

Abstract
No laughing matter, nitrous oxide’s role in stratospheric ozone depletion and as a greenhouse gas has stimulated great interest in developing and understanding its decomposition, particularly through the use of transition metal promoters. Recent advances in our understanding of the reaction pathways for N2O reduction by metal ions in the gas phase and in heterogeneous, homogeneous, and biological catalytic systems have provided provocative ideas about the structure and properties of metal-N2O adducts and derived intermediates. These ideas are likely to inform efforts to design more effective catalysts for N2O remediation.
Keywords: nitrous oxide, catalysis, small molecule activation, mechanistic analysis
Introduction
Nitrous oxide (N2O) is an important component of the Earth’s atmosphere that has garnered significant attention of late due to its environmental effects.[1,2] It is a potent greenhouse gas (~300 times greater warming potential than CO2)[3] and is involved in the depletion of stratospheric ozone.[4,5] Biological nitrification and denitrification processes within the global nitrogen cycle modulate N2O levels in the atmosphere.[6,7] The concentration of N2O is estimated to be increasing at ~0.5–0.9 parts per billion per volume per year, however, due in large part to anthropogenic sources that include manufacturing, fossil fuel use, and agricultural activities.[3]
In recognition of these facts, extensive research and development of remediation methods, especially involving heterogeneous metal-catalyzed N2O decomposition and reduction, have been pursued.[8] Biological systems form and consume N2O via reactions with metal ions encapsulated within enzymes.[6,9] Inspired by these metal-promoted processes, an important research goal has been to understand the reactivity of N2O with metal centers in widely disparate environments, including in the gas phase, on solid supports, in enzymes, and as soluble complexes in solution. These studies have provided new and provocative insights into how N2O might bind to metal ions and be activated for N-N or N-O bond scission or insertion into M-C or –H bonds. In this review, results from a selection of such studies are summarized, with specific focus on recent advances reported in the past ~10 years. The perspective is meant to complement those of previous reviews,[10] and the discussion will emphasize proposed geometries of N2O adducts and electronic effects of metal coordination to N2O.
A few general comments on the properties of N2O and its capability as a ligand to metal ions will help place the following discussion in perspective. The interatomic distances and low dipole moment of 0.161 D for N2O (point group C∞v) are consistent with primary contributions from the resonance structures shown in Fig. 1, top. The electron configuration of N2O is (1σ)2(2σ)2(3σ)2(4σ)2(5σ)2(6σ)2(1π)4(7σ)2(2π)4(3π)0,[11] with the frontier molecular orbitals (Fig. 1, bottom) comprising the highest occupied 2π set (approximately nonbonding overall) lying below the lowest unoccupied 3π* set, which is antibonding between all atoms. A potent oxidant thermodynamically, N2O is nonetheless kinetically recalcitrant toward decomposition and reduction; these kinetic barriers can be overcome through binding and activation by metal ions.[12] Yet N2O is in general a poor ligand to transition metals due to its weak sigma-donating and π-accepting capabilities, and there is no definitive X-ray crystal structure of a metal-N2O complex. A number of possible binding geometries and electronic structures in such complexes may be envisioned (Fig. 2), and evidence in favor of these various possibilities has been presented, as described in detail below.
Figure 1.

Bonding descriptions for N2O. Top: The major contributing resonance structures and interatomic distances. Bottom: The degenerate pairs of N2O frontier orbitals computed at the mPW1PW level of theory with the MIDI! basis set, visualized at a contour level of 0.02 a.u. (Computations performed and orbitals drawn by C. J. Cramer).
Figure 2.

Possible modes of bonding of N2O to a transition metal (M).
1.1. Metal-N2O Interactions in the Gas Phase or Inert Matrices
Many studies of the reactions of N2O with metal atoms or ions in the gas phase or in inert matrices have been performed with the goal of obtaining fundamental thermodynamic, kinetic, and mechanistic information potentially relevant to more complex systems. The most attention has been centered on the reaction shown in eq. 1, often because it is one step in a catalytic oxidation process involving reaction of a substrate (e.g., CO) with the MOn+ species.[13]
| eq. 1 |
In an illustrative study (see ref. [13] for others), inductively coupled plasma/selected-ion flow tube (ICP/SIFT) tandem mass spectrometry was used to measure the reactions of 46 different atomic cations (i.e., n = 1) with N2O at 295 K.[14] While in a few cases the reactions yielded simple adducts MN2O+ or the N-N cleavage products MN++ NO, the predominant route was N-O bond scission (eq. 1). Interestingly, many of the exothermic O-atom transfers proceeded with low efficiencies due either to intrinsic kinetic barriers or ones arising from the need to conserve spin;[15] because N2O and N2 are both ground state singlets, M+ and MO+ must have the same spins in order for the reaction to be spin-allowed, and this is often not the case (e.g., for M = Cr, Mn, Co, Ni, Mo, Ru).
