

Abstract
In this paper we present a fast and reliable method for generation of molecular surfaces. While the method is readily applicable to van der Waals surface generation, we shall be focusing on solvent-accessible surfaces (SAS) and solvent-excluded surfaces (SES) of a molecule. A list-based method is utilized to represent and generate the union of multiple spheres with arbitrary radii, with which we are able to produce the SAS and SES of a molecule in a very efficient way. The surface generated is represented by a quadrilateral mesh, which can be easily converted into a triangular mesh if needed. Some results will be provided to demonstrate the speed and quality of our mesh generation algorithms.
I. Introduction
Molecular surface generation is critical not only in visualization of the 3D structure of a molecule but equally importantly in studying its biochemical and biophysical properties through mathematical simulations [1]. A molecule is composed of a number of atoms, each of which is mimicked by a 3D sphere specified by a unique center and radius. The van der Waals surface is simply the union of all such spheres with their intersecting portions removed. The center of the probing sphere rolling over the van der Waals surface gives rise to the so-called solvent accessible surface (SAS) [2]. The radius of the probing sphere is chosen as the size of the solvent molecules (usually water). Another widely used surface is known as solvent excluded surface (SES), defined as the “inward-facing” part of the probing sphere as it rolls over the molecules [2, 3]. The molecular surface can be represented analytically by a list of seamless spherical surface patches.
While other types of molecular surfaces, such as those based on the level set of a Gaussian-like smoothly decaying scalar function [4, 5] or based on the surface free energy minimization [6], have been investigated, the most commonly used definition of molecular surfaces is still based on the “hard-sphere” models as defined above. Therefore, in the rest of the present paper, we will restrict ourselves to this type of molecular surfaces. The MSMS (Maximal Speed Molecular Surface), developed by Sanner et al., is one of the most popular tools for generating surface triangulation of a molecule [7]. This tool has been widely used in molecular graphics, visualization, as well as biophysical simulation. While it works very efficiently and reliably for most molecules, it does frequently fail when the size of the molecules being considered increases to tens of thousands of atoms. Additionally, the triangular meshes generated by this tool sometimes contain self-intersections or unclosed polygons at some nodes, and some of the angles in the triangulation are either too small (close to 0°) or too large (close to 180°) [5]. It is known that meshes containing too skinny triangles often cause poor approximation quality in finite or boundary element methods [8]. For these reasons, developing a reliable and high-quality mesh generation method is in great demand in molecular graphics, modeling, and simulation.
One of the recent efforts to this end is the work by Can and coworkers [9]. In their approach, the fast marching [10], a simplified level set method, was employed to find the voxels lying on the molecular surfaces. Because of the digitization of a molecule into regular grids, this approach only gives an approximation of the molecular surface but, according to the authors [9], this method is faster and more reliable than the MSMS algorithm, especially when very large molecules are taken into consideration. In terms of computational costs, the fast marching method has to traverse all voxels inside the SAS of a molecule in a voxel-by-voxel fashion. Because of this voxel-based representation, the method described in [9], as pointed out by the authors, does not perform significantly better than the simple molecule-based approach (described below).
In the present paper, we propose a new representation for a sphere (or equivalently an atom). Our method is efficient in that each sphere is represented by a number of lists, each containing a sequence of segments of voxels. The union of spheres is then calculated based on the segments instead of individual voxels. This new representation approach can speed up the calculation of the union of spheres (or van der Waals surface or SAS of a molecule) for 4~5 times on average. The SES of a molecule will be computed upon the completion of SAS generation. The rest of this paper is organized as follows. Section II gives the details of our new representation and generation methods for molecular surfaces. Results and performance analysis will be discussed in Section III, followed by a brief conclusion in Section IV.
II. Methods
Our approach is a grid-based method, meaning that the 3D space containing the molecule being considered is partitioned into regular grids, similar to a 3D digitized image with each grid point known as a voxel. The digitization rate determines the resolution of the final surface mesh – the higher the digitization rate, the denser the final mesh. With this method, each sphere (accordingly, each atom) can be represented by a number of grids (or voxels) inside the sphere. In other words, a voxel is “active” if the distance from the voxel to the center of the sphere is less than the radius of the sphere. The union of multiple spheres is hence the union of the voxels belonging to one of the spheres, as shown in Fig. 1. The computation of the union of spheres can be done simply by finding the voxels enclosed by each of the atoms in a molecule. This is so-called molecule-based approach.
Fig. 1.

Illustration of the voxel-based approach to represent and calculate the union of multiple spheres. For simplicity, we consider here only circles on a 2D plane, although the concept is readily applicable to the 3D spheres.
