

Abstract
The corrected QT (QTc) interval is a complex quantitative trait, believed to be influenced by several genetic and environmental factors. It is a strong prognostic indicator of cardiovascular mortality in patients with and without cardiac disease. More than 700 mutations have been described in 12 genes (LQT1-LQT12) involved in congenital long QT syndrome. However, the heritability (genetic contribution) of QTc interval in the general population cannot be adequately explained by these long QT syndrome genes. In order to further investigate the genetic architecture underlying QTc interval in the general population, genome-wide association studies, in which up to one million single nucleotide polymorphisms are assayed in thousands of individuals, are now being employed and have already led to the discovery of variants in seven novel loci and five loci that are known to cause congenital long or short QT syndrome. Here we show that a combined risk score using 11 of these loci explains about 10% of the heritability of QTc. Additional discovery of both common and rare variants will yield further etiological insight and accelerate clinical applications.
The QT interval and the corrected QT interval
The QT interval is a reflection of the duration of myocardial depolarization and repolarization. It is defined as the time between the onset of the QRS complex and the end of the T wave as it returns to baseline, as measured on the electrocardiogram (Figure 1). The QT interval is strongly dependent on heart rate, with 'normal' rate-corrected (QTc) values considered to be between 360 and 460 ms [1-3]. QT interval prolongation or shortening has been shown to be associated with an increased risk for life-threatening ventricular arrhythmias and sudden cardiac death (SCD) in familial congenital syndromes of long [4,5] and short QT duration [6], as well as in population-based samples with [7] and without [8,9] underlying cardiac disease. For example, Moss et al. [4] demonstrated that each 10 ms increase in QTc interval contributes to about 5% exponential increase in risk of cardiac events in patients with long QT syndrome (LQTS). Furthermore, both cardiac and non-cardiac drugs have been reported to prolong QT interval and induce arrhythmia in patients who have a QTc interval length within the reference range [10,11].
Figure 1.

The surface electrocardiogram (ECG). The ECG provides information on the electrical events occurring within the heart, and is obtained by placing electrodes on the surface of the body. The duration of the QT interval on the ECG is defined as the duration between the beginning of the QRS complex and the end of the T wave. It is a reflection of ventricular action potential duration, and represents the time during which the ventricles depolarize and repolarize.
The QTc interval is known to be influenced by genetic factors, with heritability estimates between 25% and 52% [12-14]. In the TwinsUK study, a UK-based sample of mostly female twins of European ancestry, the proportions of additive genetic influences have been estimated as 55% for resting heart rate, 60% for uncorrected QT interval, and 50% for QTc [15]. Until recently, research into genetic factors influencing QT interval was limited to candidate genes known to have a role in arrhythmogenesis, on the basis of their involvement in the congenital monogenic diseases LQTS and short QT syndrome [16-21]. However, rapid advances in biotechnology have now made genome-wide association (GWA) studies possible. In contrast to candidate gene studies in which genes are selected on the basis of known or suspected disease mechanisms, GWA studies have the potential to identify loci that have not been previously targeted as having a role in the trait or disease, thereby highlighting potentially novel biological pathways [22].
An early GWA study for QTc interval [23], based on selection of individuals from the extreme tails of the population-based QTc interval distribution, identified a common variant in the nitric oxide synthase 1 adaptor protein (NOS1AP) gene region, and this has been consistently confirmed in later studies [24-32]. Furthermore, variants in NOS1AP have since been associated with risk of SCD in two separate population-based cohorts [33,34] and in subjects with LQTS [35].
