

Summary
Defects in mitochondrial gene expression are associated with aging and disease. Mterf proteins have been implicated in modulating transcription, replication and protein synthesis. We have solved the structure of a member of this family, the human mitochondrial transcriptional terminator MTERF1, bound to dsDNA containing the termination sequence. The structure indicates that upon sequence recognition MTERF1 unwinds the DNA molecule, promoting eversion of three nucleotides. Base flipping is critical for stable binding and transcriptional termination. Additional structural and biochemical results provide insight into the DNA binding mechanism and explain how MTERF1 recognizes its target sequence. Finally, we have demonstrated that the mitochondrial pathogenic G3249A and G3244A mutations interfere with key interactions for sequence recognition, eliminating termination. Our results provide insight into the role of mterf proteins and suggest a link between mitochondrial disease and the regulation of mitochondrial transcription.
Introduction
Mitochondrial transcription is responsible for expression of 13 proteins encoded in the mitochondrial DNA as well as synthesis of two rRNAs and 22 tRNAs (Anderson et al., 1981). Transcription originates from two promoters located in the regulatory D loop, a heavy strand and a light strand promoter (HSP and LSP), that are responsible for transcription of the heavy and light strands, respectively (Asin-Cayuela and Gustafsson, 2007; Gaspari et al., 2004; Scarpulla, 2008). Initiation is dependent on the RNA polymerase, POLRMT, and two transcription factors, TFAM and TFB2M (Sologub et al., 2009; Metodiev at el., 2009), while POLRMT is sufficient for elongation. Hence, mitochondrial transcription can be regulated by modulating the expression of the genes responsible for the process itself (Scarpulla, 2006; Hock and Kralli, 2009), but some evidence suggests that transcriptional regulation can occur within the mitochondria as well, for instance in response to estrogen (Klinge, 2008). In addition, mitochondria contain their own family of dedicated transcriptional regulators: the mterf proteins. The mterf (for mitochondrial termination factor) family of proteins contains four members (MTERF1–4) all of which share homology to MTERF1, a protein originally identified as a factor responsible for mitochondrial transcriptional termination. These proteins have been implicated in mitochondrial transcription, the coordination between transcription and replication and the regulation of mitochondrial protein synthesis (Linder et al., 2005; Park et al., 2007; Roberti et al., 2009; Pellegrini et al., 2009). MTERF1 is the canonical transcriptional terminator and was originally identified as a factor responsible for terminating heavy strand transcription at a specific site at the leu-tRNA (Kruse et al., 1989), thereby modulating the ratio of mitochondrial ribosomal RNA to mRNA (Roberti et al., 2009). It was later shown that in this context, besides acting as a termination factor, MTERF1 appears to be capable of stimulating transcriptional initiation (Martin et al., 2005). Moreover, it has been proposed that MTERF1 can mediate the formation of a loop in mitochondrial DNA to promote efficient rRNA synthesis (Martin et al., 2005), although the ability of MTERF1 to bind to the HSP region is controversial (Park et al., 2007). Like other mterf proteins, MTERF1 appears to be multifunctional, and a recent report has implicated it in the control of mitochondrial replication pausing (Hyvärinen et al., 2007). All these observations are however based on in vitro experiments, and no in vivo evidence exists to support any of these proposed roles. Interestingly, the pathogenic A3243G MELAS mutation was shown to decrease the ability of MTERF1 to terminate transcription in vitro (Hess et al., 1991) and yet no obvious transcriptional alterations are observed in cells carrying this MELAS mutation (Chomyn et al. 1992). A lack of structural information on this family of proteins has contributed to maintain these uncertainties about their cellular role. Sequence alignments led to the suggestion that they are modular leucine-zipper containing proteins (Fernandez-Silva et al., 1997; Roberti et al., 2006), and that has been suggested to be the basis of their ability to bind DNA (Roberti et al., 2009).
In this paper, we have structurally characterized MTERF1 bound to its recognition sequence in the leucine tRNA and have analyzed its mechanism of binding. Our results unexpectedly show that MTERF1 does not bind DNA via leucine-zippers, but rather promotes transcriptional termination through a novel DNA binding mode. Additionally, they provide insight into how MTERF1 achieves sequence recognition, and provides clues to understand the biological roles of mterf family proteins. We have also analyzed a number of pathogenic mitochondrial DNA mutations for their ability to interfere with MTERF1 biding and transcriptional termination. We have shown that two mutations, G3249A that causes a variant of Kearns-Sayre syndrome (Seneca et al., 2001) and G3242A that is associated with an uncharacterized mitochondrial disorder (Mimaki et al., 2009), interfere with protein-DNA interactions that are key for sequence recognition by MTERF1 and as a consequence eliminate its transcriptional termination activity.
