

Abstract
A variant 35 kb upstream of the HLA-C gene (-35C/T) was previously shown to associate with HLA-C mRNA expression level and steady-state plasma HIV RNA levels. We genotyped this variant in 1,698 patients of European ancestry with HIV. Individuals with known seroconversion dates were used for disease progression analysis and those with longitudinal viral load data were used for viral load analysis. We further tested cell surface expression of HLA-C in normal donors using an HLA-C-specific antibody. We show that the -35C allele is a proxy for high HLA-C cell surface expression, and that individuals with high-expressing HLA-C alleles progress more slowly to AIDS and control viremia significantly better than individuals with low HLA-C expressing alleles. These data strongly implicate high HLA-C expression levels in more effective control of HIV-1, potentially through better antigen presentation to cytotoxic T lymphocytes or recognition and killing of infected cells by natural killer cells.
Variation at the HLA class I locus has a stronger influence on HIV-1 disease outcome than that of any other genetic locus identified so far1,2. HLA class I molecules have two fundamental roles in determining the strength of the immune response against HIV: regulation of the acquired response through presentation of antigenic epitopes to cytotoxic T lymphocytes (CTLs), and modulation of the innate response by serving as ligands for the killer cell immunoglobulin-like receptors (KIR) expressed on natural killer (NK) cells. In both regards, HLA-B has been the primary focus of research, relative to HLA-A and HLA-C, because the strongest genetic3–7 and functional8,9 associations with HIV disease outcomes have involved this locus. HLA-B*57 and HLA-B*27 confer particularly strong protection against HIV, which is thought to be primarily due to the specific HIV epitopes that are restricted by these allotypes. A subset of B*35 alleles, on the other hand, are associated with rapid AIDS progression through mechanisms that are not yet clear10. Furthermore, HLA-B alleles containing the Bw4 epitope (defined by amino acid positions 77–83) collectively show protection against HIV disease5, probably as a result of their function as ligands for the inhibitory KIR3DL17 and putatively for the activating KIR3DS1 receptors6,11 on NK cells. No other individual HLA-A or HLA-C allele, or KIR-ligand grouping of these two loci, has been reported to have nearly as great an effect on HIV as these HLA-B alleles and the Bw4 allelic grouping.
Recently, however, a scan for genetic variants that influence the control of viral load indicated that a dimorphism 35 kb upstream of the HLA-C gene (-35C/T) had one of the two strongest genome-wide effects on the level of plasma viremia in early, established HIV infection as measured by viral load set-point, although no significant association of this variant was observed with progression to a CD4 cell count of <350 (ref. 2). Notably, the -35C variant that associates with low viral load has also been shown to associate with high HLA-C mRNA levels in a codominant manner among a group of individuals of European ancestry12,13, although whether it associates with cell surface expression has not been tested. These findings suggest that certain HLA-C allotypes might have a primary role in restricting HIV replication through innate and/or acquired immune mechanisms that have previously been overlooked. Here we present data from 1,698 European American individuals, indicating that high levels of HLA-C confer strong protection early in the course of HIV infection and this early protection of high HLA-C extends to some extent into chronic infection. We propose a model in which high-expression HLA-C alleles might confer better innate and/or acquired immune responses than low-expression HLA-C alleles.
RESULTS
Effect of-35 on mean viral load
The effect of -35 genotypes on mean plasma HIV load (mVL) measurements was tested in a group of 935 seroincident European American individuals (see Online Methods). Each individual was categorized into one of three groups based on their mVL (<2,000, 2,000–10,000 and >10,000 mean viral RNA copies per ml plasma), and the frequency of each genotype (CC, CT and TT; CX = CC/CT) within these groupings was determined. Because B*57 and B*27 confer protection in our cohorts and the B*35-Px group of alleles confers susceptibility, we used these alleles as covariates in all analyses (except when the analyses are restricted to Bw6/Bw6 only, for which we used only B*35-Px as a covariate as both B*57 and B*27 are Bw4 alleles). In a comparison of the two extreme viral load groups, controllers versus non-controllers (<2,000 versus >10,000 respectively), -35C associated with protection in a codominant manner and each pair of genotypes was significantly different from one another (Table 1a). Most notably, -35CC was very protective relative to -35TT, where 62.3% of individuals with the -35CC genotype restricted the virus to mVLs of <2,000, but only 15.1% of individuals with -35TT controlled the virus to this extent (odds ratio (OR) = 0.23, P = 1 × 10−8). The group of HLA-B alleles with the Bw4 epitope is in significant positive linkage disequilibrium (LD) with the -35C single nucleotide polymorphism (SNP) (D′ = 0.52, P = 0.001), and this allelic grouping protects against HIV5, particularly the subset of Bw4 alleles with isoleucine at position 80 when combined with certain KIR3DL1 and 3DS1 alleles6,7. The protection of -35C remained as robust when we removed all individuals with one or two copies of HLA-B Bw4 from the analysis to eliminate a possible contribution of HLA-B Bw4 in the effect of -35C on mVL (Supplementary Table 1a).