Interpretation of these and other experimental results for atomic ions (n = 1) has been aided by the application of density functional theory (DFT) calculations. For example, the conversion of Fe+ to FeO+ has been examined by several authors recently,[16–18] with general agreement that N2O binds to Fe+ end-on via its O atom (η1-O) but with some disagreement concerning the specific pathway for subsequent evolution to FeO+ + N2. Using a particularly well-calibrated method, O-atom abstraction and N-O insertion paths were found to proceed from different spin states (Fig. 3).[18] The differences in the pathways are most evident from the structures of the transition states, which for the quartet (4TS1-1′) features side-on coordination of Fe to the N-O bond (η2-O,N). For the sextet (6TS1-P) the Fe is bound end-on η1-O like in the initial adduct (1), but with a shortened Fe-O and a lengthened N-O bond consistent with a route involving simple ejection of N2 via N-O bond scission. In agreement with the experimental finding that the reaction lacks an activation barrier,[19] 4TS1-1′ is 2.8 kcal/mol below the sextet starting channel and spin inversions are rate controlling (‘two-state reactivity’).[20] Related paths for the reaction in eq. 1 involving O-coordination of N2O to a metal ion have been proposed on the basis of theory for Rh+ and Pt+,[21] Ti+,[22] 4d transition metal ions (except Tc+ and Cd+),[23] and (benzene)M+ species (M = Mn+ and Co+) invoked during catalytic benzene oxidations.[24]
Figure 3.

Calculated energy profile for N2O reduction mediated by Fe+, with the numbers being relative stabilities (in kcal/mol) with respect to the separated reactants N2O + Fe+(6D). Reproduced from ref. [18] with permission from the American Chemical Society.
Gas phase reactions of N2O with metal atoms (eq. 1 with n = 0) also have been studied extensively using a variety of experimental methods.[25] Theoretical treatments[26–28] generally support end-on O-coordination of N2O to the metal atom (η1-O) with subsequent charge transfer (metal-to-N2O) and concomitant M-O-NN bending occuring along the N-O bond scission pathway. The charge transfer involves population of the LUMO of N2O (3π* in Fig. 1) and 4s/3d orbital rehybridization at the metal, with additional interactions between the N2O HOMO (2π) and the metal 3d orbitals contributing to weakening of the N-O bond. The degree of charge transfer increases along the transition metal series; indeed, the reaction of N2O with Cu is described as “proceeding via the nonadiabatic electron transfer at the crossing of the neutral Cu(4s2S1/2) + N2O(X1Σ+) and the ionic Cu+ + N2O− potential energy surfaces.”[26]
Alternative pathways for eq. 1 involving N-coordination of N2O (η1-N) to gas-phase Pt+ [29] or U+ [30] have been described. In addition, the η1-N binding mode was identified for N2O complexes of Ni, Pd, and Pt and MCl (M = Cu, Ag) embedded in solid argon at ~12 K.[31,32] The structures were assigned on the basis of analyses of IR spectra of various N2O isotopomers. For the CuCl case, DFT calculations predicted N- coordination to be more favorable than O-coordination by 8.9 kcal/mol. The authors invoke a bonding scheme involving synergistic σ-donation from the 7σ orbital of N2O into an empty σ-symmetry orbital on the metal ion and π-backbonding from metal d orbitals into the 3π* orbital of N2O (Fig. 4). While the extent of such backbonding is likely to be minimal for Cu, it can be more significant for other metals (see below).
Figure 4.
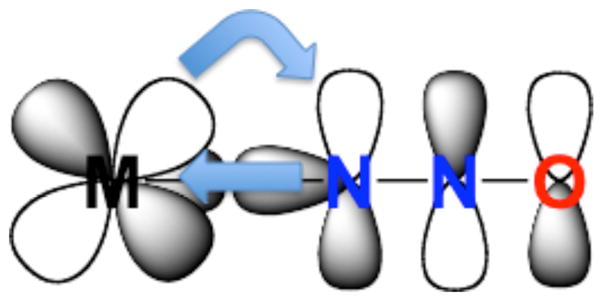
Bonding interactions for an N-coordination of N2O to a metal ion (M) showing synergistic σ-donation to M from the N2O 7σ orbital (only lobe on terminal N shown) and π-backbonding from M into the 3π* of N2O.
1.2. N2O Activation During Heterogeneous Catalysis
There has been intense interest in the development of heterogenous catalysts for abatement of N2O streams from industrial sources, particularly adipic acid and nitric acid production plants.[8,33–36] Among the efforts to understand N2O reduction by such catalysts at a molecular level, those focused on metal surfaces and iron- and copper-doped zeolites are especially widespread and have led to notably detailed insights.