A. List-based Representation of Spheres
Instead of the voxel-based method, we can also represent a sphere by a list of line segments, which is what we call list-based method, as shown in Fig. 2. In this method, each sphere is represented by line segments that are aligned in one of the three major axes (X-axis by default) and located on the grids of Y-Z plane. Each set of line segments are bounded by the corresponding sphere. If two spheres intersect, the individual sets of line segments are separated by the “bisector” between the two spheres, as shown by the dark lines in Fig. 2. All line segments with the same Y-Z coordinates are linked together forming a list. Two examples of such list are shown by arrows in red in Fig. 2 – the one on the top contains two segments and the one on the bottom contains three segments. This list-based approach can be thought of as a variant of the list processing method for distance transform as discussed in [11]. Using the list-based approach, each sphere is simplified to a list of line segments instead of a large number of voxels. The calculation of the union of multiple spheres will then reduce to the processing on a list of line segments. This is the key of our approach to speed up the computation of molecular surfaces, as explained in detail below. The data structure in C for each line segment is given as follows:
Fig. 2.

Illustration of the list-based approach to represent and calculate the union of multiple spheres. Note that the segments are separated by the “bisector” between two intersecting spheres. Each sphere has its own set of line segments. All line segments with the same y-z coordinates will be linked into a list.
typedef struct Segment SEG;
struct Segment
{
int atom;
float start;
float end;
SEG*next;
};
where the integer atom denotes the indexing number of the corresponding atom (or sphere). The start and end are the two end points for the line segment. All the segments with the same Y-Z coordinates are linked into a list using the pointer next.
B. List-based Calculation of the Union of Spheres
The union of spheres is essentially the basis of calculating the van der Waals surfaces and solvent-accessible surfaces (SES) of a molecule. In this subsection, we focus on how to compute the union of a set of spheres using the list-based representation. The steps are:
Projection of Spheres onto the Y-Z plane: Since we align the line segments along the X-axis by default, what we do first is to project all spheres onto the Y-Z plane – each sphere will be a circle on this plane. We allocate a 2D array A[j: 0~J-1][k:0~K-1] to store the occupancy information for each pixel on the Y-Z plane, where J and K are the dimensions in Y and Z axis respectively. Whenever a pixel (i, j, k) is found inside a sphere s, we add s into the array A[j][k] if s is not in the array yet. After all the spheres are projected, each entry A[j][k] in the array stores a number of integers, each of which corresponds to the sphere that intersects with the line defined by y = j and z = k (We shall denote this line as j-k below).
-
Linking the Line Segments into a List: Once the array A[j][k] is generated, we process each entry independently (therefore, our approach can be easily implemented in parallel algorithms if needed). For each of the entry A[j][k], we do the followings:
If there is no integer in an entry A[j][k], then we simply assign “null” to A[j][k], which means that no sphere intersects with the line j-k.
If there is only one integer in A[j][k], meaning that only one sphere intersects with the line j-k, then we calculate the intersection points (start and end) and assign the indexing number of the corresponding sphere to atom.
If there are two or more integers in A[j][k], we need to consider the overlapping between neighboring spheres on the line j-k. As shown in Fig. 2, when two spheres intersect, they will have their own influence zone, separated by the “bisector” between them. Using the influence zone idea, we are able to eliminate the overlapping between spheres. The detailed procedure is as follows. We initially keep an empty list in A[j][k]. Then we take each sphere into consideration – the order of the spheres being considered does not matter. Whenever a new sphere comes in, we calculate the start and end points that form a segment. Using the atom information, we can compute the “bisector” and subsequently the influence zone between the new sphere and the spheres already on the list. The influence zones are used to update the start and end values and then insert the new sphere into the current list.
Repeating the above three cases will end up with a list of non-overlapping line segments on the line j-k. We can then output the voxels covered by each line segment by visiting all segments on this line. Because we keep track of the atom for each line segment, we are able to attach the “ownership” (the atom) to each voxel found in the union of multiple spheres. This is extremely useful for the subsequent processes as discussed shortly.
From the above procedures, we are able to generate an accurate set of voxels enclosed by the union of a number of spheres (atoms). Thanks to the list-based representation, these voxels, as well as their corresponding atoms, can be found in a very efficient way, as will be demonstrated in the Results below.
C. Solvent-Excluded Surface Generation
The above list-based approach is employed to generate the solvent-accessible surface (SAS) of a molecule. The output is a number of voxels lying near the target surface. These voxels are denoted by integers (at grid points) but they can be projected onto the sphere of the associated atom – remember that we have the atom information for every voxel found. We may use the mapped positions as the centers to form a new set of spheres who share the same radius – the size of the probing sphere. This new set of spheres can be used to generate the solvent-excluded surface (SES) of the molecule. Alternatively, we can also utilize the fast marching method as discussed in [9] to find the SES of a molecule, based on the SAS results obtained in the list-based approach described above. The latter is perhaps more efficient when the sampling (the dashed squares in Fig. 1 and Fig. 2) becomes very dense and consequently the number of voxels found on the SAS of the molecule is very large, which renders the list-based method less efficient. Nevertheless, our list-based approach can still save a lot of time in generating the SAS of the molecule, upon which the generation of SES is based in the fast marching method [9].
