008) prior to analysis by liquid chromatography–tandem mass spectrometry (LC–MS/MS), exactly as previously described (Chan et al., 2006). Abundance ratios between SCL were calculated using MSQuant (http://msquant.sourceforge.net/). To ensure a high-quality dataset with minimal contaminating proteins from other subcellular compartments, proteins that were only found in the cytoplasmic fraction and never in the other two soluble fractions were used to assess PSORTb 3.0 and PA 3.0 prediction results. This dataset was also felt to be most appropriate for assessment, since our analysis had suggested that most proteins of previously unknown localization in the old version of PSORTb were most likely cytoplasmic proteins. Further details on the experimental protocols for this proteomics analysis of the subcellular fractions can be found in Supplementary Material—methods for mass spectrometry protein identification.
3 RESULTS
3.1 PSORTb 3.0: expanded predictive capabilities for all prokaryotes and localization subcategories
We present version 3.0 of PSORTb. Like the version 2 series, version 3.0 has the capability to make predictions for all Bacteria, but now makes predictions for archaea and bacteria with atypical cell wall/membrane structures as well. Users must simply select the Domain of life (Bacteria or Archaea) and, in the case of bacteria, select whether the organism is Gram-positive or Gram-negative or ‘Advanced’ (i.e. Gram-positive with an outer membrane or Gram-negative without an outer membrane). Localization predictions now include a subcategorization (see Section 2 as well as Table 1) for more precise identification of localizations (i.e. a protein may be in the outer membrane but also be a component of the flagellar machinery, so it would be classified as ‘outer membrane’, with a subcategory classification as ‘flagellar’).
3.2 PSORTb 3.0 outperforms PSORTb 2.0 and other SCL prediction tools in terms of precision and recall for bacterial proteins
The overall performance for PSORTb 3.0, calculated using 5-fold cross validation, along with the performance of other recently published bacterial SCL prediction tools tested using the homology-reduced dataset, are shown in Table 2. The SCL-specific performance values for each predictor can be found in Supplementary Tables 1 and 2. For the Gram-positive option, both PSORTb 3.0 and PSORTb 2.0 exhibit precision values >97%, whereas CELLO 2.5, Gpos-PLoc and PA 2.5 measured <95%. Overall recall values were above 90% for all benchmarked software except for PA 2.5, which seems to have an especially low recall (11.5%) for membrane proteins. For the Gram-negative option, PSORTb 3.0 still maintains the highest precision of 97.3% and the highest recall of 94.1%, where recall has increased by 8.8% compared to PSORTb 2.0. PA 2.5, which was previously shown to be comparable to PSORTb 2.0, still exhibits comparable precision (97.3%) and recall (92.0%) with this new test dataset. Although SubcellPredict and SLP-Local also show high overall precision and recall values, their precision values for the periplasmic localization prediction are under 55%. Gneg-PLoc and CELLO 2.5, having precision values <90%, also exhibit lower specificities for periplasmic localizations (56.5 and 35.2%, respectively) as well as outer membrane localizations (66.4 and 34.6%, respectively). Overall, PSORTb 3.0 appears to be the most accurate versus all other comparable methods that were tested. Compared to PSORTb 2.0, PSORTb 3.0 appears to predict more cytoplasmic proteins in particular, reflecting difficulty in identifying the localization of such proteins without an improved training dataset (since literally they have no signals to transport them to other localizations that may be detected). PSORTb 3.0 has a marked improvement over PSORTb 2.0 in recall in particular for Gram-negatives, representing a significant improvement in predictive capability for the only SCL predictor of its kind that is freely available as a standalone package.
Table 2.
Performance comparisons for Gram-positive and Gram-negative bacterial SCL prediction software
| Softwarea | Precisionb | Recallb | Accuracyb | MCCb |
|---|---|---|---|---|
| Gram-positive | ||||
| PSORTb 3.0 | 98.2 | 93.1 | 97.9 | 0.79 |
| PSORTb 2.0 | 97.0 | 90.0 | 96.8 | 0.76 |
| CELLO 2.5 | 93.7 | 93.7 | 96.9 | 0.76 |
| Gpos-PLocc | 91.2 | 90.7 | 95.5 | 0.64 |
| PA 2.5d | 90.0 | 81.8 | 90.9 | 0.57 |
| Gram-negative | ||||
| PSORTb 3.0 | 97.3 | 94.1 | 98.3 | 0.85 |
| PA 2.5 | 97.3 | 92.0 | 97.9 | 0.85 |
| PSORTb 2.0 | 95.9 | 85.3 | 96.3 | 0.69 |
| SubcellPredicte | 94.3 | 94.3 | 96.0 | 0.52 |
| SLP-Locale | 93.8 | 93.8 | 95.9 | 0.59 |
| Gneg-PLocf | 89.6 | 88.9 | 95.7 | 0.65 |
| CELLO 2.5 | 87.5 | 87.5 | 95.0 | 0.61 |
aPA 3.0 is not included in the analysis since we are unable to determine the degree of overlap between our testing dataset and the training dataset of PA 3.0.