Table 1.
effect of -35 on mean viral load
| (a) Categorical analysis using B*27, B*57 and B*35Px as covariates | |||||
|---|---|---|---|---|---|
| Genotype | VL <2,000 % (n) | VL >10,0000 % (n) | Comparison | OR (95% CI) | P value |
| CC | 62.3 (104) | 37.7 (63) | CC vs. TT | 0.23 (0.14–0.38) | 1 × 10−8 |
| CT | 33.2 (120) | 66.9 (242) | CT vs. TT | 0.55 (0.36–0.84) | 0.006 |
| TT | 15.1 (45) | 85.0 (254) | CX vs. TT | 0.42 (0.29–0.62) | 1 × 10−5 |
| CXa | 42.3 (224) | 57.7 (305) | CC vs. CT | 0.4 (0.26–0.6) | 1 × 10−5 |
| (b) Continuous variable analysis using B*27, B*57 and B*35Px as covariates | ||||||
|---|---|---|---|---|---|---|
| Genotype | N | Mean logVL | SE | Difference between genotypes | 95% CI | P value |
| CC | 188 | 2.99 | 0.12 | CT–CC (0.44) | 0.24–0.64 | <0.0001 |
| CT | 409 | 3.43 | 0.11 | TT–CC (0.71) | 0.49–0.93 | <0.0001 |
| TT | 326 | 3.70 | 0.13 | TT–CT (0.27) | 0.10–0.44 | 0.002 |
n = 923; SE, standard error; CI, confidence interval.
n = 828; OR, odds ratio; CI, confidence interval.
CX = CC/CT genotypes.
The effect of -35 genotypes on mVL as a continuous variable in which all individuals were included in the analyses (n = 923) mirrored the categorical analysis in which extreme groupings were used (Table 1b). Again, the level of protection varied as follows: -35CC > -35CT > -35TT. Confining the analysis to HLA-B Bw6/Bw6 individuals (with all individuals with Bw4 removed) showed the same trend in mVL levels across -35 genotypes as when the entire set of individuals was considered (Supplementary Table 1b). These data indicate that -35C restricts viral load, as found by others2, independently of the effects of the HLA-B locus.
Protective effect of -35 on AIDS progression
Given the large effect of -35 on mVL, we tested in a nonoverlapping HIV-seroincident European American cohort (n = 763) whether this SNP had any effect on progression to AIDS outcomes that occur during chronic infection. We used survival analysis to determine progression to three outcomes: a CD4 count of <200; AIDS-defining complication as assessed using the original 1987 definition; and death. We found that -35CC had a marginally significant protective effect on progression to a CD4 count of <200 relative to genotypes containing -35T, but a stronger protective effect on progression to AIDS and death (Table 2). Of the 763 seroconverters used in the survival analyses, viral load measurements were available for 415. Using only these 415 individuals, we determined hazard ratios in two sets of analyses where mVL either was or was not used as a covariate (the three standard HLA-B alleles were used as covariates in both sets of analyses). The addition of mVL as a covariate had virtually no effect on hazard ratio values for any of the outcomes (data not shown), suggesting that the effect of -35 on disease progression is at least somewhat independent of mVL. To eliminate the possibility of confounding of the analysis owing to population stratification, we performed survival analysis on 422 samples for which eigen coordinates were available. Hazard ratios were compared between analyses involving samples that did and did not include the eigenvalues from six significant EIGENSTRAT axes as covariates. Inclusion of the eigenvalues as covariates in the analysis had no significant effect on the hazard ratios, indicating that population stratification was not a confounding variable in our analyses (Supplementary Table 2). Overall, the protective effect of -35C alone on AIDS progression was not nearly as significant as its effect on the early outcome of viral load.