While the specifics of N2O decomposition on well-defined metal surfaces are sensitive to particular surface structure features, a general picture has been developed using experimental (angle-resolved desorption, scanning tunneling microscopy, near-edge X-ray absorption fine structure spectroscopy)[37–39] and theoretical methods.[39] As illustrated by recent theoretical studies of the reactions of N2O on Rh(111)[40,41] and Pd(110),[39,42] N2O adsorption is postulated to occur in energetically similar end-on via N (η1-N) or bent bridging modes (e.g. μ-1,3-O,N). Subsequent decomposition involves ejection of N2 (which is more thermodynamically favorable than ejection of NO) following a calculated pathway that parallels that described for metal atoms in section 1.1 (Fig. 5). Thus, electron transfer via back-bonding from a metal atom into the N2O 3π* orbital (cf. Fig. 4) induces bending of the η1-N-coordinated N2O molecule, interaction of the O atom with a nearby surface metal atom, and activation of the N-O bond for scission.[39,41]
Figure 5.

Calculated reaction paths for N2O activation and decomposition on Rh(111) into N2 and absorbed O (left) vs. NO and adsorbed N (right), where the zero energy level corresponds to the non-interacting N2O molecule in the gas phase. Reproduced from ref. [41] with permission from Elsevier, Inc.
Alternative views have been presented for other surfaces. For example, DFT calculations indicated that side-on (i.e., “parallel” like η2-N,N or –N,O) binding modes of N2O to the GaN(0001) surface were found to be energetically favored relative to end-on geometries.[43] Also, the results of a recent study of N2O decomposition on the Cu2O(111) surface showed that η1-O-coordination is favored relative to N-coordination at surface oxygen vacancy sites.[44] This switch in the favored binding mode relative to that proposed for the pure metal surfaces (N-coordination) is consistent with the expected effect of increasing the oxidation state of the metal atoms.
Iron-zeolites such as Fe-ZSM-5 are arguably the most studied heterogeneous catalysts for N2O decomposition, both because of their high activity and because they have been considered as a model system for the larger class of metal ion-doped zeolites.[45] Yet, obtaining mechanistic understanding is complicated by the fact that multiple types of iron sites are present, the relative amounts of these sites depend upon the preparation method, loading, and reaction conditions, and these different sites exhibit variable reactivity.[45–47] Essentially two models have been developed, one focused on mononuclear iron sites[48, 49] and the other on dinuclear, hydroxo/oxo-bridged iron centers;[50–52] both can be present and potentially active simultaneously under certain conditions.[53] In both models, N2O is postulated to adsorb to an FeII ion in η1-O fashion and to eliminate N2 to yield an iron-oxo intermediate. For example, the transition state for N2 production shown in Fig. 6 was calculated for the [FeII2(μ-OH)(μ-O)]+ core within ZSM-5.[52] Subsequent coupling of the resulting terminal oxo atom with the bridging one was proposed to explain the formation of molecular oxygen in catalytic N2O decomposition, a thought-provoking hypothesis in view of the interest in dioxygen production pathways at multimetallic centers in biology and catalysis.[54] Interesting similarities have been drawn between the N2O reactivity of the diiron sites in Fe-ZSM-5 and putative bis(μ-oxo)dicopper centers in Cu-ZSM-5.[55,56] Notably, the higher activity of the latter has been attributed to a more facile intramolecular O-O coupling reaction of the bridging O atoms, as seen in synthetic copper complexes.[57,58]
Figure 6.
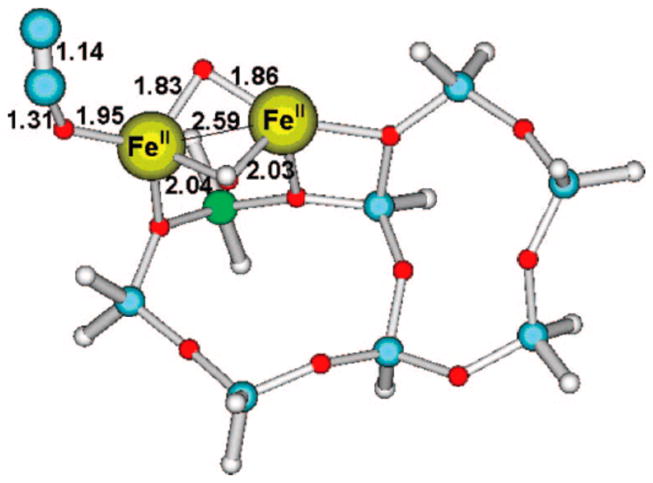
Calculated transition state structure for N2O decomposition at a [FeII2(OH)(O)]+ site in ZSM-5. Small gray balls are H, small blue balls are Si, green ball is Al, red balls are O, and large gray balls are N; bond lengths are in angstroms. Figure reproduced from ref. [52] with permission from the American Chemical Society.
1.3. N2O Activation in Biology: Nitrous Oxide Reductase
The conversion of N2O to N2 and H2O is performed in Nature by the metalloenzyme nitrous oxide reductase (N2OR) during biological denitrification, a key component of the global nitrogen cycle.[59,60] The active site contains four copper ions with a bridging (μ4) sulfide in a unique cluster geometry that has been defined in an oxidized state by X-ray crystallography[61–63] and spectroscopy[64–70] (Fig. 7). The reaction with N2O is proposed to occur with the fully reduced form (all CuI), and theoretical calculations suggest a pathway involving bent μ-1,3-O,N binding of N2O to two of the copper ions.[71,72] Concomitant electron transfer from the cluster into the π* LUMO of the bent N2O moiety is argued to induce N-O bond cleavage, which may be further facilitated by delivery of a proton(s).
