

Abstract
Purpose of review
To address the role of LKB1 and AMP-activated protein kinase (AMPK) in glucose transport, fatty acid oxidation, and metabolic adaptations in skeletal muscle.
Recent findings
Contraction-mediated skeletal muscle glucose transport is decreased in muscle-specific LKB1 knockout mice, but not in whole body AMPKα2 knockout mice or AMPKα2 inactive transgenic mice.
Chronic activation of AMPK by 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) and β-guanadinopropionic acid enhances mitochondrial function in skeletal muscle, but AICAR or exercise-induced increases in mitochondrial markers are preserved in skeletal muscles from whole body AMPKα2 or muscle-specific LKB1 knockout mice.
Pharmacological activation of AMPK increases glucose transport and fatty acid oxidation in skeletal muscle. Therefore, chronic activation of AMPK may be beneficial in the treatment of obesity and type 2 diabetes.
Summary
LKB1 and AMPK play important roles in regulating metabolism in resting and contracting skeletal muscle.
Keywords: fatty acid oxidation, glucose transport, mitochondrial biogenesis
Introduction
The AMP-activated protein kinase (AMPK) was first identified as a Ser/Thr kinase that inactivates key enzymes involved in lipid and cholesterol biosynthesis [1]. In skeletal muscle, several lines of evidence support a role of AMPK in the regulation of glucose transport. LKB1 is a Ser/Thr kinase that is thought to be a master regulator for a diverse array of cellular processes. It has been well documented that LKB1 functions in the regulation of skeletal muscle metabolic processes. Our group and others have generated mice lacking AMPK activity in skeletal muscle as well as muscle-specific LKB1 knockout mice. In this review, we discuss the potential roles of LKB1 and AMPK in regulating skeletal muscle metabolism. In particular, we discuss data pertaining to the question of whether AMPK and LKB1 are necessary for contraction-stimulated glucose transport in skeletal muscle, and whether these proteins are necessary for adaptations that occur in muscle in response to exercise training.
Structure and regulation of AMPK
AMPK is an evolutionally conserved Ser/Thr kinase that functions in the regulation of energy metabolism [2]. In mammalian tissues, AMPK consists of heterotrimeric complexes containing a catalytic α subunit and regulatory β and γ subunits. Each subunit has two or more different isoforms [2–4]. The α1 subunit is widely expressed, whereas the α2 isoform is dominant in skeletal muscle, heart, and liver [5,6]. Phosphorylation of the Thr172 site on the α1 and α2 catalytic subunits by upstream kinase(s) is essential for AMPK activation [7–9]. Recent studies have shown that LKB1 [7,10,11] and Ca2+/calmodulin kinase kinase (CaMKK) [12–14] can serve as upstream kinases for AMPK. Given that muscle-specific LKB1 knockout mice [15] and hypomorphic LKB1 knockout with a muscle LKB1 knockout [16] show ablated AMPKα2 activity in skeletal muscle, LKB1 appears to be a major AMPK kinase in skeletal muscle.
The AMPK β-subunit may function as a scaffold for the complex [17] and may also be an important regulator of glycogen metabolism via a glycogen-binding domain [18,19]. The γ-subunit has 4-cystathionine-β-synthase (CBS) domains. These domains are required for the binding of AMP or ATP, which increases or decreases AMPK activity, respectively [20]. Binding of AMP to the γ-subunit is thought to maintain AMPK activity by preventing dephosphorylation of the Thr172 site [21•].
AMPK is activated via various cellular energy stressors and signaling pathways regulating insulin sensitivity and/or glucose transport, including in vivo exercise [22,23], hypoxia [24], leptin [25], and adiponectin [26]. AMPK can be pharmacologically activated by the antidiabetic drugs metformin [27,28] and rosiglitazone [29]. AMPK activity is also increased in response to 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), which is taken up by the cell and metabolized to ZMP, an AMP analogue [2]. The activation of AMPK results in the phosphorylation of multiple downstream substrates, whose overall effects are to increase ATP production by activating pathways involved in fatty acid oxidation and glucose transport, and to simultaneously decrease ATP consumption by inhibiting pathways that lead to fatty acid, protein, and glycogen synthesis (Fig. 1) [4,30].