D. Post-processing: Mesh Smoothing
Both the SAS and SES of a molecule generated by the above procedures are represented by voxels, which can be converted into a set of quadrilaterals. However, these quadrilaterals are always aligned in one of the XY, YZ, or XZ planar orientations. For better visualization, we can smooth the quadrilateral mesh using any mesh smoothing algorithms. In particular, we can project the vertices of the mesh obtained onto the corresponding spherical patch on the SAS or SES of the molecule. Again, this becomes possible thanks to the atom information we store on each voxel found in the above algorithm.
III. Results
A. Performance Analysis
Since the algorithm proposed in [9] is not significantly better than the molecule-based method in finding the SAS of a molecule and it is claimed to be faster than other existing tools for SES generation (note that finding SAS is part of this process), we shall focus on the speed comparison between the molecule-based method and the list-based method as proposed in the present study. Fig. 3 shows the performance comparison between these two methods. The time is evaluated on a Dell workstation with a 3.00GHz processor. According to the six molecules tested, the list-based method is 4~5 times faster on average than the molecule-based method. In addition, the list-based method produces accurate “ownership” information for every voxel on the molecular surfaces. But the simple molecule-based method does not guarantee the correct “ownership” information unless we again use the “bisector” to test every voxel found. But this requires a significant amount of extra time.
Fig. 3.
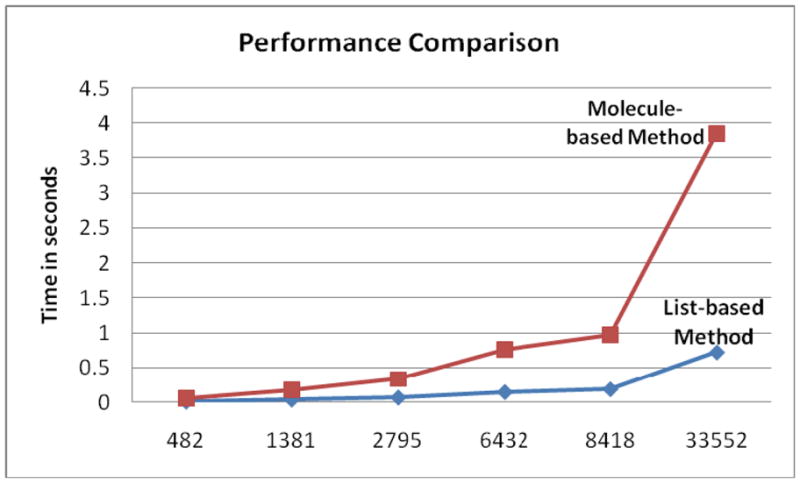
The performance comparison between the molecule-based method and the list-based method. On average, the list-based method is 4~5 times faster than the molecule-based method. The horizontal axis shows the number of atoms in the six molecules tested, which are (from left to right): 2CMP, 1CID, 2CJW, 2CEL, 2HAO, 2VBI, all taken from the protein data bank (PDB).
B. Molecular Surface Generation
In Fig. 4, we demonstrate the molecular surface generation on a small segment taken from the molecule 2CMP. The top row shows the SAS spherical patches (outside and inside views) of the atoms. Each colored patch corresponds to the contribution from one atom. The bottom row shows the generated and smoothed quadrilateral mesh of the SES surface of the molecule, which can be easily converted into a triangular mesh. The mesh is smoothed by projecting each vertex onto the corresponding spherical patch on the SES surface. From the enlarged region, we can see that the surface mesh has a very high quality in terms of angle distribution (no too small or too large angles are seen).
Fig. 4.