b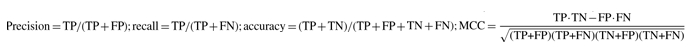 where TP is the number of true positives, FP the number of false positives, TN the number of true negatives, FN the number of false negatives and MCC the Matthew's Coefficient Constant.
where TP is the number of true positives, FP the number of false positives, TN the number of true negatives, FN the number of false negatives and MCC the Matthew's Coefficient Constant.
cSoftware also predicts periplasmic SCL. None of the testing dataset proteins received a periplasmic SCL prediction.
dSoftware only predicts cytoplasmic, membrane and extracellular categories. All proteins (including cell wall proteins) submitted to the server will receive one or more of these three localization predictions (or ‘no predictions’).
eSoftware only predicts cytoplasmic, periplasmic, and extracellular categories. All proteins (including membrane proteins) submitted to the server will receive one of these three SCL predictions.
fSoftware also predicts flagellar, fimbrium and nucleoid localizations; however, none of test dataset proteins received one of these three SCL predictions.
3.3 PSORTb 3.0 outperforms PSORTb 2.0 and other bacterial prediction software for predicting archaeal SCLs
The domain of archaea exhibits highly diverse morphologies. However, for most archaeal organisms, the basic compartments are similar to that of Gram-positive bacteria, namely cytoplasmic space, cell membrane, a proteinaceous cell wall and secreted proteins. The 5-fold cross validation results for the archaeal predictor are shown in Table 3. SCL-specific performance values for different predictors can be found in Supplementary Table 3. We compared the performance of our archaeal-specific predictor to Gram-positive bacterial SCL predictors since there is no other archaeal-specific predictor. We found that overall, Gram-positive bacterial predictors can predict archaeal cytoplasmic and membrane proteins with relatively high recall and precision, but with extracellular proteins the precision is quite low. PSORTb 3.0 is able to capture predictions for some of the archaeal-specific proteins and demonstrates superiority in performance compared to PSORTb 2.0 and to other Gram-positive bacterial SCL predictors, and now represents the first predictor specifically designed for the important domain of archaea.
Table 3.
Performance comparison for archaeal proteins between the PSORTb 3.0 archaeal option and software with Gram-positive SCL prediction capability
| Softwarea | Precision | Recall | Accuracy | MCCb |
|---|---|---|---|---|
| PSORTb 3.0 | 97.2 | 93.4 | 97.7 | 0.83 |
| PSORTb 2.0 | 95.7 | 81.0 | 94.3 | 0.59 |
| Gpos-PLocc | 92.3 | 92.3 | 96.2 | 0.65 |
| PA 2.5d | 90.0 | 77.5 | 89.6 | 0.38 |
| CELLO 2.5 | 86.5 | 86.5 | 93.2 | 0.46 |
aPA 3.0 is not included in the analysis since the exact content of the training dataset is unknown and may skew the cross-validation results.
bSee Table 2 footnotes for definitions of the four performance metrics.
cSoftware also predicts periplasmic SCL. None of the testing dataset proteins received a periplasmic SCL prediction.
dSoftware does not predict cell wall localization.