Figure 1. Activation of AMPK and metabolic consequences.
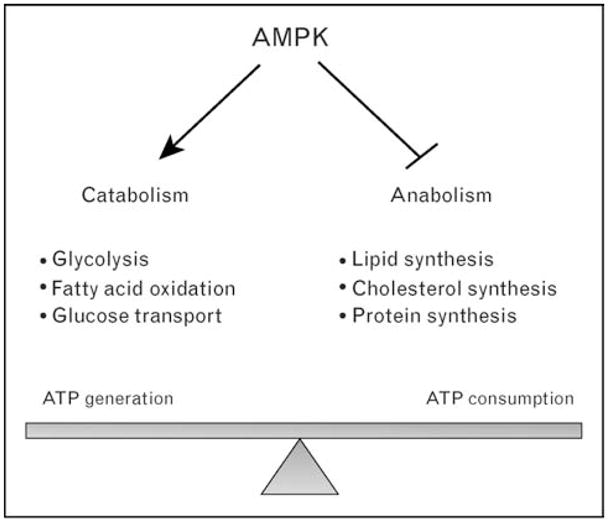
AMPK activation increases catabolic reactions, which generate ATP and inhibits anabolic reactions, which consume ATP.
Role of LKB1 and AMPK in glucose transport in skeletal muscle
Early studies from our laboratory and others provided evidence that AMPK mediates glucose transport in skeletal muscle. AICAR-induced activation of AMPK causes insulin-independent increases in skeletal muscle glucose transport [24,31]. Several studies have shown that this increase occurs concomitantly with glucose transporter 4 (GLUT4) translocation to the plasma membrane [32], similar to the effects of exercise and muscle contraction [33]. Generation of muscle-specific transgenic mice overexpressing a dominant negative form of AMPKα2, the major AMPK α subunit in skeletal muscle, and whole body knockout of AMPKα2 showed that AICAR-stimulated glucose transport was completely inhibited [34]. Collectively, these studies suggest that the activation of AMPK can lead to the activation of glucose transport in skeletal muscle. Although it is clear that AICAR-stimulated increases in skeletal muscle glucose transport are mediated by AMPKα2, there is emerging evidence that AMPK cannot be the sole mediator of contraction-stimulated glucose transport. Mu et al. [34] found only partial inhibition of contraction-mediated glucose transport in AMPKα2 dominant negative mice, and AMPKα1 and α2 knockout mice showed normal contraction-stimulated glucose transport [35]. Furthermore, we have generated muscle-specific AMPKα2 inactive transgenic mice and found that with normalization of force generation, there is no decrease in contraction-stimulated glucose transport in AMPKα2 inactive mice [36]. In addition, in-vivo measurements of contraction-stimulated glucose transport in multiple skeletal muscles (tibialis anterior, extensor digitorum longus, gastrocnemius) were completely normal in AMPKα2 inactive mice [36]. These data suggest that AMPK is not essential for contraction-stimulated glucose transport in skeletal muscle. Instead, there may be multiple, potentially redundant, signaling mechanisms mediating contraction-mediated glucose transport in skeletal muscle.
To determine the potential role of the AMPK upstream kinase, LKB1, our group has generated a muscle-specific LKB1 knockout mouse (MLKB1KO) [15]. Furthermore, Sakamoto et al. [16] have studied a hypomorphic LKB1 mouse in which whole body LKB1 protein is decreased by 70–80% and skeletal muscle LKB1 is ablated. In contrast to results from whole body AMPK (α1 and α2) knockout mice and AMPKα2 inactive transgenic mice, contraction-stimulated glucose transport was partially inhibited in these two LKB1 knockout mouse models [16,37]. Although it is not yet clear how LKB1 regulates contraction-stimulated glucose transport in skeletal muscle, decreased glucose transport cannot be explained by inactivation of AMPKα2 alone. Instead, the decrease in glucose transport could be due to decreased activity of one or more other LKB1 substrates. LKB1 is known to phosphorylate at least 12 AMPK-related protein kinases that are similar in structure and/or function to AMPK [38,39]. Although there have been no studies on the potential function of these AMPK-related kinases in regulating glucose transport in skeletal muscle, one report suggests that only some of the AMPK-related kinases (QSK, QIK, MARK2/3, and MARK4) are expressed in rat skeletal muscle, and that none of these proteins are activated by in-situ muscle contraction [40]. Interestingly, Fisher et al. [41] recently demonstrated that both muscle contraction and AICAR increase phosphorylation of the AMPK-related protein kinase (ARK) 5 in rat skeletal muscle. However, the increase in ARK5 phosphorylation was not associated with elevated enzyme activity. Thus, it is likely that contraction-stimulated glucose transport is regulated by one or more alternative downstream substrates of LKB1 (Fig. 2).