Results and discussion
Structural determination
In order to investigate how MTERF1 is able to recognize specific sequences in the mitochondrial genome as well as the mechanism by which it can modulate transcription we decided to crystallize the full-length human protein (minus the N-terminal mitochondrial localization sequence; Figure 1A) bound to the leu-tRNA sequence responsible for termination of transcription to which MTERF1 binds with high affinity (Nam and Kang, 2005). We expressed and purified human MTERF1 from E. coli cells and obtained crystals of the protein in complex with a 22-mer double stranded substrate (residues 3232 to 3253 of the human mitochondrial DNA). In order to solve the structure taking advantage of Multiwavelength Anomalous Dispersion phasing (Hendrickson and Ogata, 1997) we also crystallized MTERF1 in complex with a double stranded DNA containing eight 5-bromocytosine residues (see Experimental Procedures and Table I for data collection and refinement statistics). The native crystals diffracted to 2.2Å and contained one molecule of MTERF1 bound to DNA in the asymmetric unit. The final density was of sufficient quality to build most of the protein (residues 73 to 396; Figure 1 and 1S) and all 22 base pairs of DNA.
Figure 1. MTERF1 is a modular protein.
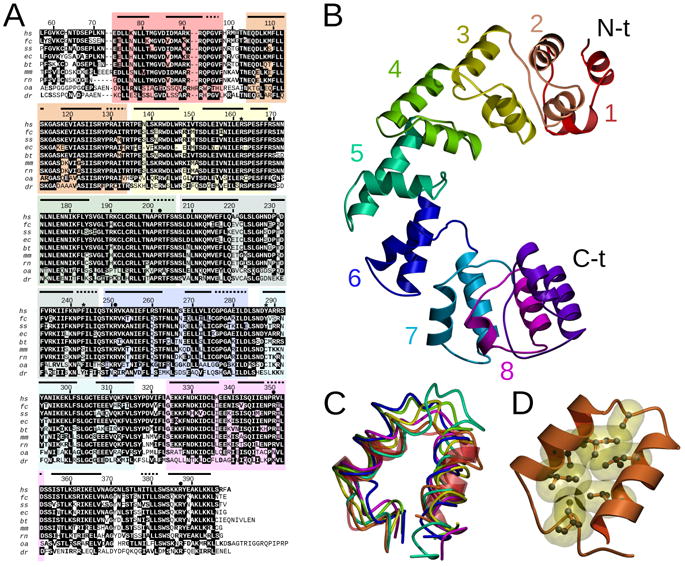
A. Amino acid alignment of MTERF1 proteins in different species. Invariant residues are white over a black background. Conserved residues are bold. The numbering corresponds to human MTERF1. Asterisks indicate residues that stack with everted nucleotides and dots indicate residues involved in sequence recognition. Horizontal black bars represent α-helices and dotted lines represent 310 helices. Hs, homo sapiens; fc, felis catus; ss, sus scrofa; ec, equus caballus; bt, bos taurus; mm, mus musculus; rn, rattus norvegicus; oa, ornithorhynchus anatinus; dr, danio rerio. B. Overview of the MTERF1 fold. The structure is shown in the absence of DNA. Mterf motifs are color coded as in A. C. Overlay of the 8 mterf motifs. The rmsd between the first (transparent red, ribbon representation) and each of the other repeats ranges between 1.0 and 3.3 Å for 24 C- atoms (the average is 2.5 Å). Each repeat is color coded as in B. D. Hydrophobic interactions stabilize each mterf repeat. The second mterf motif is shown in ribbon representation. The hydrophobic residues are shown in ball-and-stick representation with their Van der Waals surface in yellow. See also Figure 1S.
Table I.