3.4 Evaluation of PSORTb and PA 3.0 using a new proteomics-derived experimental dataset—PSORTb 3.0 has highest recall
PA 3.0, an unpublished version of bacterial SCL predictor is also available through the Proteome Analyst website (http://webdocs.cs.ualberta.ca/∼bioinfo/PA/) with updated algorithms. We wished to compare the accuracy of this predictor, but we were unable to determine the content of the software's training dataset and the degree of overlap with our testing dataset. To account for the bias associated with testing and training with the same dataset, we therefore opted to evaluate PSORTb 3.0 and PA 3.0 using an independent dataset of 171 cytoplasmic proteins from the Gram-negative organism P.aeruginosa PA01. This dataset likely contains some proteins that are part of the training dataset of one or both tools, but most of the proteins with unknown functions that are identified from the experiment were never previously characterized for their localizations before and would not have been included in any SCL predictor's training data. This experimentally generated proteomics dataset should more accurately evaluate the software's predictive capabilities for analyzing a proteome. Table 4 shows the precision and recall of each predictor, where a false positive is defined as a protein receiving an SCL prediction that is not ‘cytoplasmic’. The prediction results for PSORTb 2.0 and PA 2.5 are also shown for reference. Similar to the results derived using the literature-derived dataset, PSORTb 3.0 and PA 3.0 demonstrate higher precision and recall compared to PSORTb 2.0 and PA 2.5. However, more proteins in this dataset receive a prediction from PSORTb 3.0 than from PA 3.0, indicating that PSORTb 3.0 achieves higher recall than PA 3.0. A full list of proteins used in the proteomic analysis, and their prediction results, can be found in Supplementary Table 4.
Table 4.
Evaluation of PSORTb 3.0, PSORTb 2, PA 3.0 and PA 2.5 using an LC–MS/MS proteomics dataset of proteins found exclusively in the cytoplasmic fraction when comparing to the periplasmic and extracellular fractions of P.aeruginosa PA01
| Software | Precisiona | Recall |
|---|---|---|
| PSORTb 3.0 | 96.3 | 91.8 |
| PA 3.0 | 95.9 | 81.3 |
| PA 2.5 | 90.7 | 51.5 |
| PSORTb 2.0 | 90.3 | 54.4 |
aPrecision in this case refers to TP/(TP + FP), where FP refers to proteins predicted as SCLs other than ‘Cytoplasmic’ or ‘Unknown’.
3.5 Proteome prediction coverage is increased
Although PSORTb 3.0 exhibited higher recall compared to PSORTb 2, our main goal was to increase prediction coverage for whole bacterial proteomes while maintaining a high level of precision. Figure 1 shows the coverage results of PSORTb 2.0 compared to PSORTb 3.0. Coverage is defined as the proportion of proteins in a deduced proteome that receives a prediction from the software at a measured level of precision (see above for precision results). The proteomes analyzed were chosen to cover a wide spectrum of bacterial phyla, ranging from well-studied model organisms such as Escherichia coli to lesser studied species that previously had low predictive coverage with PSORTb 2.0. Among the species tested, on average there was a 17.1% increase in proteome prediction coverage for Gram-negative bacterial proteomes and 5.9% increase for Gram-positive bacterial proteomes. Among the selected Gram-negatives, the Aquifex aeolicus proteome achieved the highest coverage (90.5%). Helicobacter pylori obtained the highest coverage increase (23.9%), whereas P.aeruginosa PA01 only gained 10.6% of coverage increase, the lowest of the Gram-negative list. Lactobacillus johnsonii, among the list of tested Gram-positive organisms, gained 7.9% in coverage, wheras Clostridium difficile received a modest boost of 2.8% in predictive coverage. Overall, proteome prediction for all tested organisms benefitted from the performance boost from PSORTb 3.
Fig. 1.

Genome coverage prediction for PSORTb 2.0 and PSORTb 3.0 for Gram-negative and Gram-positive bacteria genomes. Chr1 denotes chromosome 1.