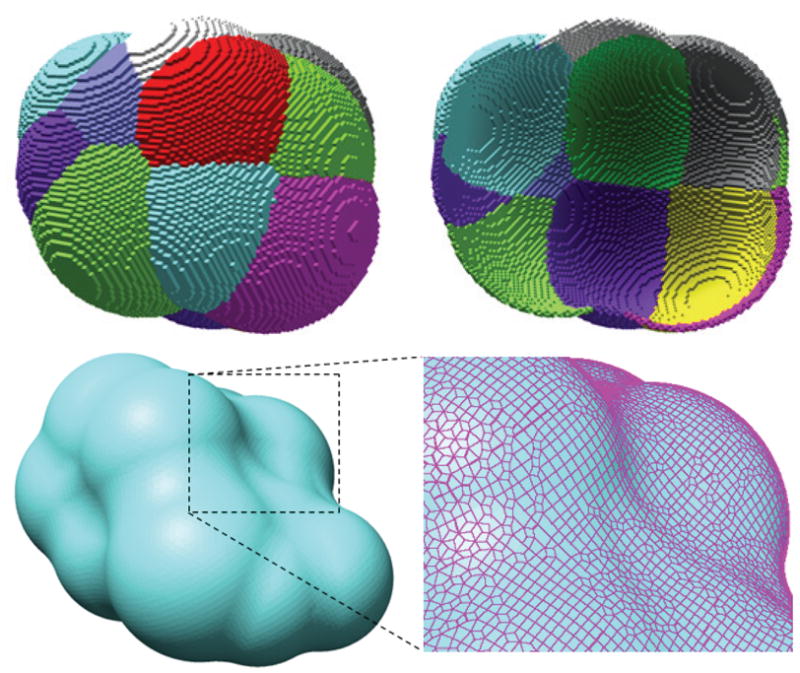
The molecular surface generation on a small segment taken from the molecule 2CMP. Top row shows the colored spherical patches on the SAS of the molecule (outside and inside views, respectively). Each of the patches is contributed from one atom. Bottom row shows the computed SES of the molecule. The surface is represented by a quadrilateral mesh, as can be seen more clearly in the enlarged region on the right. Note that a few polygon lines disappear due to some bugs in the rendering software tool (UCSF Chimera) we used.
Fig. 5 shows another example on a channeling protein called Sulfate Activating Complex, which has 8610 atoms. The generated SES mesh was smoothed using the Laplacian smoothing technique. The total time spent on both SAS and SES surface generation is less than two seconds on a Dell workstation with a 3.00GHz processor.
Fig. 5.

The molecular surface generation and smoothing on the channeling protein called Sulfate Activating Complex that has 8610 atoms. The surface mesh was smoothed using the Laplacian smoothing technique.
IV. Conclusion
In this paper, we presented a list-based approach to represent and generate the union of a set of spheres that may have different radii. This method was applied to molecules to find the solvent-accessible and solvent-excluded surfaces of a molecule. The method we proposed is very efficient in terms of speed and can produce high-quality quadrilateral or triangular meshes. In addition, it is very reliable, with the ability to handle very large molecules within a few seconds. The tool associated with the algorithms presented will be made available in a public domain. We expect that the approaches proposed here will benefit research scientists in a wide spectrum of areas by providing a fast, reliable, and high-quality surface generation tool for molecules.
Acknowledgments
This work is supported in part by a subcontract from the National Biomedical Computation Resource (NIH P41 RR08605).
References
- 1.Lu BZ, Zhou YC, Huber GA, Bond SD, Holst MJ, McCammon JA. Electrodiffusion: A continuum modeling framework for biomolecular systems with realistic spatiotemporal resolution. J Chem Phys. 2007;127:135102–19. doi: 10.1063/1.2775933. [DOI] [PubMed] [Google Scholar]
- 2.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55(3):379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 3.Connolly ML. Analytical molecular surface calculation. J Appl Cryst. 1983;16(5):548–558. [Google Scholar]
- 4.Grant JA, Pickup BT. A Gaussian description of molecular shape. J Phys Chem. 1995;99:3503–3510. [Google Scholar]
- 5.Yu Z, Holst M, Cheng Y, McCammon JA. Feature-preserving adaptive mesh generation for molecular shape modeling and simulation. Journal of Molecular Graphics and Modeling. 2008;26(8):1370–1380. doi: 10.1016/j.jmgm.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Bates PW, Wei GW, Zhao S. Minimal molecular surfaces and their applications. J Comput Chem. 2008;29(3):380–391. doi: 10.1002/jcc.20796. [DOI] [PubMed] [Google Scholar]
- 7.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 8.Huebner KH, Dewhirst D, Smith DE, Byrom TG. The Finite Element Method for Engineers. Wiley-IEEE; 2001. [Google Scholar]
- 9.Can T, Chen CI, Wang YF. Efficient molecular surface generation using level-set methods. Journal of Molecular Graphics and Modeling. 2006;25(4):442–454. doi: 10.1016/j.jmgm.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Sethian JA. Level set methods and fast marching methods: evolving interfaces in computational geometry, fluid mechanics, computer vision and materials science. 2. Cambridge University Press; 1999. [Google Scholar]
- 11.Guan W, Ma S. A list-processing approach to compute Voronoi diagrams and the Euclidean distance transform. IEEE Transaction on Pattern Analysis and Machine Intelligence. 1998;20(7):757–761. [Google Scholar]