Data collection, phasing and refinement statistics
| WT Native | WT 5BrdC | Triple mutant | |||
|---|---|---|---|---|---|
| PDB code | 3MVA | 3MVB | |||
| Data collection | |||||
| Space group | c2221 | c2221 | c2221 | ||
| Cell dimensions (Å) | 88.63, 89.71, 161.29 | 88.49, 89.93, 162.13 | 87.53, 91.43, 159.05 | ||
| α, β, γ(°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | ||
| Peak | Inflection | Remote | |||
| Wavelength | 1.0750 | 0.9195 | 0.9197 | 0.9000 | 1.0750 |
| Resolution (Å) | 2.20 | 2.80 | 2.90 | 2.80 | 2.80 |
| Rsym | 5.0 (60.2) | 9.8 (70.7) | 9.9 (76.9) | 7.7 (50.6) | 9.1 (66.1) |
| I/σI | 53.4 (2.7) | 30.8 (3.6) | 28.8 (3.6) | 37.0 (4.5) | 22.0 (2.7) |
| Completeness (%) | 99.2 (99.1) | 99.6 (100) | 99.9 (100) | 99.9 (100) | 99.4 (99.1) |
| Redundancy | 7.2 | 7.4 | 7.4 | 7.6 | 5.8 |
| Refinement | |||||
| Resolution (Å) | 2.20 | 2.80 | |||
| Unique reflections | 32806 | 16183 | |||
| Rwork/Rfree | 20.5/24.4 | 20.3/26.5 | |||
| No. atoms | 3620 | 3522 | |||
| Protein | 2606 | 2588 | |||
| DNA | 896 | 896 | |||
| Water | 118 | 38 | |||
| Mean B-factors | 66.5 | 41.2 | |||
| Protein | 65.9 | 42.1 | |||
| DNA | 68.7 | 38.8 | |||
| Water | 62.3 | 33.7 | |||
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.006 | 0.007 | |||
| Bond angles (°) | 1.24 | 1.27 | |||
Values in parenthesis are for the highest resolution shell.
Overall structure
MTERF1 adopts a fold that is very different from previously proposed models (Fernandez-Silva et al., 1997; Figure 1B). Consistent with predictions based on its primary sequence (Fernandez-Silva et al., 1997; Roberti et al., 2006), MTERF1 displays a modular architecture. Dali (Holm et al., 2008) and DejaVu (Kleywegt and Jones, 1997) were unable to identify significant structural homology with any protein in the PDB. MTERF1 is an all-alpha-helical protein composed of 19 α-helices and 7 310 helices that is structured around a motif (two α-helices followed by a 310 helix; hence referred to as the mterf motif) that is repeated throughout the structure (Figure 1B). This structural arrangement is remarkably similar to that seen in other all-alpha helical regions of proteins such as repeats of armadillo (ARM), HEAT (Groves and Barford, 1999) and PUM/PUF motifs (Lu et al., 2009). In all these cases the protein fold consists of a number of repeats of more or less conserved α-helical motifs. Interestingly, PUM/PUF motifs are the basis of RNA binding in PUMILIO proteins (Lu et al., 2009) while the HEAT motifs in DNA PK-cs have been suggested to play a role in double stranded DNA binding (Sibanda et al., 2010).
The MTERF1 fold contains 8 mterf motifs and an additional distorted motif in the C-terminus (Figure 1C). Within each motif, several hydrophobic residues create a hydrophobic core between the two α-helices (Figure 1D). The abundance of hydrophobic residues is what makes the primary sequence of MTERF1 similar to a leucine-zipper protein, but no association could be found between the predicted motifs (Fernandez-Silva et al., 1997; Roberti et al., 2006) and any differential structural feature. Hydrophobic interactions are also frequently observed between mterf motifs, but are far less abundant. This suggests a certain rigidity of the individual motifs but relative flexibility between motifs and thus of the overall fold.
DNA binding and unwinding
MTERF1 binds to the double-stranded substrate containing the termination sequence (Figure 2S) as a monomer and adopts a binding mode resulting in a remarkably large footprint on the DNA molecule. MTERF1 covers twenty base pairs even though binding does not strongly alter the curvature of the DNA duplex (Figure 2A/B). Taking advantage of its specialized architecture, MTERF1 binds along the major groove of DNA. DNA binding by MTERF1 imposes a slight bend (25°) in the DNA duplex (Figure 2C). More importantly, while the ends of the DNA duplex remain in a mostly B-DNA conformation, the DNA structure of the central part of the recognition sequence is heavily distorted. Binding by MTERF1 appears to decrease the twist of the DNA duplex, resulting in significant DNA unwinding (Figure 2D). Moreover, MTERF1 binding promotes partial duplex melting: the central part of the DNA contains three nucleotides that are everted from the double-helix (Figure 2E).
