

Abstract
Although transgenic mouse models of Alzheimer’s disease (AD) recapitulate amyloid-β (Aβ)-related pathologies and cognitive impairments, previous studies have mainly evaluated their hippocampus-dependent memory dysfunctions using behavioral tasks such as the water maze and fear conditioning. However, multiple memory systems become impaired in AD as disease progresses, and it is important to test whether other forms of memory are affected in AD models. This study was designed to use conditioned taste aversion (CTA) and contextual fear conditioning paradigms to compare the phenotypes of hippocampus-independent and dependent memory functions, respectively, in 5XFAD APP/PS1 transgenic mice that harbor five familial AD (FAD) mutations. While both types of memory were significantly impaired in 5XFAD mice, the onset of CTA memory deficits (~9 months of age) was delayed compared to that of contextual memory deficits (~6 months of age). Furthermore, 5XFAD mice genetically engineered to have reduced levels of β-site APP-cleaving enzyme 1 (BACE1+/−·5XFAD) exhibited improved CTA memory, which was equivalent to the performance of wild-type controls. Importantly, elevated levels of cerebral β-secretase-cleaved C-terminal fragment (C99) and Aβ peptides in 5XFAD mice were significantly reduced in BACE1+/−·5XFAD mice. Furthermore, Aβ deposition in the insular cortex and basolateral amygdala, two brain regions critically involved in CTA performance, was also reduced in BACE1+/−·5XFAD mice compared to 5XFAD mice. Our findings indicate that the CTA paradigm is useful for evaluating a hippocampus-independent form of memory defects in AD model mice, which is sensitive to rescue by partial reductions of the β-secretase BACE1 and consequently of cerebral Aβ.
Keywords: Alzheimer’s disease, β-secretase, knockout, implicit memory, APP transgenic
Introduction
Animal models of Alzheimer’s disease (AD) have dramatically increased our understanding of molecular and pathophysiological mechanisms of this intractable disease (LaFerla and Oddo, 2005; McGowan et al., 2006; Eriksen and Janus, 2007; Gotz and Ittner, 2008). In particular, extensive studies have tested AD-related cognitive impairments in transgenic mice that overexpress mutant forms of human amyloid precursor protein (APP), using a battery of memory assays including Morris water maze, Y-maze, fear conditioning and object or social recognition (Ashe, 2001; Kobayashi and Chen, 2005; Eriksen and Janus, 2007). These behavioral investigations have successfully characterized progressive and amyloid-β (Aβ)-dependent deficits in the hippocampus-dependent memory function in APP mice. Meanwhile, two major memory systems are distinguished: explicit memory refers to a conscious recollection of specific facts and events under the control of the hippocampus and related temporal lobe connections, while implicit memory system refers to unconscious acquisition of information and is independent of hippocampal function (Squire, 1992). Although not only explicit but also implicit memory is affected as a consequence of AD neuropathology (Carlesimo and Oscar-Berman, 1992; Jelicic et al., 1995; Meiran and Jelicic, 1995; Fleischman et al., 2005), little is known about implicit memory phenotypes in APP transgenic mice as opposed to their well documented explicit memory deficits.
Conditioned taste aversion (CTA) is a simple associative memory task, in which animals learn to avoid a novel taste such as saccharin solution (conditioned stimulus: CS) that is paired with an aversive unconditioned stimulus (US) such as malaise-inducing agents (Welzl et al., 2001; Bermudez-Rattoni, 2004). While CTA memory formation involves cortical areas including the insular cortex and subcortical regions such as the amygdala, lesions of the hippocampus exert slight or little effects on CTA performance (Yamamoto and Fujimoto, 1991; Welzl et al., 2001; Bermudez-Rattoni, 2004; Josselyn et al., 2004; Ding et al., 2008). In this study, we applied hippocampus-independent CTA paradigm to examine whether a form of implicit memory is affected in 5XFAD mice, which co-overexpress human APP and presenilin-1 (PS1) harboring five familial AD (FAD) mutations and represent an early-onset and aggressive amyloid mouse model (Oakley et al., 2006). By testing 5XFAD mice at different pathological stages with CTA and contextual fear conditioning tasks, we first compared the onset of implicit and explicit memory dysfunctions associated with AD. On the other hand, evidence is accumulating that genetic deletion of β-site APP-cleaving enzyme 1 (BACE1), which initiates the processing of APP, blocks Aβ generation and improves memory dysfunctions in AD mouse models (Ohno et al., 2004, 2006, 2007; Laird et al., 2005; for review, see Ohno, 2006, 2008). Furthermore, partial reduction of BACE1 due to its heterozygous knockout (BACE1+/−) suffices to lower cerebral Aβ levels and rescue hippocampal memory deficits in APP mice (McConlogue et al., 2007; Ohno and Kimura, 2008). Therefore, we further examined whether BACE1+/− deletion may affect hippocampus-independent memory defects of 5XFAD mice in the CTA paradigm, aiming to evaluate efficacies of partial BACE1 suppression as expected for future AD treatments with appropriate dosage of suitable β-secretase inhibitors.