Figure 2. Schematic illustration of the pathways which are thought to regulate contraction-stimulated glucose transport in skeletal muscle.
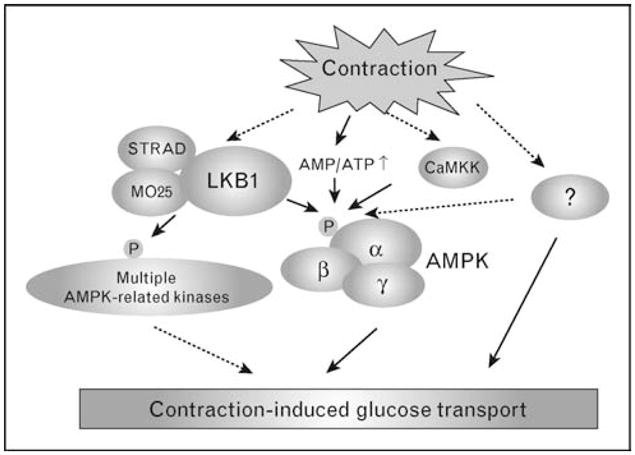
Contraction increases the [AMP]/[ATP] ratio, activates AMPK, and subsequently induces glucose transport. Studies using muscle-specifc LKB1 knockout, whole body AMPKα2 knockout, and AMPKα2 inactive transgenic mice suggest that there may be multiple pathways involved in contraction-stimulated glucose transport. Solid arrows illustrate established relationships, and dashed arrows indicate putative interactions. CaMKK, Ca2+/calmodulin kinase kinase.
Role of LKB1 and AMPK in lipid metabolism
Acute exercise results in large increases in fatty acid transport and oxidation in skeletal muscle. AMPK has been suggested to be a critical regulator of fatty acid oxidation by phosphorylating and inactivating acetyl CoA carboxylase (ACC) [22], which results in decreased production of the carnitine palmitoyltransferase I (CPT1) inhibitor, malonyl-CoA. CPT1 promotes fatty acid transport into mitochondria for subsequent oxidation [42]. Several studies [25,26,43,44] have provided evidence that AMPK activation is required for AICAR, leptin, or adiponectin-mediated fatty acid oxidation in skeletal muscle. Similarly, the effects of TNFα [45] and resistin [46] on decreased fatty acid oxidation are at least partially mediated by impaired AMPK activity in these models. Studies on mutant mice in which mutation of the γ3 subunit results in elevated AMPK activity (AMPKγ3R225) demonstrated that these mice had increased fatty acid oxidation and were protected from high fat diet-induced accumulation of intramuscular triglyceride [47]. We have found that muscle-specific LKB1 knockout mice have elevated intramuscular triglycerides [15]. Using a similar muscle-specific LKB1 knockout mouse model, Thompson et al. [48•] have shown impaired AICAR-induced fatty acid oxidation. Thus, LKB1 plays an important role in fatty acid oxidation, likely via activation of AMPK.