Figure 2. A unique DNA binding mode.
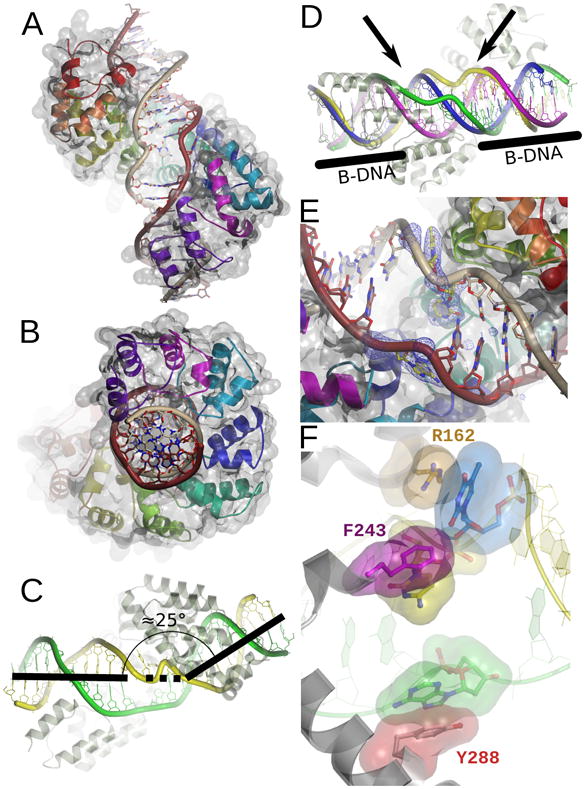
A. Global view of the protein-DNA interaction. Each repeated mterf motif is colored as in Figure 1. The light strand is brown and the heavy strand is gray. The molecular surface is rendered transparent. B. A 90° rotation of the view in A. C. MTERF1 induces a 25 bend in the DNA molecule. The light strand is green and the heavy strand is yellow. D. MTERF1 unwinds the DNA double-helix. Overlay between the DNA observed in the crystal structure (the heavy and light strands are green and yellow, respectively) and ideal B-form DNA (magenta and blue). The ends of the DNA molecule adopt B conformation (black bars). The central part of the molecule (black arrows) is unwound. E. Three nucleotides (yellow in the figure; corresponding to A3243 of the light strand and T3243 and C3242 of the heavy strand) are everted from the double-helix in the central part of the structure. A simulated annealing fo-fc electron density map is shown contoured at 3σ. F. The three everted nucleotides are stabilized by π-stacking interactions. R162 (orange), F234 (magenta) and Y288 (red) stack against each of the everted nucleotides (color coded as in B). The light strand is green and the heavy strand yellow.
Base flipping
Each of the nucleotides everted from the double-helix is stabilized by the protein via a stacking interaction (Figure 2F) and by hydrogen bonds to the base and phosphate (Figure 3A). In order to investigate this point, we constructed and purified a triple R162A/F243A/Y288A mutant in which all stacking interactions that presumably stabilize the conformation observed in the structure would be eliminated. We decided to crystallize this mutant in complex with the termination sequence. We obtained crystals that diffracted to 2.8Å (see Table I) and were able to solve the structure by molecular replacement using the wt MTERF1 structure as a model (see Experimental Procedures). Inspection of the structure showed that the triple mutant is folded exactly as WT MTERF1 (rmsd of 0.6Å for 324 C-α atoms; Figure 3B). Importantly, the protein is bound to the termination sequence in an identical manner as the wt protein: the DNA backbone in the mutant structure presents the same bend observed in the wt structure and the double-helix is equally unwound. However, removal of the stacking interactions altered the conformation of all three everted nucleotides (Figure 3C). Two of them, the adenine and thymine (green and blue in Figure 2F) are now flipped back into the double helix. A third one, the cytosine (yellow in Figure 2F), while not in the same conformation as in the wt structure, is still everted from the double helix. It is now occupying the position that the Arg162 side chain occupies in the wt structure. The cytosine base appears to be stabilized by contacts to protein backbone atoms but, unlike in the wt structure, it does not take advantage of π-π stacking interactions and therefore this conformation does not appear as favorable.