Materials and methods
Mouse lines
We used 5XFAD APP/PS1 doubly transgenic mice that co-express and co-inherit FAD mutant forms of human APP (the Swedish mutation: K670N, M671L; the Florida mutation: I716V; the London mutation: V717I) and PS1 (M146L; L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter (Oakley et al., 2006; Ohno et al., 2006). 5XFAD lines (Tg6799: B6/SJL genetic background) were maintained by crossing hemizygous transgenic mice with B6/SJL F1 breeders (Taconic, Hudson, NY). 5XFAD transgenic mice used were hemizygotes with respect to the transgene and were tested at different ages with non-transgenic wild-type littermate mice served as controls. For the other set of experiments, hemizygous 5XFAD transgenic mice (B6/SJL hybrid background) were crossbred to BACE1 homozygous knockout (BACE1−/−) mice (C57BL/6 background, The Jackson Laboratory, Bar Harbor, ME) (Cai et al., 2001; Laird et al., 2005) or C57BL/6 control mice. The resultant F1 heterozygous BACE1 knockout mice and hemizygous 5XFAD transgenic mice were further intercrossed, yielding animals with four different genotypes (wild-type, BACE1+/−, 5XFAD+/−, and BACE1+/−·5XFAD+/−) on the 75% B6 and 25% SJL background. These mice in the F2 progeny were analyzed at 9–10 months of age. All experiments were done blind with respect to the genotype of the mice, and were conducted with the approval of the Nathan Kline Institute Animal Care and Use Committee.
Conditioned taste aversion (CTA)
CTA is an associative learning task to test hippocampus-independent memory, which is induced by a single pairing of the CS (consumed substance, e.g., saccharin) and US (nausea, e.g., lithium) (Welzl et al., 2001; Bermudez-Rattoni, 2004). During the initial four-day habituation period, each mouse was placed in an individual cage and had access to water intake through two bottles (30 min per day). On the conditioning day (Day 5), mice were allowed to drink only 0.5% saccharin solution (Sigma-Aldrich, St. Louis, MO) through a single bottle for 30 min and received an i.p. injection of malaise-inducing lithium chloride (LiCl: 0.15 M, 2% body weight, Sigma-Aldrich) or saline (as unconditioned controls) 40 min after the removal of saccharin solution. One day after conditioning (Day 6), mice received a two-bottle choice test (30 min) between water presented in one bottle and 0.5% saccharin presented in another bottle: the placement of each bottle was randomized. The intake of each fluid was measured, and the percent consumption of saccharin was calculated to evaluate the CTA memory.
Contextual fear conditioning
Contextual fear conditioning is a task for hippocampus-dependent memory, in which mice learn to associate a distinct context (CS) with aversive footshocks (US) (Fanselow, 2000). Protocols were the same as those described previously (Ohno et al., 2001; Kimura and Ohno, 2009). The experiments were performed using four standard conditioning chambers, each of which was housed in a soundproof isolation cubicle and equipped with a stainless-steel grid floor connected to a solid-state shock scrambler. Each scrambler was connected to an electronic constant-current shock source that was controlled via an interface connected to a Windows XP computer running FreezeFrame software (Coulbourn Instruments, Allentown, PA). A digital camera was mounted on the steel ceiling of each chamber, and video signals were sent to the same computer for analysis. During training, mice were placed in the conditioning chamber for 3 min and then received two unsignaled footshocks (1.0 mA, 2 s) at 1 min intervals. After the last shock delivery, the mice were allowed to stay in the chamber for another 30 s and were then returned to their home cages. Contextual fear memory was evaluated by scoring freezing behavior (the absence of all movement except for that needed for breathing) for 3 min when the mice were placed back into the same conditioning chamber 1 day after training. The automated FreezeFrame system (Coulbourn Instruments), which digitizes the video signal at 4 Hz and compares movement frame by frame, was used to score the amount of freezing.
Immunoblot analysis
Hemibrain samples were taken from the mice under deep isoflurane anesthesia and were snap-frozen for biochemical assays. For Western blot analysis, each hemibrain sample was homogenized in 5 volumes of modified RIPA buffer containing 150 mM NaCl, 50 mM Tris HCl (pH 8.0), 1 mM EDTA, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Calbiochem, La Jolla, CA), and centrifuged at 10,000 g for 10 min to remove any insoluble material. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL), and 20–50 µg of protein was run on 12 or 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membrane. After blocking, membranes were probed with anti-BACE1 (1:1,000, MAB5308, Millipore, Billerica, MA), antibody that recognizes C-terminal epitope in APP (C1/6.1, kindly provided by Dr. Paul Mathews, Nathan Kline Institute) to detect full-length APP (1:1,000)/C-terminal fragments (1:500) and anti-β-actin (1:10,000, AC-15, Sigma, St. Louis, MO) as loading control, and were incubated with horseradish peroxidase-conjugated secondary IgG. Immunoblot signals were visualized by a Novex ECL chemiluminescence substrate reagent kit (Invitrogen) and were quantified by densitometric scanning and image analysis using Quantity One software (Bio-Rad Laboratories, Hercules, CA).
Enzyme-linked immunosorbent assay (ELISA) for Aβ40 and Aβ42
Sandwich Aβ ELISAs were performed as described previously (Oakley et al., 2006; Ohno et al., 2006). Briefly, each hemibrain sample was extracted in 8X cold 5 M guanidine HCl plus 50 mM Tris HCl (pH 8.0) buffer, and centrifuged at 20,000 g for 1 h at 4°C to remove insoluble material. Final guanidine HCl concentrations were below 0.1 M. Protein concentrations were determined by a BCA kit (Pierce). To quantitate total levels of cerebral Aβ(1–40) and Aβ(1–42), supernatant fractions were analyzed by well-established human Aβ40 and Aβ42 ELISA kits (KHB3481 and KHB3441, Invitrogen), respectively, according to the protocol of the manufacturer. Optical densities at 450 nm of each well were read on a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA), and sample Aβ40 and Aβ42 concentrations were determined by comparison with the respective standard curves. Aβ40 and Aβ42 values were normalized to total brain protein concentrations.