The NOS1AP variant has been estimated to explain up to only 1.5% of QTc variance [23] (Figure 2), suggesting the need for additional and larger GWA studies with the potential to detect additional common genetic variants, which are likely to be of more modest effect size. Recent efforts in this direction include meta-analyses of GWA studies of QT interval duration in population-based cohorts by a number of consortia [24-26]; these have contributed many newly associated loci to this complex trait, and have suggested a cumulative effect of individual variants on QT interval. Notably, the QTGEN [25] and QTSCD [26] consortia found that common variants in a number of genes previously known to cause congenital LQTS (KCNQ1, KCNH2, KCNE1 and KCNJ2) and short QT syndrome (SCN5A), were among the most strongly associated with QT interval in these population-based cohorts (Figure 2). Significantly, two of the novel loci contained genes with established electrophysiological function (ATP1B1 and PLN). A third locus on 16q21 was near GINS3 and NDRG4, which are genes that have been associated with myocardial repolarization in zebrafish experiments [36,37], but the remaining loci fell in or near genes with less obvious immediate biological explanations. These loci included a RING-type zinc-finger protein of unknown function (RNF207), a DNA-binding protein thought to have a role in the regulation of TNFA expression and which is related to a hereditary motor and sensory neuropathy (LITAF), and a DNA base-excision and repair gene (LIG3).
Figure 2.

Explained variance per risk gene for prolonging the QTc interval. The explained variance per risk gene is ordered along the x-axis according to year of discovery and decreasing explained variance. The green diamond represents the finding in the KORA cohort [23] (nGWA = 186, nGWA+replication = 6,612), the blue squares represent the findings of the QTGEN study [25] (n = 13,685), the red triangles those from the QTSCD study [26] (n = 15,854), and the orange circle the finding from the meta-analysis of the TwinsUK/Bright/DCCT-EDIC cohorts [24] (n = 3,558). GWA, genome-wide association.
QT interval risk model
Given that the heritability of QTc is estimated to be about 50%, how much of this can be explained by the common variants discovered so far? Based on the results of the combined analysis of the top hits of the QTGEN and QTSCD consortia, we selected the single nucleotide polymorphism (SNP) with strongest association in each of the regions (Table 1) and constructed the following risk model using these SNPs weighted by their estimated effects in the meta-analysis:
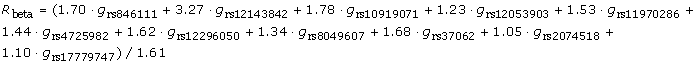 |
Table 1.
Results of 11 single nucleotide polymorphisms selected for the risk model in the combined analysis of QTSCD and QTGEN
| Locus | Chromosome | Variant | Coded allele | Beta | Standard error | P value |
|---|---|---|---|---|---|---|
| RNF207 | 1 | rs846111 | C | 1.70 | 0.21 | 3.69 × 10-16 |
| NOS1AP | 1 | rs12143842 | T | 3.27 | 0.17 | 1.88 × 10-78 |
| ATP1B1 | 1 | rs10919071 | A | 1.78 | 0.22 | 1.20 × 10-15 |
| SCN5A | 3 | rs12053903 | C | -1.23 | 0.12 | 1.0 × 10-14 |
| PLN; C6orf204 | 6 | rs11970286 | T | 1.53 | 0.15 | 2.35 × 10-24 |
| KCNH2 | 7 | rs4725982 | T | 1.44 | 0.16 | 5.0 × 10-16 |
| KCNQ1 | 11 | rs12296050 | T | 1.62 | 0.19 | 2.80 × 10-17 |
| LITAF | 16 | rs8049607 | T | 1.34 | 0.17 | 5.78 × 10-15 |
| NDRG4; CNOT1 | 16 | rs37062 | G | -1.68 | 0.16 | 3.0 × 10-25 |
| LIG3 | 17 | rs2074518 | T | -1.05 | 0.12 | 6.0 × 10-12 |
| KCNJ2 | 17 | rs17779747 | T | -1.10 | 0.16 | 6.02 × 10-12 |
The KCNE1 non-synonymous D85N variant rs1805128 (see also Figure 2) was not included in our risk score. It was genome-wide significant in the QTGEN study, but could not be confirmed in the QTSCD study and the combined analysis due to limited genotyping coverage in QTSCD.
where gSNP is the risk allele dosage of SNP, which is defined by:
(P(0 risk alleles) × 0) + (P(1 risk allele) × 1) + (P(2 risk alleles) × 2); this might be a non-integer value when the SNP is imputed, that is, it is not genotyped itself but its genotype probabilities are estimated based on linkage disequilibrium with nearby genotyped SNPs. The risk allele is defined as the allele that increases the risk of QT interval prolongation, and hence it might be different from the coded allele (for example, the risk allele of rs12053903 in SCN5A is T and not the coded allele C; Table 1). The model gives more weight to SNPs with larger effect and is standardized in such a way that the risk score lies between 0 and 22, that is, the maximum number of risk alleles.