Figure 3. Interactions of MTERF1 with DNA.
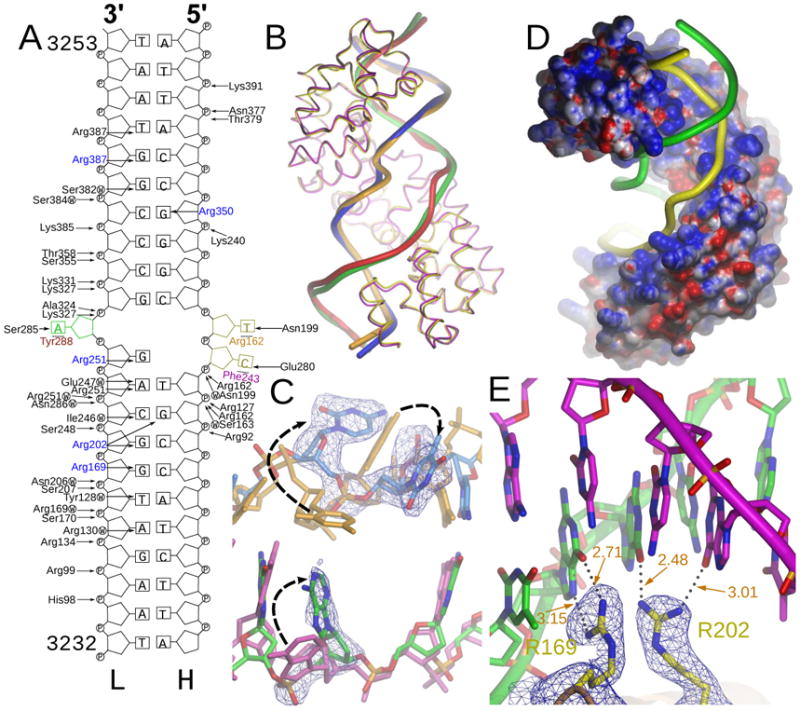
A. Scheme of the interactions between MTERF1 and the double-stranded DNA. Each interaction is listed with an arrow pointing either to the phosphate or to a DNA base. The three everted nucleotides and the residues that stack with them are colored. The five arginine residues that determine sequence specificity are shown in blue. W indicates a water mediated interaction. B. Overlay of C-α traces of WT (yellow) and triple mutant (magenta) structures. The DNA backbone is also shown for each of the DNA strands. The WT light and heavy strands are colored red and orange, respectively. The mutant DNA strands are colored green (light) and blue (heavy). C. Overlay of the central part of the DNA duplex in the WT and mutant structures. The three nucleotides that are everted in the WT structure are shown. The mutant heavy strand is blue, while the light strand is green. A simulated-annealing fo-fc omit electron density map is shown, contoured at 3σ. The DNA in the WT structure is shown as a reference. The WT heavy strand is shown in brown while the light strand is magenta. Black arrows indicate the changes in the position of the everted nucleotides that are observed upon mutation of the three stacking residues. D. Electrostatic surface potential of MTERF1. The protein surface is shown, colored between −10 kT e−1 (red) and 10 kT e−1 (blue). The DNA backbone is shown in yellow (light strand) and green (heavy strand). E. Sequence recognition by arginine residues. Five arginine residues determine sequence recognition by MTERF1. A representative example showing how R169 and R202 interact with their partner guanine residues. The R202 interaction is atypical in that only one hydrogen bond is established with the guanine base. Hydrogen-bonding distances are shown in orange. A simulated annealing fo-fc electron density map is shown contoured at 4σ. See also Figure 2S.
These observations demonstrate that stacking interactions are essential to stabilize the everted nucleotides and that the protein is actively promoting base-flipping. Furthermore, the fact that backbone distortion and unwinding is still present in the mutant structure indicates that MTERF1 binding by itself (at least in a sequence-specific context) unwinds and kinks the DNA duplex. This backbone distortion is independent of base-flipping, although by destabilizing the central base pairs it is likely essential to facilitate it. It is interesting to note that Arg162 is the only one of the three stacking residues that is universally conserved in MTERF1 proteins (Figure 1A). This suggests that differences must exist in the way MTERF1 associates with DNA in some of these species. It also perhaps suggests that the role of the three stacking residues in promoting base-flipping is not equivalent.