Role of LKB1 and AMPK in metabolic adaptation
The importance of chronic exercise for people with type 2 diabetes has been clearly established. Exercise training improves glucose homeostasis by enhancing skeletal muscle glucose transport and insulin action [49,50] and increases mitochondrial biogenesis [51–53]. These benefits are most likely related to a number of muscle adaptations in response to exercise training. AMPK has been proposed as a key molecule mediating these adaptations. Initial studies [54] have shown that chronic administration of AICAR significantly increases GLUT4 and hexokinase II, which are important for glucose transport. AICAR has also been shown to have potent effects on mitochondrial markers in skeletal muscle [44] similar to the effects of exercise training [51,55–57]. This effect was independently confirmed using another approach. Mice fed β-guanadinopropionic acid, a creatine analogue that reduces the intramuscular ATP/AMP ratio and thus results in increased AMPK activity, showed increased mitochondrial biogenesis mediated by PGC-1α and NRF-1 transcription factors [58], an adaptation that was abolished in transgenic mice overexpressing a dominant negative form of AMPKα2 [59]. Finally, decreased AMPK activity in aged human participants has been associated with decreased mitochondrial function [60••]. Taken together, these data strongly suggest that AMPK is critical for metabolic adaptations in skeletal muscle. In contrast to studies using AICAR, exercise-induced increases in GLUT4, hexokinase II, and mitochondrial markers were not abolished in whole body AMPKα2 [61••] or transgenic mice overexpressing dominant negative AMPKα2 [62••], suggesting that AMPK might be an important, but not the sole pathway regulating muscle adaptation in response to exercise training.
Exercise training results in numerous adaptations to skeletal muscle, yet the initiating signal is not fully understood. Two studies [63,64] have shown that exercise training increases LKB1 protein content, concomitant with elevated expression of the transcriptional coactivator PGC1 in rat skeletal muscle [64]. This occurs despite reportedly decreased AMPK kinase activity [63,65]. In studies of muscle-specific LKB1 knockout mice [37,66••], it was shown that LKB1 is a critical regulator of exercise capacity and basal mitochondrial function. In contrast, exercise training-induced increases in GLUT4, hexokinase II, and mitochondrial markers were preserved [66••]. Therefore, LKB1-AMPK signaling is important in skeletal muscle physiology, but there must also be additional signaling pathways that regulate skeletal muscle adaptations in response to exercise training. Potential signaling molecules that may help mediate these changes include calcium/calmodulin kinase, mitogen-activated protein kinase, calcineurin, and others.
Potential therapeutical targets of AMPK and LKB1
Given that AMPK can mediate glucose transport, fatty acid oxidation, and mitochondrial biogenesis in skeletal muscle, it is likely that chronic activation of AMPK could protect individuals from developing obesity and type 2 diabetes, similar to the effects of regular physical exercise. Although the majority of studies [28,67•] demonstrate quite consistently that AMPK activity is normal in obese patients with type 2 diabetes, one recent study [67•] proposed that exercise-induced AMPK activity and phosphorylation were impaired in these groups. In animal studies [68–71], several reports demonstrate that chronic activation of AMPK by AICAR improved insulin sensitivity in various insulin resistant models such as the ob/ob mouse, fa/fa rat, and high-fat feeding models. In fact, metformin and thiazolinediones (e.g., rosiglitazone, troglitazone, and pioglitazone) are widely used drugs in the treatment of type 2 diabetes and may function via activation of AMPK. Zhou et al. [72] demonstrated that metformin inhibits hepatic glucose production and increases glucose transport in isolated muscle via AMPK activation. Our follow-up showed that therapeutical doses of metformin increase AMPK activity in patients with type 2 diabetes, along with improved insulin sensitivity [28]. Recently, Shaw et al. [73] generated liver-specific LKB1 knockout mice, which exhibit diminished liver AMPK activity. In these animals, the blood-lowering effect of metformin in knockout mice was abolished [73].
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor γ (PPARγ) agonists that increase adiponectin secretion [74], and more recently have been found to activate muscle glucose transport mediated by AMPK [29,75]. TZDs activate AMPK in isolated rat skeletal muscle [75]. The underlying mechanism by which these drugs activate AMPK is unclear, but may be via an inhibition of the respiratory chain, leading to an increase in the intracellular AMP/ATP ratio [29]. It is also possible that the effect of TZD administration might be due to increased secretion of adipokines such as the known AMPK activator adiponectin [76,77].