Figure 7.

Proposed mechanism for the reduction of N2O by N2OR,[69–72] with the structure of the active site derived from X-ray crystallography shown in the box.
This intriguing mechanistic proposal coupled with the novelty of the [Cu4(μ-S)]n+ structure has inspired interest in developing synthetic models with which to examine the fundamental chemistry of copper-sulfur moieties supported by bio-relevant N-donor ligands.[73–75] A variety of structural motifs and electronic structures have been delineated through these efforts (Fig. 8), and heated debate about basic chemical bonding issues for several species has ensued (e.g., [Cu2S4]2+ and [Cu3S2]3+ cores).[76 – 78] To date, biologically relevant reactivity with N2O has only been seen for a complex featuring the [Cu3S2]2+ core, a localized mixed valent cluster that reduces N2O to N2 at low temperature (see section 1.4).[75]
Figure 8.

Core structural motifs identified by X-ray crystallography for copper-sulfur complexes supported by N-donor ligands (not shown).
1.4. N2O Binding and Activation by Metal Complexes in Solution
Reactions of N2O with transition metal complexes in solution are summarized in Fig. 9, and can be generally categorized as oxo-transfers involving N2 release (path A), insertions of oxygen into metal-alkyl or –hydride bonds accompanied by N2 release (path B), N-N bond scission reactions (path C), and insertions into metal-alkyl/alkyne bonds to yield products with the N2O moiety intact, as illustrated by path D (see ref. [79] for an alternate example).[2] An N2O adduct is generally thought to be a necessary first intermediate in most of these reactions, yet direct experimental supporting evidence is scarce and no such adduct has been structurally defined unambiguously by X-ray crystallography. An unusually stable complex [Ru(NH3)5(N2O)]2+ is thought to feature end-on η1-N-coordination of N2O on the basis of mechanistic, spectroscopic, and theoretical data.[80,81] A similar coordination mode was deduced from NMR data on a related Ru(II) complex.[82]
Figure 9.

Generalized summary of reactions of transition metal complexes in solution with N2O.
Recent DFT calculations on several systems that fall into the categories outlined in Fig. 9 have provided interesting mechanistic information. For example, the reactions of (dmpe)2Ru(H)2 (dmpe = Me2P(CH2)2PMe2) and (dmpe)2Ru(H)(OH) with N2O to give the O-atom insertion products (dmpe)2Ru(H)(OH) and (dmpe)2Ru(OH)2, respectively,[83] were postulated to involve nucleophilic attack of the hydride onto the terminal N-atom of N2O followed by isomerization to an O-bound Ru-ONNH intermediate.[84] Subsequent intramolecular rearrangement then yields the Ru-OH moiety and N2.
A more common postulate is that binding of N2O via η1-N-coordination is the first reaction step. For example, in a theoretical study of the reaction of Mo(NRAr)3 (R = tBu, Ar = 3,5-Me2C6H3) that proceeds according to path C, η1-N-coordination is proposed to occur in the doublet model complex (N2O)Mo(NH2)3, resulting in significant elongation of the N-N bond.[85] In studies of the incorporation of N2O into Cp2M(alkyne) (M = Ti, Zr) (path D) and with a ruthenium-nitride to yield N2 and a Ru-NO species (pathway not shown), η1-N-coordination of N2O precedes the insertion and rearrangement steps.[86, 87] Similar end-on η1-N-coordination is preferred over O-coordination in calculated pathways of reactions involving the [Fe(CN)5NO]2− ion[88] and those of complexes of early and middle transition metal ions that yield N2 and terminal or bridging metal oxo products (Fig. 10). [89] This latter conclusion is stated to be “contrary to the traditional thinking that oxygen abstraction from N2O by early or middle transition metals involves coordination of N2O via the oxygen end followed by either insertion of the metal center into the N-O bond or a direct release of the N2 molecule.”[89] A key notion reiterated in these studies is that electron donation (backbonding) by the metal ion into the π* LUMO of the η1-N-coordinated N2O underlies the preference for this binding mode and plays a key role in activating the N2O moiety for further reactions. Indeed, this idea was taken to its logical extreme in earlier calculations that showed that as the electron donating power of the metal center is increased, full reduction to the bent N2O2− moiety can occur (an N-nitrosoimide, Fig. 10).[87,90,91]
Figure 10.