Aβ immunohistochemistry
Mice were perfused with 4% paraformaldehyde in phosphate buffered saline (PBS) under deep isoflurane anesthesia. The brain was removed and sectioned coronally at 30 µm on a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany), and successive sections were stored in PBS containing 0.01% sodium azide at 4°C. For immunohistochemical analysis of amyloid deposition, the sections were stained by the avidin-biotin peroxidase complex method as described previously (Oakley et al., 2006; Ohno et al., 2007). Briefly, the sections were incubated overnight at room temperature with monoclonal anti-Aβ1–16 antibody (1:200; 6E10, Signet, Dedham, MA). The ABC kit (PK-2200, Vector Laboratories, Burlingame, CA) was utilized with 3,3’-diaminobenzidine tetrahydrochloride as a chromogen to visualize the reaction product. The sections were then mounted on charged slides, dehydrated in a series of alcohol, cleared in xylene, and covered with a coverslip. Light microscopy was conducted on an Axioskop 2 microscope equipped with an AxioCaM HRc digital camera (Zeiss, Munich, Germany) for capturing images.
Statistical analysis
The significance of differences between the groups was determined by a one-way ANOVA and post-hoc Fisher’s PLSD tests were performed when appropriate. Data were presented as mean ± SEM and the level of significance was set for p value less than 0.05.
Results
CTA and contextual memory deficits in 5XFAD mice
Pairing of the novel taste saccharin (CS) with malaise induced by LiCl (US) resulted in significant reductions in percent saccharin intake during the choice test given 1 day after conditioning in 5XFAD mice at 6–7 months of age (F1,24 = 19.58, P < 0.05) as well as wild-type littermates (F1,28 = 32.91, P < 0.05) compared to their respective saline-treated (unconditioned) controls (Fig. 1A). The relative amount of saccharin intake during testing was indistinguishable between two groups (F1,34 = 0.07, P > 0.05), indicating normal CTA performance of 5XFAD mice at this age. In contrast, LiCl-conditioned 5XFAD mice at 9–10 months (F1,21 = 2.23, P > 0.05) or 12–15 months (F1,17 = 1.01, P > 0.05) of age failed to acquire an aversion to saccharin solution and showed a preference for saccharin to a level that was not significantly different from that of unconditioned subjects (Fig. 1B and 1C). Consequently, the CTA memory was significantly impaired in 5XFAD mice at 9–10 months (F1,27 = 5.08, P < 0.05) and 12–15 months (F1,18 = 10.08, P < 0.05) of ages as compared with their respective wild-type littermate controls, which exhibited robust avoidance of saccharin solution after conditioning with LiCl (9–10-month-old wild-type, F1,23 = 44.00, P < 0.05; 12–15-month-old wild-type, F1,18 = 67.64, P < 0.05). On the other hand, the natural preference for saccharin solution, as observed in unconditioned control animals that received saline instead of LiCl after saccharin exposure, was not different between 5XFAD and wild-type control mice at any ages tested. Therefore, it is unlikely that the ability to perceive the taste of 0.5% saccharin may be affected in 5XFAD mice. Furthermore, a trend toward increases rather than decreases in the absolute amount of saccharin intake during conditioning was observed in 5XFAD mice compared to wild-type controls (data not shown). Together, these data indicate that the poor CTA performance of 5XFAD mice is not due to changes in their taste perception, baseline preference to saccharin solution or weaker presentation of the CS but reflects specific impairments in CTA memory function.
Fig. 1.

Age-dependent impairments of conditioned taste aversion (CTA) in 5XFAD mice. (A–C) 5XFAD mice at 6–7 months (A), 9–10 months (B) and 12–15 months (C) of ages, and their respective wild-type littermate mice received LiCl (conditioned group) or saline (unconditioned group) after saccharin intake. While 5XFAD mice at 6–7 months of age exhibit significant reductions in percent saccharin consumption 1 day after conditioning compared to unconditioned controls (*P < 0.05 vs. saline), 5XFAD mice at 9–10 and 12–15 months of ages show preference for saccharin solution to levels not significantly different from that of unconditioned subjects and their CTA performance is significantly lower than that of wild-type controls (#P < 0.05). n = 8–18 mice per group. All data are presented as mean ± SEM.
To compare changes in the CTA memory and hippocampus-dependent explicit memory performances of 5XFAD mice, we next applied contextual fear conditioning (Fig. 2). Wild-type mice exhibited a robust conditioned fear response as assessed by freezing (the absence of all but respiratory movements) when placed back into the conditioning chamber 1 day after training with two CS–US parings. While 5XFAD mice at 3–4 months of age showed contextual freezing that was indistinguishable from that of wild-type controls (F1,26 = 0.004, P > 0.05) (Fig. 2A), 5XFAD mice at 6–7 months (F1,39 = 6.19, P < 0.05) and 12–13 months (F1,22 = 5.09, P < 0.05) of ages exhibited significantly lower levels of freezing compared to their respective wild-type littermates (Fig. 2B and 2C). Therefore, 5XFAD mice also showed age-dependent impairments in contextual fear memory. However, it should be noted that the onset of hippocampus-independent CTA memory deficits (i.e., 9–10 months of age) is delayed compared with that of explicit memory deficits as tested by contextual conditioning (i.e., 6–7 months of age) in 5XFAD mice.
Fig. 2.

Age-dependent impairments of contextual fear conditioning in 5XFAD mice. (A–C) 5XFAD mice at 3–4 months (A), 6–7 months (B) and 12–13 months (C) of ages, and their respective wild-type littermate mice were trained with two CS-US pairings for contextual fear conditioning. 5XFAD mice at 6–7 and 12–13 months of ages, but not at 3–4 months of age, show significantly lower levels of contextual freezing than wild-type controls (*P < 0.05) when tested 1 day after training. n = 11–23 mice per group. All data are presented as mean ± SEM.