3.6 PSORTb 3.0 and PA 3.0 make complementary predictions—a combined analysis with both methods has the highest coverage overall
Since PA 3.0 was the only comparable program to PSORTb in terms of precision and ability to not force predictions (i.e. have an ‘unknown’ prediction category), and has been validated using the proteomics dataset to have better performance compared to PA 2.5, we examined the prediction results for combining PSORTb 3.0 and PA 3.0. We tested this on several Gram-positive and Gram-negative bacterial genomes, including both model organisms and lesser-studied species. The results are shown in Figure 2. In combination, the two predictors were capable of generating predictions for about 80–95% of all bacterial proteins encoded in the selected bacterial genomes, which exhibits an impressive increase versus the previous predictive capability of PSORTb 2.0 (57–75%) (Gardy et al., 2005) and PA 2.5 (67–76%) (Lu et al., 2004). On average, 52.5% of the proteins in each genome-derived proteome received consensus SCL predictions from the two predictors. About 20–30% of the genes were predicted by either PA 3.0 or PSORTb 3.0 but not both programs, which shows a significant level of complementarities for two very precise predictors. Of the cases with different predictions (5–10%), we found that over half of these predictions consist of neighboring localizations (e.g. cytoplasmic versus cytoplasmic membrane). Upon manual inspection, these likely reflect the nature of peripheral membrane proteins that could not be detected as such by each predictor alone. For example, one program predicted cytoplasmic and the other predicted cytoplasmic membrane. For such membrane-associated proteins, technically both programs could be considered correct. For Gram-positive bacterial proteomes, although PA 3.0 does not predict a ‘cell wall’ SCL, many of the PSORTb-predicted cell wall proteins received ‘membrane’ or ‘extracellular’ predictions by PA, which does reflect the fact that many of them are membrane-anchored and protrude into the extracellular space. Taking these points into consideration, only ∼2.5–5% of the predictions appear to disagree, reflecting an expected level of error given the precision of each method. Taken together, it appears that combining the two methods notably increases genome prediction coverage indicating that the two methods are complementary and should be used together when possible.
Fig. 2.

Genome prediction coverage results from combining PSORTb 3.0 and PA 3.0 output. The majority of the ‘disagreement’ cases are boundary localizations (membrane prediction and a neighboring compartment). This likely reflects the true nature of the proteins. Only a small fraction of the disagreements (2.5–5% of the deduced proteome) are non-boundary cases.
4 DISCUSSION
The new version of PSORTb was created with the following improvements in mind: refining localization prediction, implementing archaeal SCL prediction capabilities, increasing software recall and proteome prediction coverage while maintaining high precision, and ensuring user-friendly software installation as well as usage. We found it necessary to implement subcategory localizations for several reasons. First of all, we have anecdotally observed that effector proteins secreted by Types III and IV secretion systems were predicted as cytoplasmic proteins by PSORTb 2.0 and most other SCL predictors due to the fact that their final destination is the host cell cytoplasm and likely contain properties similar to cytoplasmic proteins. Second, for structural proteins that are parts of a bacterial organellar apparatus, it would be more informative to note the apparatus itself as localization in addition to the main subcellular compartment currently assigned by PSORTb. Although the initial BLAST-based approach may be limited in capturing only effector proteins with enough sequence similarity to each other, we hope to further expand the dataset of effector proteins for training as they are identified. Having a subcategory localization detection allows PSORTb to give these types of proteins a more refined localization annotation, for example: ‘extracellular—T3SS (Type III secretion apparatus)’ rather than just the misleading classification of ‘extracellular’.
We have built the first SCL predictor specific for the domain of archaea and assessed its performance with a dataset of archaeal proteins. Although Gram-positive bacterial predictors seem to perform quite well for archaeal cytoplasmic and membrane proteins, the low recall values show that a bacterial only training dataset fails to predict archaeal cell wall and extracellular proteins well. Because of the unique nature of archaeal cell walls, which usually consist of a proteinaceous S-layer rather than peptidoglycan found in bacteria, proteins that reside in this localization can be quite different from cell wall proteins of Gram-positive organisms. If the training dataset does not contain representative properties for its localization category, no software would be able to generate highly accurate predictions for that particular category or that particular species. To further improve prediction for archaeal proteomes, we suspect that a more extensive training dataset needs to be collected for cell wall and secreted proteins in particular.
We have also added the capability to handle predictions for the four possible different types of bacteria: Gram-positive with and without an outer membrane, and Gram-negative with and without an outer membrane. As the diversity of bacteria being studied increases through metagenomics and other larger scale studies, having such capability to handle the diversity found in this domain of life will become increasingly important. Future research should focus on increasing the ability of SCL predictors to handle more specialized types of bacteria and archaea with atypical cell structures.
Most high-throughput mass spectrometry-based proteomic studies of subcellular fractions tend to include proteins from other subcellular compartments, due to some degree of cell lysis (Rey et al., 2005a). We were able to generate a relatively small dataset of highly reliant cytoplasmic identifications by eliminating any proteins that were found also in periplasmic or extracellular fractions in a proteome-scale analysis. While this approach will miss a lot of potential cytoplasmic proteins, this dataset is of high specificity and contains proteins that are not part of any SCL predictor's training dataset. For the other localizations, however, it is much more difficult to obtain relatively contaminant-free fraction samples, due to the fact that highly abundant cytoplasmic proteins (such as ribosomal proteins and molecular chaperone GroEL) tend to contaminate other fractions at such high levels. Further improvements in protein sample preparation for the non-cytoplasmic fractions are needed if we want to use this approach to validate software precision for the other SCLs.