Table 2.
Effect of -35 on AIDS progression
| Outcome | Genotype | n | HR (95% CI) | P value |
|---|---|---|---|---|
| CD4 <200 | CC vs. TT | 114 vs. 251 | 0.62 (0.43–0.90) | 0.01 |
| CT vs. TT | 367 vs. 251 | 0.89 (0.71–1.12 | 0.31 | |
| CC vs. CT | 114 vs. 367 | 0.70 (0.50–0.99) | 0.05 | |
| CXa vs. TT | 481 vs. 251 | 0.84 (0.67–1.04) | 0.11 | |
| AIDS 1987 | CC vs. TT | 117 vs. 265 | 0.52 (0.35–0.78) | 0.001 |
| CT vs. TT | 381 vs. 265 | 0.68 (0.53–0.86) | 0.002 | |
| CC vs. CT | 117 vs. 381 | 0.75 (0.51–1.10) | 0.14 | |
| CX vs. TT | 498 vs. 265 | 0.65 (0.51–0.82) | 0.0003 | |
| Death | CC vs. TT | 117 vs. 265 | 0.43 (0.26–0.70) | 0.001 |
| CT vs. TT | 381 vs. 265 | 0.64 (0.49–0.85) | 0.002 | |
| CC vs. CT | 117 vs. 381 | 0.64 (0.40–1.02) | 0.06 | |
| CX vs. TT | 498 vs. 265 | 0.60 (0.46–0.79) | 0.0003 |
n = 763; HR, hazard ratio; CI, confidence interval. Age of seroconversion, B*27, B*57 and B*35Px were used as covariates and data stratified by cohort.
CX = CC/CT genotypes.
HLA-C protein expression correlates with -35 genotype
A previous scan to determine inter-individual fluctuation in levels of mRNA expression showed that HLA-C mRNA expression differs across European American lymphoblastoid cell lines12,13, and this variation correlates with the -35 genotype2. To confirm the specificity of HLA-C microarray probes, and because HLA mRNA in immortalized cell lines might not reflect the levels of surface protein on primary cells, we investigated HLA-C transcript and surface protein expression in freshly isolated peripheral blood lymphocytes (PBLs). Transcript levels were analyzed in four donors typed for HLA class I where HLA-C could be specifically amplified (and other class I molecules were not). Three donors were selected that were heterozygous at the -35 SNP and one donor was homozygous for -35T, all of which could be amplified by the same HLA-C-specific PCR reaction and typed by restriction digestion. In all three -35CT donors, the HLA-C allele residing on the -35C haplotype dominated the pool of PCR products amplified relative to that on the -35T haplotype; in the TT donor, Cw*0701 and Cw*0303 were expressed at levels not significantly different from each other (Fig. 1).
Figure 1.
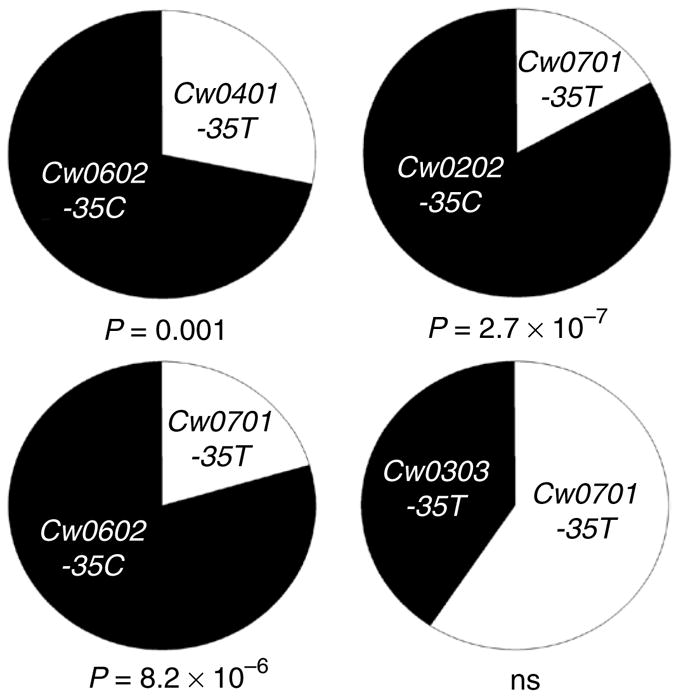
Level of mRNA expression varies among HLA-C alleles and correlates with the -35 SNP. HLA-C was amplified from the cDNA of peripheral blood lymphocytes and the relative contribution of each allele was established by cloning and restriction digestion of PCR products. In donors heterozygous at the -35 genotype, HLA-C alleles in LD with the -35C genotype are in excess compared to the HLA-C allele that is in LD with -35T. The numbers of clones identified were as follows: Donor 1, Cw*0602 (-35C) 43 clones and Cw*0401 (-35T) 17 clones; Donor 2, Cw*0202 (-35C) 49 clones and Cw*0701 (-35T) 10 clones; Donor 3, Cw*0602 (-35C) 46 clones and Cw*0701 (-35T) 12 clones; Donor 4, Cw*0303 (-35T ) 20 clones and Cw*0701 (-35T) 29 clones. Statistical analysis was performed using two-tailed t-test with binomial distribution.