BACE1+/− deletion rescues CTA memory deficits in 5XFAD mice
We examined whether partial β-secretase suppression due to BACE1+/− mutation would ameliorate the CTA memory deficits observed in 5XFAD mice at 9–10 months of age (Fig. 3). We confirmed that whereas wild-type mice showed significant reductions in percent saccharin consumption compared to their unconditioned control subjects (F1,30 = 31.74, P < 0.05), 5XFAD mice failed to exhibit significant CTA to saccharin solution 1 day after conditioning with LiCl (F1,31 = 3.19, P > 0.05). Importantly, levels of CTA performance were significantly different between four groups of mice when the amount of percent saccharin intake in conditioned animals was analyzed by a one-way ANOVA (F3,85 = 3.72, P < 0.05). As a consequence, post-hoc Fisher’s PLSD test revealed that 5XFAD mice exhibited significantly lower CTA memory as compared to wild-type controls (P < 0.05), while BACE1+/−·5XFAD bigenic mice showed significantly higher levels of CTA performance than did 5XFAD mice (P < 0.05). Robust avoidance of saccharin solution after conditioning was restored to wild-type control levels in BACE1+/−·5XFAD mice (F1,32 = 71.56, P < 0.05). Meanwhile, BACE1+/− mice exhibited normal CTA to saccharin solution (F1,33 = 15.09, P < 0.05). The absence of significant differences in relative saccharin consumption in unconditioned saline controls between four genotype groups demonstrated no changes in baseline preference for saccharin or taste perception, favoring the notion that BACE1+/− deletion specifically rescues CTA memory dysfunction in 5XFAD mice.
Fig. 3.

Effects of heterozygous BACE1 deletion on impairments of conditioned taste aversion (CTA) in 5XFAD mice. Mice received LiCl (conditioned group) or saline (unconditioned group) after saccharin intake. Only 5XFAD mice show preference for saccharin solution to levels not significantly different from that of unconditioned subjects, whereas the other three groups exhibit significant reductions in percent saccharin consumption 1 day after conditioning compared to unconditioned controls (*P < 0.05 vs. saline). Note that 5XFAD mice at 9–10 months of ages show significantly lower levels of CTA to saccharin compared with wild-type littermate controls, while BACE1+/−·5XFAD are rescued completely back to wild-type levels of CTA performance (#P < 0.05 vs. 5XFAD). n = 10–25 mice per group. All data are presented as mean ± SEM.
BACE1+/− deletion reduces Aβ and C99 in 5XFAD mice
To elucidate the biochemical basis for the improvements of CTA memory in BACE1+/−·5XFAD mice, we measured levels of BACE1, full-length APP and its β-cleaved fragments at 9 months of age (Figs. 4 and 5). Immunoblot analysis of hemibrain homogenates (Fig. 4A) demonstrated that BACE1 levels in 5XFAD mice were significantly increased up to ~200% of those found in wild-type mice (F2,21 = 14.19, P < 0.05) (Fig. 4B). Due to this upregulation, the remaining levels of BACE1 in BACE1+/−·5XFAD bigenic brains were equivalent to those of wild-type controls, although heterozygous knockout reduced cerebral BACE1 expression by ~50% in 5XFAD mice in concordance with the reduction in gene copy number (P < 0.05). On the other hand, transgene-derived overexpression of human APP in 5XFAD mice was approximately fourfold relative to endogenous levels of mouse APP found in wild-type mice (F2,22 = 42.10, P < 0.05), while BACE1+/− genotype did not affect the APP overexpression associated with 5XFAD transgenes (Fig. 4C). As shown in Fig. 4A and 4D, the level of β-secretase-cleaved C-terminal fragment (C99) was dramatically elevated in 5XFAD mouse brains as opposed to wild-type control brains that showed only background levels of C99 immunoreactive bands. Notably, cerebral C99 levels were significantly reduced in BACE1+/−·5XFAD mice compared to 5XFAD mice (F2,21 = 17.19, P < 0.05). Therefore, ~50% reductions in BACE1 expression were effective in suppressing the β-cleavage of APP in 5XFAD mice, resulting in ~40% lower levels of C99.
Fig. 4.

Effects of heterozygous BACE1 deletion on levels of BACE1, full-length APP and C99 in brains of 5XFAD mice. (A) Immunoblot analysis of protein extracts from hemibrain homogenates of wild-type mice and 5XFAD mice with BACE1+/+ or BACE1+/− genotype at 9 months of age. (B–D) Intensities of immunoreactive bands on blots for BACE1 (B), full-length APP (C) and C99 (D) were quantified by phosphorimaging and expressed as percentage of 5XFAD levels. BACE1+/− genotype produces ~50% elimination of BACE1 expression and results in ~40% reductions in C99 without affecting APP overexpression in 5XFAD mice. Note that since BACE1 expression is significantly elevated in 5XFAD brains, levels of BACE1 that are equivalent to those of wild-type controls remain in BACE1+/−·5XFAD bigenic brains. *P < 0.05 (vs. wild-type controls), #P < 0.05 (vs. 5XFAD). n = 7–9 mice per group. All data are presented as mean ± SEM.
Fig. 5.

Effects of heterozygous BACE1 deletion on Aβ levels in brains of 5XFAD mice. (A–B) Levels of total Aβ40 (A) and Aβ42 (B) were quantified by sandwich ELISAs of guanidine extracts of hemibrain samples and expressed as percentage of 9-month-old 5XFAD mice. Note that excessive levels of Aβ40 and Aβ42 are significantly reduced in BACE1+/−·5XFAD mice at 9 months of age compared with 5XFAD littermate controls (by ~65% and ~45%, respectively; #P < 0.05), while their residual Aβ levels are indistinguishable from those of 6-momth-old 5XFAD mice (N.S.: not significant). n = 7–9 mice per group. All data are presented as mean ± SEM.