This model was then validated in an independent sample of 2,838 twins from the TwinsUK cohort; part of this sample (n = 1,048) had been analyzed in a GWA study on QTc interval [24]. We adjusted QT interval for the effects of RR interval, age, sex, height, body mass index, hypertension and QT-interval-influencing drugs, and used the non-standardized residuals for the genetic analyses. The twin cohort consisted of 2,144 dizygotic twins (that is, 1,072 pairs) and 694 singletons, including 478 monozygotic twins of which the mean residual QTc interval of both twins was used to optimize information.
The effect of the risk model on QTc was estimated using linear regression while correcting the standard error of the regression coefficient for the twin relations [38,39]. The risk model was highly significantly associated with QTc interval (P = 2.0 × 10-31) and explained 4.7% of the phenotypic variance. Figure 3 shows that the length of the QTc interval increases with increasing genetic risk score, meaning that a larger number of risk alleles indeed predicts a longer QTc interval. For instance, individuals with a high genetic risk score of 15, which roughly corresponds to 15 (out of 22) risk alleles, have a QTc interval of 422.4 ± 3.3 ms, which is, on average, 17.6 ms longer than individuals with a low risk score of 6 (mean QTc = 404.8 ms).
Figure 3.

Correlation between genetic risk score and QT interval. The bars show the distribution of the risk score classes (left axis). In pink, a plot of the risk score versus unstandardized QT residual is given, and the blue line shows the means of the unstandardized QT residual within risk score classes (right axis). Error bars represent the standard errors of the means.
Future directions
In summary, the QTc genetic risk model based on the effects of the 11 genome-wide significant SNPs identified in the combined analysis of QTGEN and QTSCD was strongly associated with QT interval in our independent cohort consisting of 2,838 twins from the TwinsUK cohort. However, all these variants together explain only about 5% of the total variance in QTc, and hence about 10% of the heritability of QTc [15].
There are a number of possible explanations for this [40,41]. First, GWA studies rely on the 'common disease, common variant' hypothesis [42], which suggests that genetic influences on many common diseases will be at least partly attributable to a limited number of common allelic variants present in more than 10% of the population. As discussed, GWA studies have successfully identified such variants for QTc interval [23-26]. However, to avoid false-positive findings, they have used extreme significance thresholds to reliably identify these associations, potentially missing many common variants of small effect that did not reach the genome-wide significance level. Detection of these additional novel variants will require huge sample sizes. To this end, the three existing consortia [24-26] and additional studies recently merged into one QT Interval International GWAS Consortium (QT-IGC).
Second, many important disease-causing variants may in fact be rare (that is, <5% or even <1%) and are unlikely to be detected through the GWA approach [43]. These rare variants may exert relatively strong phenotypic effects in the individuals carrying them, and may be more valuable in individualized risk stratification, given their greater predictive value [41]. The current GWA studies lack power to identify such rare variants with modest effect sizes.
While GWA studies have identified several novel determinants of QT interval, very few functional variants have been identified. There is increasing evidence that many of the functional variants that underlie associations in GWA studies exert their effects through gene regulation rather than changing gene products. Additional resequencing of the genomic region of interest may be needed to identify the 'causal' variant followed by subsequent functional annotation studies to ascertain the clinical implications of these variants on arrhythmias and SCD. Progress towards finding these causal variants will likely increase the amount of heritability that can be explained. Information on lower frequency alleles emerging from projects such as the 1,000 Genomes project [44] and the Personal Genome Project [45] will be used to produce even more comprehensive GWA arrays, and will facilitate the investigation of the lower frequency variants without the need for de novo sequencing. The use of next-generation sequencing platforms, which provide high-volume sequence data with costs for resequencing exonic regions of the genome now approaching those for GWA studies, will also no doubt play a role in achieving this goal.