DNA backbone interactions and unspecific DNA binding
MTERF1 establishes numerous interactions with the DNA duplex. The backbone of both strands is bound along positively charged grooves in the protein surface (Figure 3D). Most of the interactions that are observed in the structure are electrostatic in nature and are established with the phosphate groups of the DNA strands (Figure 3A). This type of interaction does not impart any sequence specificity. Each of the mterf motifs contributes DNA backbone interactions. The interactions with the light strand appear to be much more numerous (Figure 3A), consistent with the reported stronger affinity of the protein for the mitochondrial light strand (Nam and Kang, 2005).
The number of sequence-independent interactions suggested that MTERF1 should be able to bind double stranded DNA regardless of sequence. To investigate this point, we performed isothermal titration calorimetry (ITC) experiments. As expected, MTERF1 was able to bind to its specific recognition sequence and did so with near 1:1 stoichiometry (Figure 4A/D), further indicating specific binding. Binding was significantly affected by the salt concentration, consistent with the number of protein-DNA electrostatic interactions. Because of the tendency of the protein to aggregate at lower salt concentrations in the absence of DNA we performed all subsequent experiments at 200 mM KCl, even though at this salt concentration MTERF1 exhibits weaker binding (see Experimental Procedures and Figure 4D). Our measurements also demonstrate that MTERF1 is capable of binding a double stranded DNA of arbitrary sequence, although with significantly lower affinity than for its specific recognition sequence (Figure 4D and 3SA). Importantly, the lower DNA to protein ratio of binding indicates that MTERF1 does not preferentially associate with the DNA duplex in a particular conformation, consistent with the lack of sequence specificity. Instead, it can bind to different regions of the duplex and as a consequence more than one MTERF1 molecule can simultaneously bind the same substrate molecule. To eliminate the possibility that accidental sequence similarity with the wt sequence could result in residual specific binding and affect the measurements, we repeated them with two additional substrates: a homopolymeric DNA (Figure 3SC) and a completely random sequence (not shown). The results led in both cases to identical conclusions (Figure 4D).
Figure 4. DNA binding measurements.
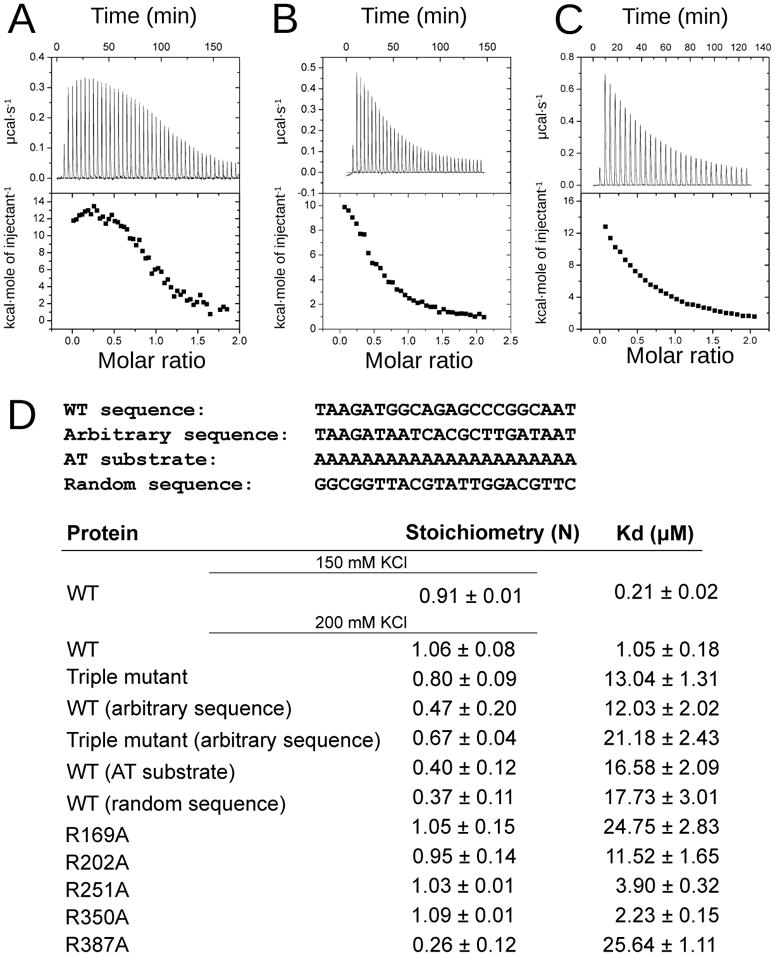
ITC data from the titration of the leu-tRNA MTERF1 binding sequence into wt MTERF1 (A), the triple R162A-F243A-Y288A mutant (B) or the R387A mutant (C). D. Summary of the observed binding constants. The sequence of each substrate (one of the strands) is indicated at the top of the table. See also Figure 3S.