Furthermore, sandwich ELISAs revealed that BACE1+/− ablation also significantly lowered excessive levels of Aβ40 and Aβ42 found in 9-month-old 5XFAD mice (Aβ40, F1,16 = 11.89, P < 0.05; Aβ42, F1,16 = 6.92, P < 0.05) (Fig. 5A and 5B, respectively). It should be noted that the residual levels of Aβ40 and Aβ42 in BACE1+/−·5XFAD mouse brains at 9 months of age were indistinguishable from the respective Aβ values of 6-momth-old 5XFAD brains. Importantly, these data demonstrate that partial BACE1 ablation in 5XFAD mice at 9 months of age was sufficient to reduce cerebral Aβ40 and Aβ42 (by ~65% and ~45%, respectively) to below-threshold levels equivalent to those of 6-month-old 5XFAD mice whose CTA performance was normal (Fig. 1), and consequently manifested a clear rescue from the CTA memory impairment (Fig. 3).
To investigate the relationship between regional Aβ accumulation and changes in CTA performance, we further conducted Aβ immunostaining (Fig. 6). Massive Aβ deposition was observed in brains of 5XFAD mice at 9 months of age including the insular cortex (Fig. 6C1–C3) and basolateral amygdala (Fig. 6G1–G3), both of which have been reported to play crucial roles in mediating CTA memory formation (Yamamoto and Fujimoto, 1991; Welzl et al., 2001; Bermudez-Rattoni, 2004; Josselyn et al., 2004). Amyloid plaque pathology was suppressed in both brain regions of BACE1+/−·5XFAD mice compared to those of 5XFAD littermate control mice, as shown in Fig. 6D1–D3 for the insular cortex and in Fig. 6H1–H3 for the amygdala. Therefore, immunohistochemistry revealed that partial inhibition of BACE1 resulted in reduced Aβ concentrations in the brain structures responsible for CTA in BACE1+/−·5XFAD mice at 9 months of age consistent with their CTA memory improvements.
Fig. 6.
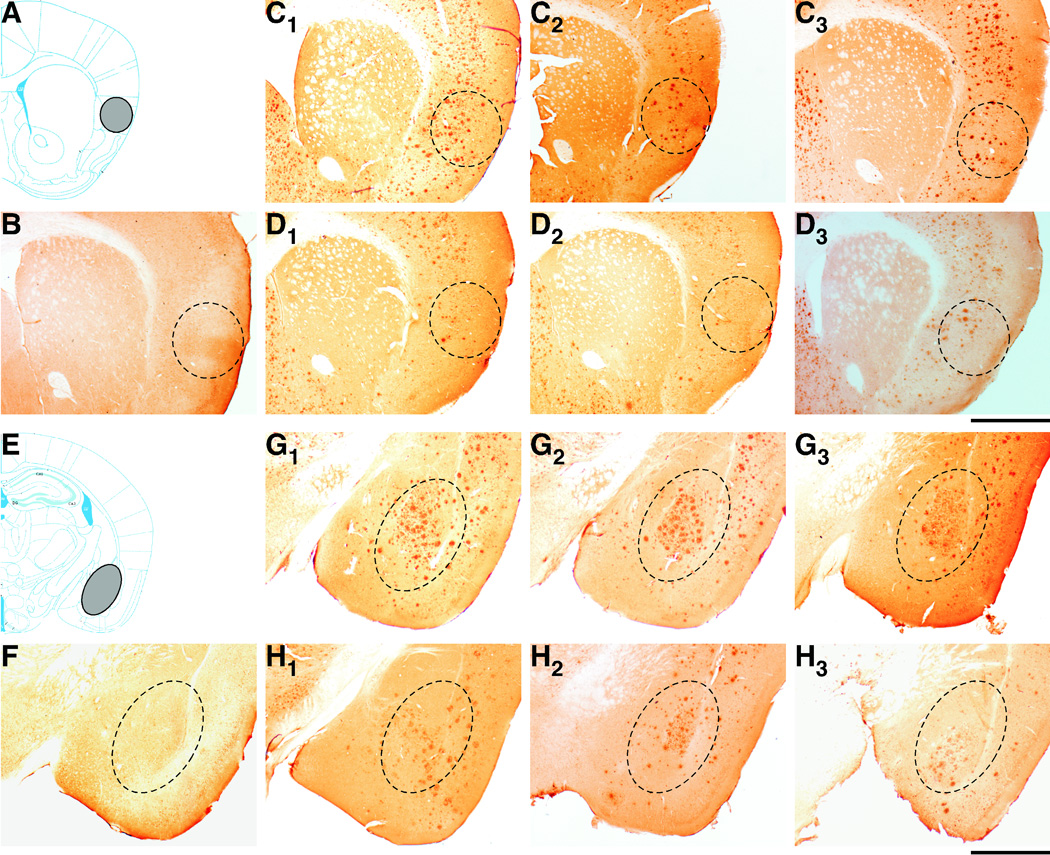
Effects of heterozygous BACE1 deletion on amyloid deposition in the insular cortex and basolateral amygdala of 5XFAD mice. (A, E) Diagrams illustrating the location of the insular cortex (A) and basolateral amygdala (E) (grey shaded areas). Schematic drawings of coronal sections were adapted from the mouse brain atlas of Franklin and Paxinos (2008). (B–D and F–H) Brain sections from wild-type control (B, F), 5XFAD (C, G) and BACE1+/−·5XFAD (D, H) mice at 9 months of age were immunostained with the 6E10 anti-Aβ antibody (n = 3 mice per group). Three different mice are presented for each of the 5XFAD (C1–C3, G1–G3) and BACE1+/−·5XFAD (D1–D3, H1–H3) genotypes. Shown are photomicrographs of the insular cortex (B–D) and basolateral amygdala (F–H) (areas within dashed ovals). Note that BACE1+/−·5XFAD brain sections exhibit lower levels of Aβ deposits in both brain regions than those of 5XFAD sections. Scale bar = 1 mm.