The problem of missing heritability may also be partially solved using an approach whereby many of the hits from a GWA study are followed up, rather than the current practice of carrying out meta-analyses and extensive follow-up of only the top ranked hits. This approach was successfully employed in a recent GWA study of celiac disease [46,47]. By taking advantage of the ever-decreasing price of genotyping, one might simultaneously follow up in a large replication sample, for example, 1,536 loci, a typical panel for one common platform, in a single experiment.
To date, the primary study population of published GWA studies has been of European origin. Therefore, there is also a need to extend association analyses to diverse non-European populations to confirm association signals identified thus far, as well as to potentially identify novel association signals [48,49] and etiological pathways.
Analyzing existing QT GWA study datasets with computational tools and pathway databases rather than considering only genes or gene variants may well further increase our understanding of the genetic architecture of this complex trait. Future and existing QT GWA study results have and will continue to identify important and potentially novel biochemical pathways for pathophysiology and therapeutics. Results have already pointed toward a greater emphasis on ion channels, which have long been known to be involved in congenital LQTS, and more recently to the nitric oxide pathway. Indeed a recent study found that SNPs in the NOS1AP gene modify the QT, prolonging effects of certain drugs [50].
Newly identified risk genes can therefore potentially advance drug development by highlighting novel therapeutic targets, or refocusing existing efforts for drug development to target, for example, the ion channel gene pathways. Furthermore, genetic profiling might advance drug development by identifying participants most likely to benefit from, or least likely to experience adverse effects of, a targeted therapeutic approach.
Due to the generally small effect sizes of the markers identified through GWA studies, much of the genetic data generated will not be of great value in isolation, but should rather be interpreted within the context of a predictive score, ideally complemented with information on non-genetic/environmental risk exposures, to allow targeted medical intervention before the onset of symptoms. The viability of this application might be limited, however, because the currently identified genes only explain a small proportion of the heritability. This reflects the complexity of translating markers identified through population studies into reliable predictors at an individual level. The diagnostic utility of genetic profiling also appears to be limited in other common complex diseases and traits. For example, a 54-locus genetic profile for the highly heritable trait height could predict only 4 to 6% of variation in height compared with 40% by traditional predictions based on parental height [51]. In fact, although GWA studies have been very successful in identifying specific loci and/or genomic regions that contribute to QTc and many other phenotypes, there has been some disappointment that only a small proportion of the heritability of many conditions has been accounted for [52,53]. However, it is important to remember that the main goal of GWA studies has never been disease prediction, but rather the discovery of biological pathways underlying polygenic disease or traits.
Despite the problems of 'missing heritability', associated loci identified from GWA studies can yield, and are already yielding, important insights into disease etiology, as well as potential drug targets. In the context of QT interval, the novel implication of a biochemical pathway such as the nitric oxide pathway in repolarization and arrhythmogenesis has already led to the suggestion that it is no longer sufficient to focus on the electrical properties of the heart when attempting to link genetic variation to cardiac arrhythmias. Rather, scientists and clinicians should now also consider electrical remodeling in response to environmental factors which can be controlled by the expression and activity of signaling molecules such as NOS1AP.
Abbreviations
GWA: genome-wide association; LQTS: long QT syndrome; NOS1AP: nitric oxide synthase adaptor protein; QTc: corrected QT; SCD: sudden cardiac death; SNP: single nucleotide polymorphism.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IMN and HS carried out statistical analysis and interpretation of the data. YJ and IMN drafted the manuscript, which was critically revised by YJ, IMN and HS. YJ, HS and TDS obtained funding.
Contributor Information
Yalda Jamshidi, Email: y.jamshidi@sgul.ac.uk.
Ilja M Nolte, Email: i.m.nolte@epi.umcg.nl.
Timothy D Spector, Email: tim.spector@kcl.ac.uk.
Harold Snieder, Email: h.snieder@epi.umcg.nl.
Acknowledgements
The work was partly funded by the British Heart Foundation, project grant no. 06/094.