For surface protein analysis, we used the monoclonal antibody (mAb) DT9, which has been reported to recognize HLA-C but not HLA-A or HLA-B allotypes14. We characterized DT9 mAb reactivity using several methods (Supplementary Fig. 1). To verify its specificity for HLA-C, we tested the binding of DT9 to beads coated with 96 of the HLA class I allotypes that are common in European Americans (Supplementary Fig. 1a). DT9 consistently bound all HLA-C allotypes, and it bound strongly to HLA-A*8001 and B*7301 and weakly to B*1301, B*3501 and B*4006. From surface- biotinylated peripheral blood lymphocytes, DT9 immunoprecipitates only 45-kDa classical HLA class I antigens, without significant amounts of the smaller nonclassical HLA class I antigens15 (Supplementary Fig. 1b). Both the DT9 and MEM-E/08 mAbs bind HLA-E on transfected cells that are otherwise negative for class I expression, but only mAb DT9 binds to peripheral blood lymphocytes, consistent with the level of HLA-E on the primary cells being below that detected by flow cytometry (Supplementary Fig. 1c). Furthermore, acid-induced conformational changes show that DT9 recognizes only properly folded HLA-C molecules (Supplementary Fig. 1d).
We selected 50 normal donors on the basis of their HLA-C genotype distribution, excluding any HLA-A and HLA-B allotypes recognized by DT9. Binding of DT9 to freshly isolated CD3+ cells varied between individuals by more than tenfold (Supplementary Fig. 2a–d). Relative levels of HLA-C expression between donors correlated significantly with the -35 genotype (Fig. 2a). For individuals with the -35TT genotype, 17 times more binding of mAb DT9 than its isotype control was detected on average. In comparison, for individuals with -35CX, an average of 30 times more DT9 binding was detected. Assuming that binding of our mAbs increases linearly between these levels of HLA-C, we conclude that -35CX individuals express an average of 1.75 times more HLA-C molecules than donors with -35TT genotypes.
Figure 2.

Variation in the level of surface HLA-C protein correlates with -35 genotype. HLA-C molecules on CD3+ T lymphocytes from 50 normal individuals were measured by flow cytometry using the DT9 mAb and grouped according to -35 genotype (a) and HLA-C allele (b). The -35 allele (C or T) that is in LD with the corresponding HLA-C allele is shown below each HLA-C allele name along the x axis and the -35 genotype of each donor is indicated by shading of the individual data points.
Grouping individuals by the eight most frequent HLA-C alleles typed in our sample population (rather than by -35 genotype) showed that HLA-C expression levels also varied across HLA-C allotypes (Fig. 2b). Most of the donors were heterozygous at the HLA-C locus, so variation across donors within a given HLA-C allele grouping is probably due in part to the expression level of the coexpressed HLA-C allotype. This was especially apparent for the Cw*07 subtypes, in which heterozygotes who had one -35T (the Cw*07 allele) and one -35C allele (depicted in Fig. 2b by half-shaded circles) had higher DT9 binding values than those with two -35T alleles (solid circles). In general, a trend was observed in which the alleles in positive LD with -35T tend to have the lowest expression and those with -35C had the highest (for LD values, see Supplementary Table 3).