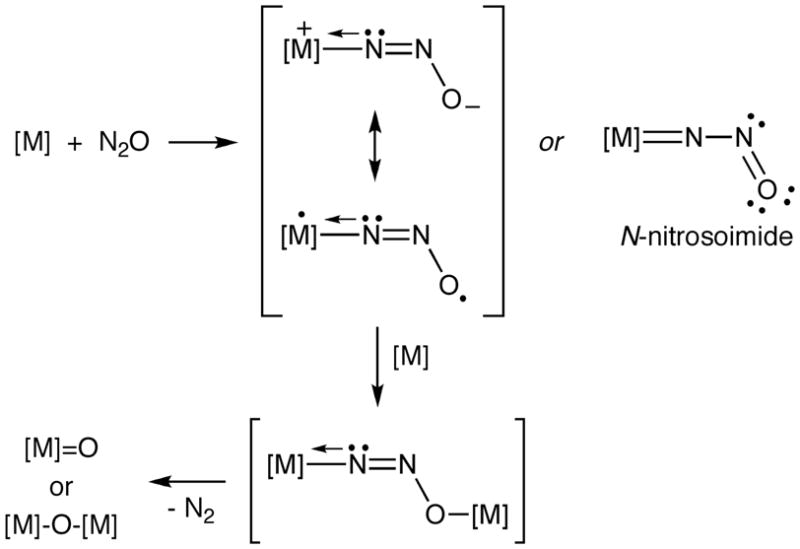
General mechanism proposed[89] for reactions of transition metal complexes ([M]) with N2O proceeding according to path A (Figure 9), including a previously suggested[90] N-nitrosoimide bonding alternative.
A quite different mechanism has been proposed for the reaction of N2O with the late transition metal complex [(Me3tacn)3Cu3S2]2+ (Me3tacn = N,N,N-trimethyl-1,4,7-triazacyclonane).[75] Experimental and theoretical data support dissociation of a CuI fragment to yield a dicopper(I,II) species that binds N2O via μ-1,1-O-coordination and induces N-O bond scission by the calculated transition state shown in Fig. 11. This pathway for a sulfur-bridged multicopper complex differs from that invoking μ-1,3-O,N-coordination proposed for the copper cluster active site in the enzyme N2OR.[71,72]
Figure 11.

Reaction coordinate for N–O bond cleavage computed at the M06L DFT level for the dicopper species derived from [(Me3tacn)3Cu3S2]2+. Free energies are in kcal/mol and selected bond distances are in angstroms. Carbon and hydrogen atoms of Me3tacn ligands are not shown for clarity. Free energies correspond to values calculated after correcting for solvation. Color key for atoms: green = Cu, blue = N, yellow = S, and red = O. Figure reproduced from ref. [75] with permission from the American Chemical Society.
1.5. Conclusion and Perspective
Mechanistic studies with a predominant use of DFT calculations have provided many interesting insights into how N2O might bind and be activated toward decomposition and/or reduction at transition metal sites in a range of environments, including in the gas phase, on solid supports, in biology, and as complexes in homogeneous solution. The various pathways postulated reflect the diversity of the systems that are capable of reacting with N2O and the myriad ways that N2O can interact with a metal center(s). Nonetheless, common elements have emerged. For example, it has become evident that N-coordination is likely in many systems, particularly those that are electron rich, and that backbonding from the metal d orbital manifold into the N2O LUMO (3π* in the free molecule) is a critical component of the bonding that has important consequences for subsequent reactions.
In general, however, experiment lags behind theory; there is a paucity of definitive evidence in support of many of the fascinating hypotheses put forward on the basis of calculations. Unmet goals include the X-ray crystallographic characterization of a metal-N2O adduct, the direct observation via spectroscopy of intermediates along an N2O reduction path(s), and the preparation of a structurally accurate and functionally competent synthetic model of the N2OR active site cluster. Addressing these and other related goals continues to be important in future research that is motivated by the significance of N2O as an atmospheric pollutant and greenhouse gas.
Acknowledgments
I thank the NIH (GM47365) for financial support of the research performed in my laboratory on copper bioinorganic chemistry, as well as each of the excellent collaborators and coworkers who have contributed to the research cited herein.
Biography

William B. Tolman obtained a B.A. in chemistry from Wesleyan University, Middletown, CT (1983) and a Ph.D. in chemistry from the University of California, Berkeley (1987). After a postdoctoral period at the Massachusetts Institute of Technology, he joined the faculty at the University of Minnesota in 1990, where he is now a Distinguished McKnight University and Lee Irvin Smith Professor and Chair of the Department of Chemistry. His research encompasses synthetic bioinorganic chemistry focused on modeling copper metalloprotein active sites and polymerization catalysis focused on the synthesis of biodegradable materials derived from renewable resources.