References
- Kobza R, Roos M, Niggli B, Abacherli R, Lupi GA, Frey F, Schmid JJ, Erne P. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6:652–657. doi: 10.1016/j.hrthm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116:714–720. doi: 10.1161/CIRCULATIONAHA.106.676551. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Moss AJ, Zareba W. QT interval: how to measure it and what is 'normal'. J Cardiovasc Electrophysiol. 2006;17:333–336. doi: 10.1111/j.1540-8167.2006.00408.x. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Schwartz PJ, Crampton RS, Locati E, Carleen E. The long QT syndrome: a prospective international study. Circulation. 1985;71:17–21. doi: 10.1161/01.cir.71.1.17. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A Jr. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Wolf S. QT interval prolongation as predictor of sudden death in patients with myocardial infarction. Circulation. 1978;57:1074–1077. doi: 10.1161/01.cir.57.6.1074. [DOI] [PubMed] [Google Scholar]
- Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QT interval variables from 24 hour electrocardiography and the two year risk of sudden death. Br Heart J. 1993;70:43–48. doi: 10.1136/hrt.70.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten EG, Dekker JM, Meppelink P, Kok FJ, Vandenbroucke JP, Pool J. QT interval prolongation predicts cardiovascular mortality in an apparently healthy population. Circulation. 1991;84:1516–1523. doi: 10.1161/01.cir.84.4.1516. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, Escande D, Franz M, Malik M, Moss A, Shah R. The potential for QT prolongation and proarrhythmia by non-antiarrhythmic drugs: clinical and regulatory implications. Report on a policy conference of the European Society of Cardiology. Eur Heart J. 2000;21:1216–1231. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- Mutikainen S, Ortega-Alonso A, Alen M, Kaprio J, Karjalainen J, Rantanen T, Kujala UM. Genetic influences on resting electrocardiographic variables in older women: a twin study. Ann Noninvasive Electrocardiol. 2009;14:57–64. doi: 10.1111/j.1542-474X.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Larson MG, Corey DC, Benjamin EJ, Herbert AG, Levy D, D'Agostino RB, O'Donnell CJ. QT interval is a heritable quantitative trait with evidence of linkage to chromosome 3 in a genome-wide linkage analysis: The Framingham Heart Study. Heart Rhythm. 2005;2:277–284. doi: 10.1016/j.hrthm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Russell MW, Law I, Sholinsky P, Fabsitz RR. Heritability of ECG measurements in adult male twins. J Electrocardiol. 1998;30(Suppl):64–68. doi: 10.1016/S0022-0736(98)80034-4. [DOI] [PubMed] [Google Scholar]
- Dalageorgou C, Ge D, Jamshidi Y, Nolte IM, Riese H, Savelieva I, Carter ND, Spector TD, Snieder H. Heritability of QT interval: how much is explained by genes for resting heart rate? J Cardiovasc Electrophysiol. 2008;19:386–391. doi: 10.1111/j.1540-8167.2007.01030.x. [DOI] [PubMed] [Google Scholar]
- Pietila E, Fodstad H, Niskasaari E, Laitinen PP, Swan H, Savolainen M, Kesaniemi YA, Kontula K, Huikuri HV. Association between HERG K897T polymorphism and QT interval in middle-aged Finnish women. J Am Coll Cardiol. 2002;40:511–514. doi: 10.1016/S0735-1097(02)01979-4. [DOI] [PubMed] [Google Scholar]
- Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, Camargo AL, Drake JA, Benjamin EJ, Levy D, D'Agostino RB Sr, Hirschhorn JN, O'Donnell CJ. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation. 2007;116:1128–1136. doi: 10.1161/CIRCULATIONAHA.107.710780. [DOI] [PubMed] [Google Scholar]
- Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, Akyol M, Huth C, Schopfer-Wendels A, Kuch B, Steinbeck G, Holle R, Näbauer M, Wichmann HE, Meitinger T, Kääb S. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96:693–701. doi: 10.1161/01.RES.0000161077.53751.e6. [DOI] [PubMed] [Google Scholar]