The possibility that variations in DT9 binding were the result of differences in affinity for HLA-C allotypes was countered by consistent DT9 binding to the 16 different HLA-C allotypes available as single-antigen coated beads (Supplementary Fig. 1a). Similar binding of the mAbs BBM.1 and W6/32 confirm a consistent level of HLA class I on each of these beads16. BBM.1 recognizes β2m, so it would be predicted to have the same affinity for all HLA class I allotypes17, whereas W6/32 recognizes a conformationally dependent epitope of all HLA class I allotypes, so that it controls against degradation of the bead-bound antigen18.
Overall, these data show differential expression of HLA-C allo-types and a close correlation between allotypic expression and the -35 genotype. Thus, the association of -35 with outcomes from HIV infection might be due to the level of HLA-C expression on HIV-infected target cells.
Effect of specific HLA-C alleles on mean viral load
Although both HLA-A and HLA-B are downregulated by the HIV nef protein, HLA-C is not19, so the cognate expression patterns of HLA-C might be particularly meaningful in HIV pathogenesis. Given the association of the -35 SNP with outcomes of HIV infection and the correlation between HLA-C surface expression and -35 genotype, we tested whether alleles in LD with -35C individually associate with protection and those in LD with -35T with susceptibility in the controller versus non-controller groups (Fig. 3). In this analysis we used only alleles with a frequency >5% and included all samples (n = 1,239) for which HLA-C typing and viral load measurements were available. The HLA-C alleles that are in positive LD with -35C each had lower (more protective) odds ratios than those in positive LD with -35T, and four of the alleles showed significant associations individually. In fact, every -35C-associated HLA-C allele was more common in the controller group than in the non-controller group, except for Cw*0501 (which was slightly more common in the non-controller group; data not shown). Conversely, every -35T-associated HLA-C allele was more common in the non-controller group than the controller group. This is noteworthy given that the alternative HLA-C allele for each individual (which can be expressed at a high or low level) in each allelic category was not taken into consideration. The consistent over-representation of -35C-associated HLA-C alleles among the controllers and -35T-associated HLA-C alleles among the non-controllers strongly supports a model in which the general characteristic of higher cell surface expression is a protective determinant against HIV and provides a functional explanation for the effect of -35 on HIV outcomes.
Figure 3.

Highly expressed HLA-C allotypes control HIV to a greater extent than HLA C allotypes with lower expression. Odds ratios are based on the frequency of HLA-C alleles in the VL <2,000 group versus the >10,000 group. -35 LD with the respective HLA-C allele is indicated on the x axis.
The proposed protective effect of high-expression HLA-C alleles appears to be independent of the antigenic peptides presented by the specific allotypes, as the various HLA-C alleles within either the -35C or T grouping bind, and present to T cells, very distinct antigenic epitope motifs20. Also, variants that encode the two distinct binding supertypes of HLA-C defined by Ser77 and Asn77 (ref. 21) are found for multiple HLA-C alleles on both the -35C and -35T haplotypes (Fig. 4). As the classification of the supertypes for HLA-C is based on in silico analysis, however, there is no direct evidence to negate the possibility that the -35 SNP marks alleles that present similar peptides. Nevertheless, phylogenetic analysis indicates that -35C is present on quite distantly related lineages of HLA-C alleles and even closely related HLA-C alleles can differ with regard to the presence of -35C. It is therefore unlikely that the protective effect of -35C is associated with a particular functional grouping of HLA-C alleles based on the type of epitopes they present.
Figure 4.

The phylogram shows neighbor-joining relationships among full-length HLA-C alleles estimated using Kimura’s 2-parameter substitution model. Numbers above nodes indicate bootstrap recovery of that node in 500 bootstrap replications and the scale bar indicates the number of substitutions per site in a given interval along branches. HLA-C alleles known to be in strong LD with C at −35kb are indicated with a solid triangle while the two functional supertypes containing either Asn77 or Ser77 are distinguished by a ‘-N’ suffix on the designation of alleles containing an Asn residue. Strong bootstrap support (up to 100%) indicates that alleles associated with either C or T at -35 group together in multiple clades. Likewise, alleles of both functional supertypes are grouped together in multiple clades (bootstrap support up to 98%).