References
- 1.Trogler WC. Coord Chem Rev. 1999;187:303–327. [Google Scholar]
- 2.Leont’ev AV, Fomicheva OA, Proskurnina MV, Zefirov NS. Russ Chem Rev. 2001;70:91–104. [Google Scholar]
- 3.2009 US Greenhouse Gas Inventory Report. Environmental Protection Agency; ( http://tinyurl.com/emissionsreport) [Google Scholar]
- 4.Prather M. Science. 1998;279:1339–1341. doi: 10.1126/science.279.5355.1339. [DOI] [PubMed] [Google Scholar]
- 5.Ravishankara AR, Daniel JS, Portmann RW. Science. 2009;326:123–125. doi: 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- 6.Eady RR, Hasnain SS. In: Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Vol. 8. Elsevier; Amsterdam: 2004. pp. 759–786. [Google Scholar]
- 7.Trogler WC. J Chem Educ. 1995;72:973–976. [Google Scholar]
- 8.Kapteijn F, Rodriguez-Mirasol J, Moulijn JA. Appl Catal B: Environ. 1996;9:25–64. [Google Scholar]
- 9.Chen P, Gorelsky SI, Ghosh S, Solomon EI. Angew Chem Int Ed. 2004;43:4132–4140. doi: 10.1002/anie.200301734. [DOI] [PubMed] [Google Scholar]
- 10.Lee D-H, Mondal B, Karlin KD. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; Weinheim: 2006. pp. 43–79. [Google Scholar]
- 11.Tuan DFT, Hoffmann R. Inorg Chem. 1985;24:871–876. [Google Scholar]
- 12.An alternative method involving reaction with sterically encumbered Lewis acid/base pairs has recently been reported: Otten E, Neu RC, Stephan DW. J Am Chem Soc. 2009;131:9918–9919. doi: 10.1021/ja904377v.
- 13.Böhme DK, Schwarz H. Angew Chem Int Ed. 2005;44:2336–2354. doi: 10.1002/anie.200461698. [DOI] [PubMed] [Google Scholar]
- 14.Lavrov V, Blagojevic V, Koyanagi G, Orlova G, Bohme D. J Phys Chem A. 2004;108:5610–5624. [Google Scholar]
- 15.Schwarz H. Int J Mass Spectrom. 2004;237:75–105. [Google Scholar]
- 16.Blagojevic V, Orlova G, Böhme DK. J Am Chem Soc. 2005;127:3545–3555. doi: 10.1021/ja044950m. [DOI] [PubMed] [Google Scholar]
- 17.Rondinelli F, Russo N, Toscano M. Inorg Chem. 2007;46:7489–7493. doi: 10.1021/ic7007045. [DOI] [PubMed] [Google Scholar]
- 18.Zhao L, Wang Y, Guo W, Shan H, Lu X, Yang T. J Phys Chem A. 2008;112:5676–5683. doi: 10.1021/jp800511h. [DOI] [PubMed] [Google Scholar]
- 19.Armentrout PB, Halle LF, Beauchamp JL. J Chem Phys. 1982;76:2449–2457. [Google Scholar]
- 20.Schroder D, Shaik S, Schwarz H. Acc Chem Res. 2000;33:139–145. doi: 10.1021/ar990028j. [DOI] [PubMed] [Google Scholar]
- 21.Rondinelli F, Russo N, Toscano M. J Chem Theory Comput. 2008;4:1886–1890. doi: 10.1021/ct800199b. [DOI] [PubMed] [Google Scholar]
- 22.Lu L, Liu X, Wang Y, Wang H. J Mol Struc Theochem. 2006;774:59–65. [Google Scholar]
- 23.Yang X, Wang Y, Geng Z, Liu Z. Chem Phys Lett. 2006;430:265–270. [Google Scholar]
- 24.Zhao L, Liu Z, Guo W, Zhang L, Zhang F, Zhu H, Shan H. Phys Chem Chem Phys. 2009;11:4219–4229. doi: 10.1039/b901019j. [DOI] [PubMed] [Google Scholar]
- 25.Campbell ML. J Phys Chem A. 2003;107:3048–3053. and references cited therein. [Google Scholar]
- 26.Kryachko ES, Tishchenko O, Nguyen MT. Int J Quantum Chem. 2002;89:329–340. [Google Scholar]
- 27.Stirling A. J Am Chem Soc. 2002;124:4058–4067. doi: 10.1021/ja0118946. [DOI] [PubMed] [Google Scholar]
- 28.Delabie A, Vinckier C, Flock M, Pierloot K. J Phys Chem A. 2001;105:5479–5485. [Google Scholar]
- 29.Wang Y, Wang Q, Geng Z, Si Y, Zhang J, Li H, Zhang Q. Chem Phys Lett. 2008;460:13–17. [Google Scholar]
- 30.Alikhani ME, Michelini MDC, Russo N, Silvi B. J Phys Chem A. 2008;112:12966–12974. doi: 10.1021/jp803269j. [DOI] [PubMed] [Google Scholar]
- 31.Jin X, Wang G, Zhou M. J Phys Chem A. 2006;110:8017–8022. doi: 10.1021/jp061557p. [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Jin X, Chen M, Zhou M. Chem Phys Lett. 2006;420:130–134. [Google Scholar]
- 33.Pérez-Ramírez J, Kapteijn F, Schöffel K, Moulijn JA. Appl Catal B-Environ. 2003;44:117–151. [Google Scholar]