- Bezzina CR, Verkerk AO, Busjahn A, Jeron A, Erdmann J, Koopmann TT, Bhuiyan ZA, Wilders R, Mannens MM, Tan HL, Luft FC, Schunkert H, Wilde AA. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36. doi: 10.1016/S0008-6363(03)00342-0. [DOI] [PubMed] [Google Scholar]
- Gouas L, Nicaud V, Chaouch S, Berthet M, Forhan A, Tichet J, Tiret L, Balkau B, Guicheney P. Confirmation of associations between ion channel gene SNPs and QTc interval duration in healthy subjects. Eur J Hum Genet. 2007;15:974–979. doi: 10.1038/sj.ejhg.5201866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–1222. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–731. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marbán E, O'Donnell CJ, Hirschhorn JN, Kääb S, Spooner PM, Meitinger T, Chakravarti A. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- Nolte IM, Wallace C, Newhouse SJ, Waggott D, Fu J, Soranzo N, Gwilliam R, Deloukas P, Savelieva I, Zheng D, Dalageorgou C, Farrall M, Samani NJ, Connell J, Brown M, Dominiczak A, Lathrop M, Zeggini E, Wain LV. for the Wellcome Trust Case Control Consortium, DCCT/EDIC Research Group; Newton-Cheh C, Eijgelsheim M, Rice K, de Bakker PI. for the QTGEN consortium; Pfeufer A, Sanna S, Arking DE. for the QTSCD consortium; Asselbergs FW, Spector TD, Carter ND, Jeffery S. Common genetic variation near the phospholamban gene is associated with cardiac repolarisation: meta-analysis of three genome-wide association studies. PLoS ONE. 2009;4:e6138. doi: 10.1371/journal.pone.0006138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, Fuchsberger C, Ehret GB, Orru M, Pattaro C, Kottgen A, Perz S, Usala G, Barbalic M, Li M, Pütz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WH, Mühleisen TW, Dei M, Happle C, Möhlenkamp S, Crisponi L, Erbel R, Jöckel KH, Naitza S, Steinbeck G. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitakari OT, Blom-Nyholm J, Koskinen TA, Kahonen M, Viikari JS, Lehtimaki T. Common variation in NOS1AP and KCNH2 genes and QT interval duration in young adults. The Cardiovascular Risk in Young Finns Study. Ann Med. 2009;41:144–151. doi: 10.1080/07853890802392529. [DOI] [PubMed] [Google Scholar]
- Eijgelsheim M, Aarnoudse AL, Rivadeneira F, Kors JA, Witteman JC, Hofman A, van Duijn CM, Uitterlinden AG, Stricker BH. Identification of a common variant at the NOS1AP locus strongly associated to QT-interval duration. Hum Mol Genet. 2009;18:347–357. doi: 10.1093/hmg/ddn341. [DOI] [PubMed] [Google Scholar]
- Tobin MD, Kahonen M, Braund P, Nieminen T, Hajat C, Tomaszewski M, Viik J, Lehtinen R, Ng GA, Macfarlane PW, Burton PR, Lehtimäki T, Samani NJ. Gender and effects of a common genetic variant in the NOS1 regulator NOS1AP on cardiac repolarization in 3761 individuals from two independent populations. Int J Epidemiol. 2008;37:1132–1141. doi: 10.1093/ije/dyn091. [DOI] [PubMed] [Google Scholar]
- Lehtinen AB, Newton-Cheh C, Ziegler JT, Langefeld CD, Freedman BI, Daniel KR, Herrington DM, Bowden DW. Association of NOS1AP genetic variants with QT interval duration in families from the Diabetes Heart Study. Diabetes. 2008;57:1108–1114. doi: 10.2337/db07-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, Uitterlinden AG, Witteman JC, Stricker BH. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10–16. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- Post W, Shen H, Damcott C, Arking DE, Kao WH, Sack PA, Ryan KA, Chakravarti A, Mitchell BD, Shuldiner AR. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered. 2007;64:214–219. doi: 10.1159/000103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijgelsheim M, Newton-Cheh C, Aarnoudse AL, van NC, Witteman JC, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in NOS1AP is associated with sudden cardiac death: evidence from the Rotterdam Study. Hum Mol Genet. 