Discussion
The two major findings of this study are: (i) 5XFAD transgenic mice exhibit impairments in hippocampus-independent CTA memory whose onset is delayed as compared to that of explicit memory declines tested by hippocampus-dependent contextual fear conditioning, and (ii) partial reductions of the Alzheimer’s β-secretase BACE1, and consequently of cerebral Aβ, can rescue CTA memory deficits in the 5XFAD mouse model of AD.
Comparison of CTA and contextual memory deficits in 5XFAD mice
5XFAD transgenic mice co-overexpress human APP and PS1 containing five FAD mutations, and recapitulate many features of AD-related pathophysiological changes in an accelerated fashion (Oakley et al., 2006; Ohno et al., 2006, 2007). In this AD model, the Swedish APP mutation increases the production of total Aβ while the other four mutations specifically increase the production of Aβ42. Consistent with the dramatically accelerated Aβ42 production due to a combination of multiple FAD mutations, visible amyloid deposition starts to occur in 5XFAD mice as early as ~2 months of age and Aβ pathology increases rapidly with age, spreading to fill much of the hippocampus and cortex by 6 months. Furthermore, 5XFAD mice exhibit pronounced loss of large pyramidal neurons by 9 months of age in the cortical layer 5 and subiculum (the same regions with the greatest amyloid burden), a feature that is absent in the majority of APP transgenic mouse models. In the present study, we compared an aspect of implicit and explicit memory phenotypes in 5XFAD mice by applying hippocampus-independent CTA and hippocampus-dependent contextual fear conditioning paradigms to this model at different pathological stages: 3–4 months of age with moderate Aβ deposition, 6–7 months of age with massive Aβ deposition, and 9< months of age with severer Aβ deposition, marked synaptic degeneration and neuron loss.
Recent studies reported that CTA performance was impaired in APP transgenic mice such as TgCRND8 and APPswe/ΔE9 mice (Janus et al., 2004; Pistell et al., 2008). However, the progression of this type of AD-associated memory decline remains to be fully investigated, especially, by comparison with that of explicit memory failure. Here, we demonstrated that 5XFAD mice show age-dependent defects in CTA memory, which start to occur at ~9 months of age. The confirmation of CTA deficits across different APP transgenic mouse models increases the validity of this behavioral assay for evaluating AD-related alterations in a form of implicit memory function. On the other hand, explicit memory function as assessed by contextual fear conditioning began to decline in 5XFAD mice at ~6 months of age, which is consistent with the onset of their hippocampal synaptic dysfunctions such as reduced levels of basal synaptic transmission and deficient long-term potentiation (LTP: a measure of synaptic plasticity representing a cellular basis for learning and memory) at Schaffer collateral-CA1 pathways (Kimura and Ohno, 2009). Furthermore, we previously reported that hippocampus-dependent spatial learning in the Morris water maze task also deteriorates in 5XFAD mice by 6 months of age (Ohno et al., 2006). Taken collectively, it is likely that 5XFAD mice exhibit the delayed onset of hippocampus-independent CTA memory deficits relative to hippocampal synaptic and explicit memory dysfunctions, presumably reflecting their different sensitivities to increases in cerebral Aβ levels. Further studies are needed to investigate the precise relationship between emergences of explicit or implicit memory deficits and progressive increases of Aβ concentrations in discrete brain regions that are responsible for each type of memory functions in AD mouse models. Nevertheless, our results are in agreement with clinical observations that implicit or unconscious forms of memory function remain relatively intact or are less severely affected as compared with explicit or conscious forms of memory in demented AD patients (Carlesimo and Oscar-Berman, 1992; Meiran and Jelicic, 1995; Fleischman et al., 2005).
Beneficial effects of partial BACE1 deletion on CTA memory deficits in 5XFAD mice
Previous studies comparing APP transgenic versus non-transgenic control mice could not unequivocally exclude the possibility that APP overexpression rather than excess Aβ levels might cause impairments in CTA memory performances in TgCRND8 and APPswe/ΔE9 mice (Janus et al., 2004; Pistell et al., 2008). By crossing 5XFAD mice with BACE1+/− knockout mice, we clearly demonstrated that partial BACE1 reduction rescues CTA memory deficits in 5XFAD mice without affecting their APP transgene expression levels in brains. Therefore, it appears that merely overexpressing FAD mutant forms of APP is not the cause of CTA memory defects found in the 5XFAD model. To our knowledge, this is the first demonstration that BACE1 suppression is beneficial for a form of implicit memory deficiency in animal models of AD.