Nucleotide eversion is essential for stable DNA binding
The difference in binding affinity between unspecific and specific binding can be rationalized from the crystal structure. In a sequence specific context as observed in the structure, MTERF1 binds DNA in a distorted conformation, where each of the three nucleotides everted from the double-helix is stabilized by the protein via a stacking interaction (Figure 2F) and, in addition, by hydrogen bonds to the base and phosphate (Figure 3A). It was therefore tempting to speculate that while MTERF1 can bind to any double stranded DNA molecule by virtue of its electrostatic surface, the distorted conformation observed in the crystal structure is only adopted upon recognition of a specific sequence and is responsible for stable specific binding. In order to investigate this, we performed DNA binding measurements using the triple R162A/F243A/Y288A mutant to analyze its ability to bind to the termination sequence. As expected from the structure, the affinity for DNA of the triple mutant is significantly reduced (Figure 4B/D) and is similar to that of wt MTERF1 for an unspecific DNA sequence (Figure 4D), indicating that nucleotide eversion is essential for stable binding. Furthermore, the triple mutation does not seem to affect binding to the oligonucleotide of arbitrary sequence in a significant way (Figure 4D and 3SB), strongly supporting the idea that base eversion is only a feature of sequence-specific binding. Importantly, since the triple mutant structure indicates that bending and unwinding of the DNA still take place, the increase in affinity can be entirely attributed to the ability of the enzyme to promote base-flipping.
Mechanism of sequence recognition
The observed binding stoichiometry when titrating the triple mutant (Figure 4D) implies that despite lower binding affinity the mutant still conserves site-specificity (i.e., the stoichiometry is still close to 1). Moreover, the structure indicates that it binds DNA just like the wt protein. This is also consistent with the fact that the stacking residues would appear to contribute little to sequence recognition. Because most of the protein-DNA interactions observed in the structure are to the DNA backbone, there are only a handful of interactions that appear capable of discriminating against a particular sequence. Sequence recognition thus seems to be mediated by specific hydrogen bonding to the DNA major groove. Five arginine residues (blue in Figure 3A) establish base interactions that are likely to determine sequence-specificity. Three of these (Arg 169, Arg350 and Arg387) simultaneously hydrogen-bond to N7 and O6 of a guanine base (Figure 3E). Arg202 hydrogen bonds to two adjacent guanines in opposite strands (Figure 3E), while Arg 251 hydrogen bonds to O6 of a single guanine and to N7 of the adjacent adenine. These arginine residues (black dots in Figure 1A) are conserved in MTERF1, as are the nucleotides that they recognize in the mitochondrial DNA (Figure 2S). Interestingly, these arginines are not conserved in other mterf proteins (Figure 1S), consistent with their different sequence specificity.
To assess the role of these residues in determining sequence specificity, we decided to construct individual arginine to alanine substitutions. ITC measurements (Figure 4D) allowed us to conclude that these residues play a role in sequence recognition. Interestingly, only R387A appears to have completely lost specific binding (Figure 4C/D). All other mutants appear to preserve some sequence specificity but, with the possible exception of R350A, they all show lower binding affinity for the termination sequence than the wt protein. This suggests that some of these residues might play an important role in determining the conformations observed in the crystal structure, and also indicates that the importance of each of these residues for DNA binding and sequence recognition is not equal, prompting us to analyze their ability to promote transcriptional termination.