DISCUSSION
Certain host genetic variants have been shown to exert their effects on HIV-induced disease at specific times after infection22,23, including certain alleles of the HLA-B locus4,24. Using two independent, non-overlapping cohorts, we show that the -35 variant associates strongly with HIV outcomes during the early phase of infection by influencing steady-state viral load, and to a weaker extent with the very late phase of infection by influencing time to death. These temporal data implicate two at least partially distinct mechanisms of HIV restriction associated with the -35 variant. In support of this model, a preliminary analysis suggests that the early effect on viral load is not modulated by the presence or absence of activating KIR2DS, whereas the late effect might be dependent on the presence or absence of KIR2DS (Supplementary Tables 4a,b). Very few well-powered natural history cohorts of seroincident patients before initiation of highly active antiretroviral therapy are available for study, and we have not been able to replicate in an independent cohort our findings regarding the potential modulating effect of KIR2DS in late chronic disease. Thus, these data remain intriguing, but unsubstantiated.
We have shown that surface expression of HLA-C varies significantly across -35 genotypes and that the HLA-C alleles that are in positive LD with -35C are expressed at a higher level than those that are in LD with -35T. However, the level of HLA-C expression is not bimodal; rather there is a continuum of expression that cannot be attributed completely to zygosity of -35 genotypes (Fig. 2b). It will be necessary to probe further into expression levels of the different HLA-C allotypes, for example by studying expression on cells from HLA-C homozygotes, to determine precisely the order of expression levels among the HLA-C allotypes. The expression data argue against the -35 variant having a sole, direct effect on HLA-C expression, but rather indicates a more complex regulatory mechanism of HLA-C expression in which -35 might simply mark by LD the true regulatory variant(s). Nevertheless, viral load analysis of 43 SNPs in the coding region of exons 2–3 and three more SNPs in the 5′UTR region show that -35 is still the variant that best associates with both viral load control and HLA-C expression (Supplementary Table 5). The most parsimonious model from the data presented in this report is that the level of HLA-C expression is responsible for the genetic effects on early viral load outcomes to HIV infection and that -35 is serving as a reliable proxy for HLA-C expression levels in European American individuals.
High levels of HLA-C expression during early HIV infection may provide protection by enhancing antigen presentation to CTLs in an acquired immune response. Another possible mechanism to explain the protective effect of high HLA-C expression relates to the interactions between inhibitory KIR2DLs and their HLA-C ligands. Several studies have shown that high expression of MHC class I–specific inhibitory receptors on an NK cell during its maturation is necessary for arming the cell to respond to aberrant targets when it matures25–28. Accordingly, higher surface expression of inhibitory KIRs that recognize self–MHC class I augments the activity of mature NK cells27, so it is likely that higher expression of ligands for KIRs during NK cell maturation also better arms the effector cell for stronger activity when faced with an appropriate target cell. The inhibitory KIR2DL1 and KIR2DL2/L3 receptors recognize the dimorphic C1 and C2 HLA-C allotypes, and these inhibitory KIRs are present in the genomes of nearly all individuals29. Thus, the differential effect of low- versus high-expressing HLA-C types on mVL might relate to greater KIR2DL recognition of highly expressed HLA-C molecules during NK cell maturation, preparing the NK cell for a more effective response against aberrant target cells. That is, more interaction between inhibitory KIRs and their self–MHC class I ligands during NK cell development (which would occur when class I is present at a high level) would result in stronger responses upon the loss or alteration of cognate ligands associated with viral infection. This scenario cannot be tested in a genetic association study, as the genes for the inhibitory KIRs for both HLA-C groupings are present in nearly all individuals, but functional approaches could appropriately address whether protection conferred by high HLA-C expression involves enhanced activity of NK cells through a licensing process25,26. Regardless of the mechanism through which high HLA-C provides protection, HIV Nef variants derived from patients with the protective -35CC genotype seem indirectly to counteract an enhanced HLA-C–mediated immune control by manipulating MHC class II antigen presentation and helper T-cell function (F. Kirchhoff, personal communication). These data suggest that high levels of HLA-C expression exert selection pressure on the virus.