As AMPK has emerged as a promising target for treating obesity and type 2 diabetes, there has been considerable effort to develop new small molecules to activate AMPK. Recently, A-769662 was identified as a novel AMPK activator [78]. Treatment of ob/ob mice with A-769662 protected these mice from weight gain, hyperglycemia, and hyperlipidemia via a largely liver-specific mechanism.
Conclusion
Numerous studies performed over the past several years have clearly shown that activation of AMPK results in an acute increase in catabolic reactions such as glucose and fatty acid oxidation, and a rapid inhibition of anabolic processes such as lipid and cholesterol biosynthesis. Activation of AMPK is also associated with altered transcriptional regulation, which may act as an additional contributor in the long-term prevention of obesity and type 2 diabetes. Skeletal muscle LKB1, an upstream kinase of AMPK and several other AMPK-related protein kinases, has been shown to regulate contraction-stimulated glucose transport. The underlying mechanisms, which appear to be AMPK-independent, still need to be elucidated. In addition, the question whether LKB1 is useful as a therapeutical target for the treatment of type 2 diabetes is not clear as the role of LKB1 in other tissues such as adipocytes or pancreas has not been studied. AMPK activation using a novel small molecule results in improved glucose homeostasis, which is an important clinical component in treating type 2 diabetes. Recent findings suggest that chronic activation of AMPK may assist in preventing obesity and type 2 diabetes.
Acknowledgments
The authors would like to acknowledge Michael F. Hirshman and Julie A. Ripley for their editorial contributions. This work was supported by National Institutes of Health (NIH) grants to L.J. Goodyear (AR45670 and DK68626). H.J. Koh is supported by the American Physical Society (APS) Fellowship in Physiological Genomics and J. Brandauer by a fellowship from NIH (T32-DK07260-29).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 339–340).
- 1.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 3.Carling D. The AMP-activated protein kinase cascade: a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Kemp BE, Mitchelhill KI, Stapleton D, et al. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci. 1999;24:22–25. doi: 10.1016/s0968-0004(98)01340-1. [DOI] [PubMed] [Google Scholar]
- 5.Stapleton D, Mitchelhill KI, Gao G, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 6.Woods A, Azzout-Marniche D, Foretz M, et al. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol Cell Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crute BE, Seefeld K, Gamble J, et al. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 9.Stein SC, Woods A, Jones NA, et al. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345 (Pt 3):437–443. [PMC free article] [PubMed] [Google Scholar]
- 10.Hong SP, Leiper FC, Woods A, et al. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Woods A, Dickerson K, Heath R, et al. C(Ca2+)/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Hurley RL, Anderson KA, Franzone JM, et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 15.Koh HJ, Arnolds DE, Fujii N, et al. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakamoto K, McCarthy A, Smith D, et al. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woods A, Salt I, Scott J, et al. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996;397:347–351. doi: 10.1016/s0014-5793(96)01209-4. [DOI] [PubMed] [Google Scholar]
- 18.Hudson ER, Pan DA, James J, et al. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol. 2003;13:861–866. doi: 10.1016/s0960-9822(03)00249-5. [DOI] [PubMed] [Google Scholar]
- 19.Polekhina G, Gupta A, Michell BJ, et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- 20.Cheung PC, Salt IP, Davies SP, et al. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346 (Pt 3):659–669. [PMC free article] [PubMed] [Google Scholar]