- 34.Alini S, Basile F, Blasioli S, Rinaldi C, Vaccari A. Appl Catal B-Environ. 2007;70:323–329. and references cited therein. [Google Scholar]
- 35.Jiang H, Wang H, Liang F, Werth S, Schiestel T, Caro J. Angew Chem Int Ed. 2009;48:2983–2986. doi: 10.1002/anie.200804582. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 36.Kondratenko E, Ovsitser O. Angew Chem Int Ed. 2008;47:3227–3229. doi: 10.1002/anie.200705324. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 37.Matsushima T. Prog Surf Sci. 2007;82:435–477. [Google Scholar]
- 38.Matsushima T. Surf Sci. 2009;603:1415–1426. [Google Scholar]
- 39.Kang D. Bull Kor Chem Soc. 2007;28:2369–2376. and references cited therein. [Google Scholar]
- 40.Paul JF, Pérez-Ramírez J, Arnple F, Ricart JM. J Phys Chem B. 2004;108:17921–17927. [Google Scholar]
- 41.Ricart JM, Ample F, Clotet A, Curulla D, Niemantsverdriet JW, Paul JF, Pérez-Ramírez J. J Catal. 2005;232:179–185. [Google Scholar]
- 42.Kokalj A. Surf Sci. 2003;532:213–220. [Google Scholar]
- 43.Hu C, Chen Y, Li J, Zhang Y. Chem Phys Lett. 2007;438:213–217. [Google Scholar]
- 44.Chen WK, Sun BZ, Wang X, Lu CH. J Theo Comp Chem. 2008;7:263–276. [Google Scholar]
- 45.Zecchina A, Rivallan M, Berlier G, Lamberti C, Ricchiardi G. Phys Chem Chem Phys. 2007;9:3483–3499. doi: 10.1039/b703445h. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 46.Sun K, Xia H, Feng Z, van Santen R, Hensen E, Li C. J Catal. 2008;254:383–396. [Google Scholar]
- 47.Long J, Wang X, Ding Z, Zhang Z, Lin H, Dai W, Fu X. J Catal. 2009;264:163–174. [Google Scholar]
- 48.Heyden A, Peters B, Bell AT, Keil FJ. J Phys Chem B. 2005;109:1857–1873. doi: 10.1021/jp040549a. [DOI] [PubMed] [Google Scholar]
- 49.Heyden A, Hansen N, Bell AT, Keil FJ. J Phys Chem B. 2006;110:17096–17114. doi: 10.1021/jp062814t. [DOI] [PubMed] [Google Scholar]
- 50.Hansen N, Heyden A, Bell AT, Keil FJ. J Phys Chem C. 2007;111:2092–2101. [Google Scholar]
- 51.Fellah MF, Onal I. Catal Today. 2008;137:410–417. [Google Scholar]
- 52.Guesmi H, Berthomieu D, Kiwi-Minsker L. J Phys Chem C. 2008;112:20319–20328. and references cited therein. [Google Scholar]
- 53.Hansen N, Heyden A, Bell AT, Keil FJ. J Catal. 2007;248:213–225. [Google Scholar]
- 54.Betley TA, Wu Q, Van Voorhis T, Nocera DG. Inorg Chem. 2008;47:1849–1861. doi: 10.1021/ic701972n. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 55.Smeets PJ, Sels BF, van Teeffelen RM, Leeman H, Hensen EJM, Schoonheydt RA. J Catal. 2008;256:183–191. [Google Scholar]
- 56.Smeets PJ, Groothaert MH, van Teeffelen RM, Leeman H, Hensen EJM, Schoonheydt RA. J Catal. 2007;245:358–368. [Google Scholar]
- 57.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Jr, Que L, Jr, Zuberbühler AD, Tolman WB. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 58.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 59.Zumft WG, Kroneck PMH. Adv Microb Phys. 2007;52:107–227. doi: 10.1016/S0065-2911(06)52003-X. [DOI] [PubMed] [Google Scholar]
- 60.Solomon E, Sarangi R, Woertink J, Augustine A, Yoon J, Ghosh S. Acc Chem Res. 2007;40:581–591. doi: 10.1021/ar600060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown K, Tegon M, Prudencio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. Nat Struct Biol. 2000;7:191–195. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]
- 62.Brown K, Djinovic-Carugo K, Haltia T, Cabrito I, Saraste M, Moura JJG, Moura I, Tegoni M, Cambillau C. J Biol Chem. 2000;275:41133–41136. doi: 10.1074/jbc.M008617200. [DOI] [PubMed] [Google Scholar]
- 63.Paraskevopoulos K, Antonyuk SV, Sawers RG, Eady RR, Hasnain SS. J Mol Biol. 2006;362:55–65. doi: 10.1016/j.jmb.2006.06.064. [DOI] [PubMed] [Google Scholar]