2009;18:4213–4218. doi: 10.1093/hmg/ddp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marbán E, Spooner PM, Burke GL, Chakravarti A. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940–951. doi: 10.1161/CIRCULATIONAHA.108.791723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL Jr. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan DJ, Kim AM, Winterfield JR, Jones IL, Pfeufer A, Sanna S, Arking DE, Amsterdam AH, Sabeh KM, Mably JD, Rosenbaum DS, Peterson RT, Chakravarti A, Kääb S, Roden DM, MacRae CA. Drug-sensitized zebrafish screen identifies multiple genes, including GINS3, as regulators of myocardial repolarization. Circulation. 2009;120:553–559. doi: 10.1161/CIRCULATIONAHA.108.821082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Jia H, Garrity DM, Tompkins K, Batts L, Appel B, Zhong TP, Baldwin HS. Ndrg4 is required for normal myocyte proliferation during early cardiac development in zebrafish. Dev Biol. 2008;317:486–496. doi: 10.1016/j.ydbio.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber PJ. Proceedings of the Fifth Berkeley Symposium on Mathematical Statististics and Probability: 21 June to 21 July 1965. Vol. 1. Berkeley. Berkeley: University of California Press; 1967. The behavior of maximum likelihood estimates under non-standard conditions; pp. 221–233. [Google Scholar]
- White H. Maximum likelihood estimation of misspecified models. Econometrica. 1982;50:1–26. doi: 10.2307/1912526. [DOI] [Google Scholar]
- Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA. 2008;299:1335–1344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Nolte IM, McCaffery JM, Snieder H. In: Handbook of Behavioral Medicine: Methods and Applications. Steptoe A, editor. New York: Springer; 2010. Candidate gene and genome-wide association studies in behavioral medicine. [Google Scholar]
- 1000 Genomes. A Deep Catalog of Human Genetic Variation. http://www.1000genomes.org
- Personal Genome Project. http://www.personalgenomes.org
- Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, Gwilliam R, Takeuchi F, McLaren WM, Holmes GK, Howdle PD, Walters JR, Sanders DS, Playford RJ, Trynka G, Mulder CJ, Mearin ML, Verbeek WH, Trimble V, Stevens FM, O'Morain C, Kennedy NP, Kelleher D, Pennington DJ, Strachan DP, McArdle WL. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Zhernakova A, Romanos J, Franke L, Hunt KA, Turner G, Bruinenberg M, Heap GA, Platteel M, Ryan AW, de Kovel C, Holmes GK, Howdle PD, Walters JR, Sanders DS, Mulder CJ, Mearin ML, Verbeek WH, Trimble V, Stevens FM, Kelleher D, Barisani D, Bardella MT, McManus R, van Heel DA, Wijmenga C. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–1083. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatti C, Service SK, Hartikainen AL, Pouta A, Ripatti S, Brodsky J, Jones CG, Zaitlen NA, Varilo T, Kaakinen M, Sovio U, Ruokonen A, Laitinen J, Jakkula E, Coin L, Hoggart C, Collins A, Turunen H, Gabriel S, Elliot P, McCarthy MI, Daly MJ, Järvelin MR, Freimer NB, Peltonen L. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noord C, Aarnoudse AJ, Eijgelsheim M, Sturkenboom MC, Straus SM, Hofman A, Kors JA, Newton-Cheh C, Witteman JC, Stricker BH. Calcium channel blockers, NOS1AP, and heart-rate-corrected QT prolongation. Pharmacogenet Genomics. 2009;19:260–266. doi: 10.1097/FPC.0b013e328324e556. [DOI] [PubMed] [Google Scholar]
- Aulchenko YS, Struchalin MV, Belonogova NM, Axenovich TI, Weedon MN, Hofman A, Uitterlinden AG, Kayser M, Oostra BA, van Duijn CM, Janssens AC, Borodin PM. Predicting human height by Victorian and genomic methods. Eur J Hum Genet. 2009;17:1070–1075. doi: 10.1038/ejhg.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]