Our results strongly implicate HLA-C as a major determinant in both early and late outcomes of HIV infection. No individual HLA-C allotype appears to be clearly better than others in terms of controlling HIV through antigen presentation to CTLs, unlike HLA-B allotypes8. However, higher levels of HLA-C on target cell surfaces might generally result in more effective antigen presentation to CTLs or enhance NK cell activity, thus boosting the immune system and leading to better viral control by the host.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contracts HHSN261200800001E, N02-CP-55504, R01-DA04334 and R01-DA12568. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This research was partially funded by a grant from the Bill & Melinda Gates Foundation as part of the Collaboration for AIDS Vaccine Discovery. We would also like to acknowledge the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation, and the SCOPE study was funded by the UL1 RR024131 (Clinical and Translational Sciences Award) and P30 AI27763 (Center for AIDS Research) grants. R.A. is funded by the Cambridge Center for Trophoblast Research. We also thank R. Fernando and the Anthony Nolan Research Institute for the Luminex analysis.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
Author contributions are listed in alphabetical order. Project conception and supervision: M.C.; study rationale: A.M., D.G., D.V.M., J.F., M.C.; data analysis: D.G., Y.Q.; data interpretation and manuscript preparation: M.C., R.A, R.T.; genotyping/sequencing: M.P.M.; R.T.; X.G.; HLA-C expression characterization: G.O.C., R.A., R.T., V.M.; phylogenetic analysis: C.O.H.; clinical samples and data: A.T., B.D.W., G.D.K., J.J.G., J.M., J.N.M., S.B., S.G.D.; manuscript editing: all authors.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 2.Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao X, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 4.Gao X, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med. 2005;11:1290–1292. doi: 10.1038/nm1333. [DOI] [PubMed] [Google Scholar]
- 5.Flores-Villanueva PO, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci USA. 2001;98:5140–5145. doi: 10.1073/pnas.071548198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin MP, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 7.Martin MP, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–740. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiepiela P, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 9.Alter G, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–3036. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin X, et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+-T-cell responses for groups of HIV-1-infected individuals with different HLA-B*35 genotypes. J Virol. 2002;76:12603–12610. doi: 10.1128/JVI.76.24.12603-12610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi Y, et al. KIR/HLA pleiotropism: protection against both HIV and opportunistic infections. PLoS Pathog. 2006;2:e79. doi: 10.1371/journal.ppat.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stranger BE, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stranger BE, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braud VM, Allan DS, Wilson D, McMichael AJ. TAP- and tapasin-dependent HLA-E surface expression correlates with the binding of an MHC class I leader peptide. Curr Biol. 1998;8:1–10. doi: 10.1016/s0960-9822(98)70014-4. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu Y, Geraghty DE, Koller BH, Orr HT, DeMars R. Transfer and expression of three cloned human non-HLA-A,B,C class I major histocompatibility complex genes in mutant lymphoblastoid cells. Proc Natl Acad Sci USA. 1988;85:227–231. doi: 10.1073/pnas.85.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apps R, et al. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology. 2009;127:26–39. doi: 10.1111/j.1365-2567.2008.03019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brodsky FM, Bodmer WF, Parham P. Characterization of a monoclonal anti-beta 2-microglobulin antibody and its use in the genetic and biochemical analysis of major histocompatibility antigens. Eur J Immunol. 1979;9:536–545. doi: 10.1002/eji.1830090709. [DOI] [PubMed] [Google Scholar]
- 18.Barnstable CJ, et al. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell. 1978;14:9–20. doi: 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- 19.Cohen GB, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 20.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 21.Doytchinova IA, Guan P, Flower DR. Identifiying human MHC supertypes using bioinformatic methods. J Immunol. 2004;172:4314–4323. doi: 10.4049/jimmunol.172.7.4314. [DOI] [PubMed] [Google Scholar]
- 22.Martin MP, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science. 1998;282:1907–1911. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- 23.Smith MW, et al. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 24.Altfeld M, et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS. 2003;17:2581–2591. doi: 10.1097/00002030-200312050-00005. [DOI] [PubMed] [Google Scholar]
- 25.Fernandez NC, et al. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 2005;105:4416–4423. doi: 10.1182/blood-2004-08-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 27.Yawata M, et al. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med. 2006;203:633–645. doi: 10.1084/jem.20051884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anfossi N, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Winter CC, Long EO. A single amino acid in the p58 killer cell inhibitory receptor controls the ability of natural killer cells to discriminate between the two groups of HLA-C allotypes. J Immunol. 1997;158:4026–4028. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.