We show that with the addition of new training data, PSORTb's recall and coverage improved and the performance remains ahead of other comparable bacterial SCL prediction software. This demonstrates that the effect of increasing training data size on improving such a prediction tool is still an effective way to increase predictive accuracy. By combining PA and PSORTb, two of the most accurate SCL predictors, we can now predict localizations for 80–95% of most bacterial proteomes. Efforts to further improve prediction capabilities should focus on developing approaches to tackle the last 5–20% of the proteomes. Preliminary analysis suggests that these are likely to be uncharacterized genes that are either common to a smaller subset of prokaryotic classes or unique to particular strains. A combined effort of small-scale as well as refined high-throughput experimental approaches, continual data mining from literature and algorithm improvement will be required to determine the localization of these proteins. The significant number of cases where PSORTb and PA predicted localizations to neighboring compartments highlights the need to further refine the SCL classification and identification of peripheral membrane proteins, which include proteins attached to the inner or outer membrane via a single α-helix, a lipid moiety or covalently linked to an integral membrane protein. Although LocateP and Augur begin to deal with this issue, such refinement should eventually be incorporated into whole-genome SCL analyzing software.
5 CONCLUSION
In summary, PSORTb 3.0 continues to be the most precise SCL predictor of its kind and now has notably increased recall and predictive coverage. It is also the most flexible SCL prediction software for prokaryotes, with both an online web server (with associated email client for larger jobs) as well as an open source standalone version with simplified installation procedure, which allows it to be easily used locally or incorporated into any existing bioinformatics analysis pipeline. With the added predictive capability of archaeal protein SCL prediction, predictions for bacteria with atypical cell morphologies and the addition of new predictive subcategories, this represents the first SCL predictor designed to handle a diverse range of all prokaryotes and handle prokaryotic subcategory localizations. Our results show that this tool can be effectively complemented by PA 3.0, generating an impressively high number of SCL predictions for proteomes at high precision. This new version of PSORTb, as well as the datasets used to train the software, will serve as a useful resource for bioinformaticists and the greater microbiology community.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Francis Lim for protein sample processing for mass spectrometry analysis, as well as Kurt McMillan and Yifeng Liu for providing PA 3.0 whole genome prediction results as well as helpful discussions. We thank Shannan Ho Sui for constructive feedback. L.J.F. is the Canada Research Chair in Quantitative Proteomics. L.J.F. and F.S.L.B. are Michael Smith Foundation for Health Research Scholar and Senior Scholar, respectively. F.S.L.B. is also a Canadian Institutes of Health Research New Investigator.
Funding: Natural Sciences and Engineering Research Council of Canada, with the support of the Cystic Fibrosis Foundation and Simon Fraser University Community Trust; BC Proteomics Network (to N.Y.Y. in part by a training grant); Postgraduate Scholarship from the Natural Sciences and Engineering Research Council of Canada (to J.R.W.); Canadian Institutes of Health Research and Michael Smith Foundation for Health Research Strategic Training Program in Bioinformatics Graduate Award (to J.R.W.).
Conflict of Interest: none declared.