- 21•.Sanders MJ, Grondin PO, Hegarty BD, et al. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. The authors proposed the mechanism of AMP to activate AMPK by two distinct pathways: direct allosteric activation, and inhibition of protein phosphatase 2C to dephosphorylate Thr172 on AMPK α subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 23.Fujii N, Hayashi T, Hirshman MF, et al. Exercise induces isoform-specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochem Biophys Res Commun. 2000;273:1150–1155. doi: 10.1006/bbrc.2000.3073. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi T, Hirshman MF, Kurth EJ, et al. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- 25.Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 26.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 27.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 28.Musi N, Hirshman MF, Nygren J, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 29.Fryer LG, Parbu-Patel A, Carling D. The antidiabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 30.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 31.Bergeron R, Russell RR, III, Young LH, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol. 1999;276:E938–E944. doi: 10.1152/ajpendo.1999.276.5.E938. [DOI] [PubMed] [Google Scholar]
- 32.Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–1671. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi T, Wojtaszewski JF, Goodyear LJ. Exercise regulation of glucose transport in skeletal muscle. Am J Physiol. 1997;273:E1039–E1051. doi: 10.1152/ajpendo.1997.273.6.E1039. [DOI] [PubMed] [Google Scholar]
- 34.Mu J, Brozinick JT, Jr, Valladares O, et al. A role for AMP-activated protein kinase in contraction-and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen SB, Viollet B, Andreelli F, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 36.Fujii N, Hirshman MF, Kane EM, et al. AMP-activated protein kinase {alpha}2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. J Biol Chem. 2005;280:39033–39041. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- 37.Hirshman MF, Koh HJ, Goodyear LJ. LKB1 in muscle is critical for exercise capacity and partially regulates glucose transport. Diabetes. 2006;55 (Suppl 1):A13. [Google Scholar]
- 38.Lizcano JM, Goransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaleel M, McBride A, Lizcano JM, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–1423. doi: 10.1016/j.febslet.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab. 2004;287:E310–E317. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- 41.Fisher JS, Ju JS, Oppelt PJ, et al. Muscle contractions, AICAR, and insulin cause phosphorylation of an AMPK-related kinase. Am J Physiol Endocrinol Metab. 2005;289:E986–E992. doi: 10.1152/ajpendo.00335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- 43.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 44.Winder WW, Holmes BF, Rubink DS, et al. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 45.Steinberg GR, Michell BJ, van Denderen BJ, et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signalling. Cell Metab. 2006;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Palanivel R, Sweeney G. Regulation of fatty acid uptake and metabolism in L6 skeletal muscle cells by resistin. FEBS Lett. 2005;579:5049–5054. doi: 10.1016/j.febslet.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Barnes BR, Marklund S, Steiler TL, et al. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004;279:38441–38447. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- 48•.Thomson DM, Brown JD, Fillmore N, et al. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab. 2007;293:E1572–E1579. doi: 10.1152/ajpendo.00371.2007. As in [15], lack of LKB1 results in impaired fatty acid oxidation and elevated intramuscular triglycerides in skeletal muscle. [DOI] [PubMed] [Google Scholar]
- 49.Holloszy JO. Exercise-induced increase in muscle insulin sensitivity. J Appl Physiol. 2005;99:338–343. doi: 10.1152/japplphysiol.00123.2005. [DOI] [PubMed] [Google Scholar]
- 50.Zierath JR. Invited review: exercise training-induced changes in insulin signaling in skeletal muscle. J Appl Physiol. 2002;93:773–781. doi: 10.1152/japplphysiol.00126.2002. [DOI] [PubMed] [Google Scholar]
- 51.Holloszy JO. Biochemical adaptations in muscle: effects of exercise on mitochondrial oxygen uptake and respiratory activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- 52.Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Ann Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- 53.Gollnick PD, Armstrong RB, Saltin B, et al. Effect of training on enzyme activity and fiber composition of human skeletal muscle. J Appl Physiol. 1973;34:107–111. doi: 10.1152/jappl.1973.34.1.107. [DOI] [PubMed] [Google Scholar]
- 54.Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- 55.Goodyear LJ, Hirshman MF, Smith RJ, Horton ES. Glucose transporter number, activity, and isoform content in plasma membranes of red and white skeletal muscle. Am J Physiol. 1991;261:E556–E561. doi: 10.1152/ajpendo.1991.261.5.E556. [DOI] [PubMed] [Google Scholar]