Importantly, the present study as well as others has shown that BACE1 expression levels in 5XFAD mouse brains are significantly elevated up to ~200% of those found in wild-type controls (Ohno et al., 2007; Zhao et al., 2007), which is consistent with clinical findings that BACE1 levels and activity are increased in sporadic AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Yang et al., 2003; Li et al., 2004; Zacchetti et al., 2007; Zhao et al., 2007). Since BACE1 mRNA levels are unchanged, it has been proposed that Aβ accumulation induces posttranscriptional upregulation of BACE1 in plaque-surrounding neurons in 5XFAD mice and the BACE1 elevation further accelerates Aβ generation (Zhao et al., 2007). Recent studies have started to reveal a couple of key signaling pathways that are responsible for increasing BACE1 expression and activities in AD: e.g., caspase-3-dependent inactivation of GGA3 leading to BACE1 protein stabilization (Tesco et al., 2007), changes in microRNA expression profiles (Hebert et al., 2008; Wang et al., 2008) and increased phosphorylation of the translation initiation factor elF2α (O'Connor et al., 2008). Interestingly, our results indicated that while heterozygous gene knockout reduces cerebral BACE1 expression by ~50% in 5XFAD mice in concordance with the reduction in gene copy number, levels of BACE1 that remain in BACE1+/−·5XFAD bigenic brains are equivalent to those of wild-type controls. Therefore, it is reasonable to speculate that ablation of a single BACE1 allele is not sufficient to prevent the mechanisms underlying posttranscriptional elevation of BACE1 in 5XFAD mice at 9 months of age, which results in maintaining wild-type levels of this enzyme in BACE1+/−·5XFAD mice. Further study is needed to explore how the candidate signaling molecules that are hypothesized to mediate BACE1 elevation in AD are altered in BACE1+/−·5XFAD mice compared to 5XFAD mice at different ages.
Of particular interest, the present results indicated that partial suppression of BACE1 due to heterozygous gene knockout significantly reduces cerebral Aβ40 and Aβ42 levels (by ~65% and ~45%, respectively) and ameliorates CTA memory deficits in 5XFAD mice at 9 months of age. Furthermore, BACE1+/− deletion also reduced Aβ deposition in the insular cortex and basolateral amygdala of 5XFAD mice, two brain regions critically involved in the processing of CTA. Therefore, it is conceivable that partial reductions in Aβ accumulation in these structures, at least in part, account for the improved CTA memory performance of BACE1+/−·5XFAD mice. On the other hand, we recently reported that heterozygous BACE1 deletion lowers Aβ levels by approximately 60% in 6-month-old 5XFAD mice and also rescues their deficits in hippocampus-dependent explicit memories such as contextual memory in the fear conditioning and spatial working memory in the spontaneous alternation Y-maze paradigm (Ohno and Kimura, 2008). Our findings are consistent with the data from other laboratories that knocking down of hippocampal BACE1 by injections of lentiviral-vectored siRNAs specifically targeting the BACE1 enzyme (Singer et al., 2005) and chronic immunization with BACE1 ectodomain (Chang et al., 2007) produce ~35–60% reductions in Aβ concentrations in APP751Swe·Lon transgenic or Tg2576 mice, resulting in better hippocampus-dependent spatial memory performances in the water maze as compared to APP mice with control treatments. Collectively, different lines of evidence from mouse model studies strongly support the idea that partial suppression of BACE1 can impact the β-cleavage of APP to reduce cerebral Aβ levels and is effective in ameliorating AD-associated cognitive dysfunctions including implicit as well as explicit components of memories.
It should be noted that we tested the impact of BACE1+/− deletion in 5XFAD mice at 9–10 months of age, which corresponded to the onset of CTA memory impairment found in 5XFAD mice. At this age, BACE1+/− mutation significantly reduced Aβ40 and Aβ42 in 5XFAD brains to levels equivalent to those of 6-month-old 5XFAD mice with BACE1+/+ genotype, which showed impaired contextual memory but normal CTA memory performance. Therefore, it is reasonable to speculate that ~50% BACE1 reductions due to heterozygous ablation suffices to decrease Aβ peptides to below-threshold levels allowing the prevention of CTA memory dysfunction in 5XFAD mice at 9–10 months of age. However, since BACE1 activities are only partially suppressed in BACE1+/−·5XFAD mice, they slowly but continuously accumulate Aβ peptides in brains afterwards. In particular, it has been reported that the impact of BACE1+/− deletion on Aβ concentrations becomes weaker and eventually disappears in APP transgenic mice as disease progresses into the more severe pathological stages (Laird et al., 2005; McConlogue et al., 2007). Together, it will be important to investigate how efficacies of partial BACE1 reduction may alter as disease further develops in 5XFAD mice, especially focusing on correlations between changes in their cerebral Aβ levels and implicit as well as explicit memory performances.
It is also important to note that BACE1+/− deletion partially suppressed build-up of the β-secretase-cleaved C-terminal fragment C99 (by ~40%) in 5XFAD mouse brains. Transgenic overexpression or central administration of the potentially amyloidogenic C99 has been shown to cause memory deficits in a broad range of behavioral tasks (Nalbantoglu et al., 1997; Song et al., 1998; Berger-Sweeney et al., 1999; Choi et al., 2001; Lee et al., 2006). Therefore, it is likely that partial BACE1 inhibition may benefit explicit and implicit memories by reducing brain levels of neurotoxic C99 as well as Aβ peptides in 5XFAD transgenic mice.
In summary, this study demonstrates that the CTA represents a useful behavioral assay for testing hippocampus-independent implicit memory declines associated with AD. In particular, since CTA and more widely used explicit memory assays such as contextual fear conditioning and water maze are different in the detection sensitivity of the onset of their impairments in 5XFAD mice, combining CTA with hippocampus-dependent paradigms would be important for characterizing behavioral phenotypes of AD mouse models and provide a unique opportunity to evaluate the effects of potential therapeutic interventions. Our results clearly show that BACE1+/− ablation can improve CTA memory deficits found in 5XFAD mice, providing a comprehensive experimental framework for the efficacy of partially β-secretase-inhibiting and Aβ/C99-reducing approaches for the treatment of AD-related deficits in multiple memory systems including a form of implicit memory failure.
Acknowledgements
This work was supported by grants from National Institute of Mental Health (R01 MH067251) and Alzheimer’s Association (IIRG-08-91231) to M.O.