- 64.Rasmussen T, Berks BC, Sanders-Loehr J, Dooley DM, Zumft WG, Thomson AJ. Biochemistry. 2000;39:12753–12756. doi: 10.1021/bi001811i. [DOI] [PubMed] [Google Scholar]
- 65.Alvarez ML, Ai J, Zumft W, Sanders-Loehr J, Dooley DM. J Am Chem Soc. 2001;123:576–587. doi: 10.1021/ja994322i. [DOI] [PubMed] [Google Scholar]
- 66.Chen P, DeBeer George S, Cabrito I, Antholine WE, Moura JJG, Moura I, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2002;124:744–745. doi: 10.1021/ja0169623. [DOI] [PubMed] [Google Scholar]
- 67.Chen P, Cabrito I, Moura JJG, Moura I, Solomon EI. J Am Chem Soc. 2002;124:10497–10507. doi: 10.1021/ja0205028. [DOI] [PubMed] [Google Scholar]
- 68.Oganesyan VS, Rasmussen T, Fairhurst S, Thomson AJ. Dalton Trans. 2004:996–1002. doi: 10.1039/b313913a. [DOI] [PubMed] [Google Scholar]
- 69.Ghosh S, Gorelsky SI, DeBeerGeorge S, Chan JM, Cabrito I, Dooley DM, Moura JJG, Moura I, Solomon EI. J Am Chem Soc. 2007;129:3955–3965. doi: 10.1021/ja068059e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujita K, Chan JM, Bollinger JA, Alvarez ML, Dooley DM. J Inorg Biochem. 2007;101:1836–1844. doi: 10.1016/j.jinorgbio.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 71.Chen P, Gorelsky SI, Ghosh S, Solomon EI. Angew Chem Int Ed. 2004;43:4132–4140. doi: 10.1002/anie.200301734. [DOI] [PubMed] [Google Scholar]
- 72.Gorelsky SI, Ghosh S, Solomon EI. J Am Chem Soc. 2006;128:278–290. doi: 10.1021/ja055856o. [DOI] [PubMed] [Google Scholar]
- 73.York JT, Bar-Nahum I, Tolman WB. Inorg Chim Acta. 2008;361:885–893. doi: 10.1016/j.ica.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarangi R, York JT, Helton ME, Fujisawa K, Karlin KD, Tolman WB, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2008;130:676–686. doi: 10.1021/ja0762745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bar-Nahum I, Gupta A, Huber S, Ertem M, Cramer C, Tolman W. J Am Chem Soc. 2009;131:2812–2814. doi: 10.1021/ja808917k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mealli C, Ienco A, Poduska A, Hoffmann R. Angew Chem Int Ed. 2008;47:2864–2868. doi: 10.1002/anie.200705296. [DOI] [PubMed] [Google Scholar]
- 77.Carrasco R, Aullón G, Alvarez S. Chem Eur J. 2009;15:536–546. doi: 10.1002/chem.200800914. [DOI] [PubMed] [Google Scholar]
- 78.Alvarez S, Hoffmann R, Mealli C. Chem Eur J. 2009;15:8358–8373. doi: 10.1002/chem.200900239. [DOI] [PubMed] [Google Scholar]
- 79.Labahn T, Mandel A, Magull J. Z Anorg Allg Chem. 1999;625:1273–1277. [Google Scholar]
- 80.Bottomley F, Brooks WVF. Inorganic Chemistry. 1977;16:501–502. and references cited therein. [Google Scholar]
- 81.Paulat F, Kuschel T, Näther C, Praneeth V, Sander O, Lehnert N. Inorg Chem. 2004;43:6979–6994. doi: 10.1021/ic049302i. [DOI] [PubMed] [Google Scholar]
- 82.Pamplin CB, Ma ESF, Safari N, Rettig SJ, James BR. J Am Chem Soc. 2001;123:8596–8597. doi: 10.1021/ja0106319. [DOI] [PubMed] [Google Scholar]
- 83.Kaplan AW, Bergman RG. Organometallics. 1998;17:5072–5085. [Google Scholar]
- 84.Yu H, Jia G, Lin Z. Organometallics. 2008;27:3825–3833. [Google Scholar]
- 85.Khoroshun D, Musaev D, Morokuma K. Organometallics. 1999;18:5653–5660. [Google Scholar]
- 86.Yu H, Jia G, Lin Z. Organometallics. 2007;26:6769–6777. [Google Scholar]
- 87.Walstrom A, Pink M, Fan H, Tomaszewski J, Caulton KG. Inorg Chem. 2007;46:7704–7706. doi: 10.1021/ic700789y. [DOI] [PubMed] [Google Scholar]
- 88.Olabe JA, Estiu GL. Inorg Chem. 2003;42:4873–4880. doi: 10.1021/ic0261880. [DOI] [PubMed] [Google Scholar]
- 89.Yu H, Jia G, Lin Z. Organometallics. 2009;28:1158–1164. [Google Scholar]
- 90.Fan H, Caulton KG. Polyhedron. 2007;26:4731–4736. [Google Scholar]
- 91.Lee J, Pink M, Tomaszewski J, Fan H, Caulton KG. J Am Chem Soc. 2007;129:8706–8707. doi: 10.1021/ja071452f. [DOI] [PubMed] [Google Scholar]