- 56.Ploug T, Stallknecht BM, Pedersen O, et al. Effect of endurance training on glucose transport capacity and glucose transporter expression in rat skeletal muscle. Am J Physiol Endocrinol Metab. 1990;259:E778–E786. doi: 10.1152/ajpendo.1990.259.6.E778. [DOI] [PubMed] [Google Scholar]
- 57.Baldwin KM, Winder WW, Terjung RL, Holloszy JO. Glycolytic enzymes in different types of skeletal muscle: adaption of exercise. Am J Physiol. 1973;225:962–966. doi: 10.1152/ajplegacy.1973.225.4.962. [DOI] [PubMed] [Google Scholar]
- 58.Bergeron R, Ren JM, Cadman KS, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 59.Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60••.Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. This study showed that the acute stimulation of AMPK by AICAR or exercise was impaired in old rats, which results in impaired mitochondrial function. Chronic activation of AMPK by AICAR or exercise blunts aging-associated reductions in AMPK activity and mitochondrial function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61••.Jorgensen SB, Treebak JT, Viollet B, et al. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab. 2007;292:E331–E339. doi: 10.1152/ajpendo.00243.2006. Using the whole body AMPKα2 knockout mice, the authors suggested that AMPK is required for mitochondrial function in basal state but is not necessary for muscle adaptation in response to chronic exercise. [DOI] [PubMed] [Google Scholar]
- 62••.Rockl KS, Hirshman MF, Brandauer J, et al. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–2069. doi: 10.2337/db07-0255. Consistent with the study [ 61••], the authors proposed that AMPK is not necessary for exercise-induced increases in mitochondrial markers but is required for training-induced fiber type shift and hexokinase II expression. [DOI] [PubMed] [Google Scholar]
- 63.Taylor EB, Hurst D, Greenwood LJ, et al. Endurance training increases LKB1 and MO25 protein but not AMP-activated protein kinase kinase activity in skeletal muscle. Am J Physiol Endocrinol Metab. 2004;287:E1082–E1089. doi: 10.1152/ajpendo.00179.2004. [DOI] [PubMed] [Google Scholar]
- 64.Sriwijitkamol A, Ivy JL, Christ-Roberts C, et al. LKB1–AMPK signaling in muscle from obese insulin-resistant Zucker rats and effects of training. Am J Physiol Endocrinol Metab. 2006;290:E925–E932. doi: 10.1152/ajpendo.00429.2005. [DOI] [PubMed] [Google Scholar]
- 65.Hurst D, Taylor EB, Cline TD, et al. AMP-activated protein kinase kinase activity and phosphorylation of AMP-activated protein kinase in contracting muscle of sedentary and endurance trained rats. Am J Physiol Endocrinol Metab. 2005;289:E710–E715. doi: 10.1152/ajpendo.00155.2005. [DOI] [PubMed] [Google Scholar]
- 66••.Thomson DM, Porter BB, Tall JH, et al. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab. 2007;292:E196–E202. doi: 10.1152/ajpendo.00366.2006. Consistent with the study [37], the authors showed that the lack of LKB1 results in impaired mitochondrial function and exercise capacity. [DOI] [PubMed] [Google Scholar]
- 67•.Sriwijitkamol A, Coletta DK, Wajcberg E, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. This study suggested that patients with obesity and type 2 diabetes impair exercise-stimulated AMPK activity but normal increase in PGC1α. This study proposed the essential of exercise for patients with obese and type 2 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song XM, Fiedler M, Galuska D, et al. 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia. 2002;45:56–65. doi: 10.1007/s125-002-8245-8. [DOI] [PubMed] [Google Scholar]
- 69.Iglesias MA, Ye JM, Frangioudakis G, et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–2894. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- 70.Bergeron R, Previs SF, Cline GW, et al. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50:1076–1082. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- 71.Buhl ES, Jessen N, Pold R, et al. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51:2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 72.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miyazaki Y, Mahankali A, Matsuda M, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87:2784–2791. doi: 10.1210/jcem.87.6.8567. [DOI] [PubMed] [Google Scholar]
- 75.Yonemitsu S, Nishimura H, Shintani M, et al. Troglitazone induces GLUT4 translocation in L6 myotubes. Diabetes. 2001;50:1093–1101. doi: 10.2337/diabetes.50.5.1093. [DOI] [PubMed] [Google Scholar]
- 76.Tomas E, Tsao TS, Saha AK, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99:16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 78.Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]