References
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer's disease. Learn. Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, McPhie DL, Arters JA, Greenan J, Oster-Granite ML, Neve RL. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res. Mol. Brain Res. 1999;66:150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- Bermudez-Rattoni F. Molecular mechanisms of taste-recognition memory. Nat. Rev. Neurosci. 2004;5:209–217. doi: 10.1038/nrn1344. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Carlesimo GA, Oscar-Berman M. Memory deficits in Alzheimer's patients: a comprehensive review. Neuropsychol. Rev. 1992;3:119–169. doi: 10.1007/BF01108841. [DOI] [PubMed] [Google Scholar]
- Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J. Amyloid-β reduction by memapsin 2 (β-secretase) immunization. FASEB J. 2007;21:3184–3196. doi: 10.1096/fj.06-7993com. [DOI] [PubMed] [Google Scholar]
- Choi SH, Park CH, Koo JW, Seo JH, Kim HS, Jeong SJ, Lee JH, Kim SS, Suh YH. Memory impairment and cholinergic dysfunction by centrally administered Aβ and carboxyl-terminal fragment of Alzheimer's APP in mice. FASEB J. 2001;15:1816–1818. doi: 10.1096/fj.00-0859fje. [DOI] [PubMed] [Google Scholar]
- Ding HK, Teixeira CM, Frankland PW. Inactivation of the anterior cingulate cortex blocks expression of remote, but not recent, conditioned taste aversion memory. Learn. Mem. 2008;15:290–293. doi: 10.1101/lm.905008. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav. Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Contextual fear, gestalt memories, and the hippocampus. Behav. Brain Res. 2000;110:73–81. doi: 10.1016/s0166-4328(99)00186-2. [DOI] [PubMed] [Google Scholar]
- Fleischman DA, Wilson RS, Gabrieli JD, Schneider JA, Bienias JL, Bennett DA. Implicit memory and Alzheimer's disease neuropathology. Brain. 2005;128:2006–2015. doi: 10.1093/brain/awh559. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. New York: Academic Press; 2008. [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor β-secretase in Alzheimer's disease. Ann. Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Janus C, Welzl H, Hanna A, Lovasic L, Lane N, St George-Hyslop P, Westaway D. Impaired conditioned taste aversion learning in APP transgenic mice. Neurobiol. Aging. 2004;25:1213–1219. doi: 10.1016/j.neurobiolaging.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Jelicic M, Bonebakker AE, Bonke B. Implicit memory performance of patients with Alzheimer's disease: a brief review. Int. Psychogeriatr. 1995;7:385–392. doi: 10.1017/s1041610295002134. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol. Learn. Mem. 2004;82:159–163. doi: 10.1016/j.nlm.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Kimura R, Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Oddo S. Alzheimer's disease: Aβ, tau and synaptic dysfunction. Trends Mol. Med. 2005;11:170–176. doi: 10.1016/j.molmed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Im JY, Song JS, Lee SH, Lee HJ, Ha HY, Koh JY, Gwag BJ, Yang SD, Paik SG, Han PL. Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol. Dis. 2006;22:10–24. doi: 10.1016/j.nbd.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc. Natl. Acad. Sci. U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J. Biol. Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Meiran N, Jelicic M. Implicit memory in Alzheimer's disease: a meta-analysis. Neuropsychology. 1995;9:291–303. [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. Genetic and pharmacological basis for therapeutic inhibition of β- and γ-secretases in mouse models of Alzheimer's memory deficits. Rev. Neurosci. 2006;17:429–454. doi: 10.1515/revneuro.2006.17.4.429. [DOI] [PubMed] [Google Scholar]
- Ohno M. β-Secretase as a prime therapeutic target for Alzheimer’s disease: a perspective from mouse model studies. In: Araki W, editor. Recent Advances in the Biology of Secretases, Key Proteases in Alzheimer’s Disease. Kerala: Research Signpost; 2008. pp. 1–25. [Google Scholar]
- Ohno M, Kimura R. Impacts of partial reduction of BACE1 on synaptic and memory dysfunction in Alzheimer’s transgenic mice. Alzheimer's & Dementia. 2008;4 Suppl 2:T233–T234. [Google Scholar]
- Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ. Inducible, pharmacogenetic approaches to the study of learning and memory. Nat. Neurosci. 2001;4:1238–1243. doi: 10.1038/nn771. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur. J. Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol. Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistell PJ, Zhu M, Ingram DK. Acquisition of conditioned taste aversion is impaired in the amyloid precursor protein/presenilin 1 mouse model of Alzheimer's disease. Neuroscience. 2008;152:594–600. doi: 10.1016/j.neuroscience.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat. Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Song DK, Won MH, Jung JS, Lee JC, Kang TC, Suh HW, Huh SO, Paek SH, Kim YH, Kim SH, Suh YH. Behavioral and neuropathologic changes induced by central injection of carboxyl-terminal fragment of β-amyloid precursor protein in mice. J. Neurochem. 1998;71:875–878. doi: 10.1046/j.1471-4159.1998.71020875.x. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzl H, D'Adamo P, Lipp HP. Conditioned taste aversion as a learning and memory paradigm. Behav. Brain Res. 2001;125:205–213. doi: 10.1016/s0166-4328(01)00302-3. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Fujimoto Y. Brain mechanisms of taste aversion learning in the rat. Brain Res. Bull. 1991;27:403–406. doi: 10.1016/0361-9230(91)90133-5. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zacchetti D, Chieregatti E, Bettegazzi B, Mihailovich M, Sousa VL, Grohovaz F, Meldolesi J. BACE1 Expression and Activity: Relevance in Alzheimer's Disease. Neurodegener. Dis. 2007;4:117–126. doi: 10.1159/000101836. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J. Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]