REFERENCES
- Arnold R, et al. Sequence-based prediction of type III secreted proteins. PLoS Pathog. 2009;5:e1000376. doi: 10.1371/journal.ppat.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billion A, et al. Augur—a computational pipeline for whole genome microbial surface protein prediction and classification. Bioinformatics. 2006;22:2819–2820. doi: 10.1093/bioinformatics/btl466. [DOI] [PubMed] [Google Scholar]
- Bulashevska A, Eils R. Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinformatics. 2006;7:298. doi: 10.1186/1471-2105-7-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan QW, et al. Quantitative comparison of caste differences in honeybee hemolymph. Mol. Cell. Proteomics. 2006;5:2252–2262. doi: 10.1074/mcp.M600197-MCP200. [DOI] [PubMed] [Google Scholar]
- Chan QW, Foster LJ. Changes in protein expression during honey bee larval development. Genome Biol. 2008;9:R156. doi: 10.1186/gb-2008-9-10-r156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JM, et al. PSLDoc: protein subcellular localization prediction based on gapped-dipeptides and probabilistic latent semantic analysis. Proteins. 2008;72:693–710. doi: 10.1002/prot.21944. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Large-scale predictions of gram-negative bacterial protein subcellular locations. J. Proteome Res. 2006;5:3420–3428. doi: 10.1021/pr060404b. [DOI] [PubMed] [Google Scholar]
- de Champdoré M, et al. Proteins from extremophiles as stable tools for advanced biotechnological applications of high social interest. J. R. Soc. Interface. 2007;4:183–191. doi: 10.1098/rsif.2006.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy JL, et al. PSORTb 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Brinkman F.SL. Methods for predicting bacterial protein subcellular localization. Nat. Rev. Microbiol. 2006;4:741–751. doi: 10.1038/nrmicro1494. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Lu Z, et al. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics. 2004;20:547–556. doi: 10.1093/bioinformatics/btg447. [DOI] [PubMed] [Google Scholar]
- Matias Y, et al. ESA 1998. Berlin/Heidelberg: Springer; 1998. Augmenting suffix trees with applications; pp. 67–78. [Google Scholar]
- Matsuda S, et al. A novel representation of protein sequences for prediction of subcellular location using support vector machines. Protein Sci. 2005;14:2804–2813. doi: 10.1110/ps.051597405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melli G, et al. Proceedings of LBM-2007. CEUR-WS.org; 2007. Recognition of Multi-sentence n-ary Subcellular Localization Mentions in Biomedical Abstracts. [Google Scholar]
- Miyata M, Ogaki H. Cytoskeleton of mollicutes. J. Mol. Microbiol. Biotechnol. 2006;11:256–264. doi: 10.1159/000094059. [DOI] [PubMed] [Google Scholar]
- Nair R, Rost B. Sequence conserved for subcellular localization. Protein Sci. 2002;11:2836–2847. doi: 10.1110/ps.0207402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, et al. Using AdaBoost for the prediction of subcellular location of prokaryotic and eukaryotic proteins. Mol. Divers. 2008;12:41–45. doi: 10.1007/s11030-008-9073-0. [DOI] [PubMed] [Google Scholar]
- Rashid M, et al. Support vector machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinformatics. 2007;8:337. doi: 10.1186/1471-2105-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, et al. Assessing the precision of high-throughput computational and laboratory approaches for the genome-wide identification of protein subcellular localization in bacteria. BMC Genomics. 2005a;6:162. doi: 10.1186/1471-2164-6-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, et al. PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 2005b;33:D164–168. doi: 10.1093/nar/gki027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudrala R, et al. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009;5:e1000375. doi: 10.1371/journal.ppat.1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HB, Chou KC. Gpos-PLoc: an ensemble classifier for predicting subcellular localization of Gram-positive bacterial proteins. Protein Eng. Des. Sel. 2007;20:39–46. doi: 10.1093/protein/gzl053. [DOI] [PubMed] [Google Scholar]
- Stover CK, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Su EC, et al. Protein subcellular localization prediction based on compartment-specific features and structure conservation. BMC Bioinformatics. 2007;8:330. doi: 10.1186/1471-2105-8-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BG, Murray RG. Isolation and characterization of the plasma membrane and the outer membrane of Deinococcus radiodurans strain Sark. Can. J. Microbiol. 1981;27:729–734. doi: 10.1139/m81-111. [DOI] [PubMed] [Google Scholar]
- Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- Viklund H, Elofsson A. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004;13:1908–1917. doi: 10.1110/ps.04625404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. SIGMOD 1994. New York, USA: ACM; 1994. Combinatorial Pattern Discovery for Scientific Data: Some Preliminary Results; pp. 115–125. [Google Scholar]
- Winsor GL, et al. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 2008;37:D483–D488. doi: 10.1093/nar/gkn861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, et al. The Universal Protein Resource (Uniprot): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, et al. A phylogeny-driven genomic encyclopedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CS, et al. Prediction of protein subcellular localization. Proteins. 2006;64:643–651. doi: 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Zhou M, et al. LocateP: genome-scale subcellular-location predictor for bacterial proteins. BMC Bioinformatics. 2008;9:173. doi: 10.1186/1471-2105-9-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.