Implications for transcriptional termination
To analyze the functional importance of the different protein-DNA interactions we studied the ability of the different proteins to promote transcriptional termination in a reconstituted in vitro system. We adapted the assay utilized by Asin-Cayuela and colleagues (Asin-Cayuela et al., 2005) and generated a substrate for run-off transcription from the HSP where the termination sequence (the 22-mer sequence used for crystallization) has been inserted 100 bp downstream from the promoter (see Experimental Procedures). As can be seen in Figure 5A, TFAM, TFB2M and POLRMT generate a unique run-off transcription product on this substrate, but addition of MTERF1 results in appearance of a specific termination product. Our results are essentially equivalent to previously published data (Asin-Cayuela et al., 2005), suggesting that our purified MTERF1 is active and confirming that, at least in vitro, MTERF1 only displays moderate termination activity. It has however been previously shown that the termination activity of MTERF1 is substantially stronger when the termination sequence is inverted as to simulate termination of LSP-initiated transcription (Asin-Cayuela et al., 2005). We decided to analyze transcriptional termination in this orientation as well. As can be seen in Figure 5B, termination in this orientation is indeed much more robust, perhaps in agreement with the observed pattern of interactions and the stronger affinity of MTERF1 for the light strand (Nam and Kang, 2005). In contrast, as expected from the binding data, no termination was observed with the triple mutant even when a substantial excess of protein was added to the reaction (Figure 5A/B, RFY). Unexpectedly, none of the five arginine mutants were able to promote termination (Figure 5A/B/D). The higher signal observed in the reverse orientation allowed us to quantify the termination activity. The results (Figure 5C) indicate that while wt MTERF1 achieved an average of near 75% termination over the course of a twenty minute reaction, the triple mutant only supported residual termination (<5%). The arginine mutations, irrespective of DNA binding affinity, were equally unable to support termination (<5%), with only some termination activity observed for R350A (17.7%).
Figure 5. Termination activity of WT and mutant MTERF1.
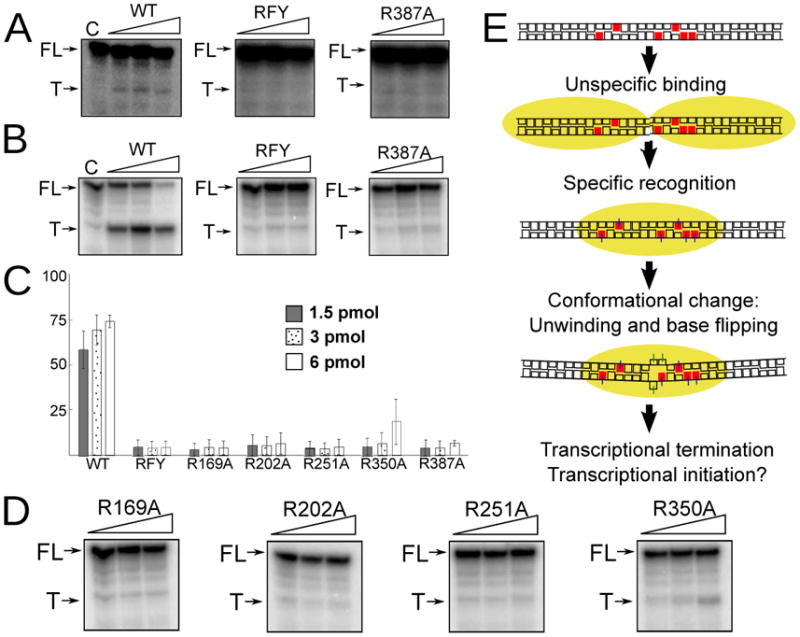
A. In vitro termination activity. WT MTERF1 and the different mutants were assayed for their ability to terminate transcription from the HSP promoter in vitro (see Experimental Procedures). The termination sequence was cloned in either the forward (A) or reverse orientation (B). The results are equivalent on both orientations and show clear termination for wt MTERF1 but only residual termination for the triple (RFY) mutant and the R387A mutation. FL, full-length run-off transcription. T, termination product. C, control lane without MTERF1. C. Quantification of the termination activity. The bar graph shows the percent termination observed in in vitro termination experiments with the termination sequence in the reverse orientation. Values correspond to the mean ± SD of at least three independent experiments. D. Termination activity of the remaining arginine mutants. E. Model depicting the events leading to specific DNA binding by MTERF1 (represented as a yellow oval).
This supports the functional importance of the main interactions suggested by the crystal structure. Since the triple mutation solely affects nucleotide eversion, our data clearly demonstrate the critical importance of base-flipping for transcriptional termination. They also indicate that while there is a correlation between DNA binding affinity and termination activity, the role of some residues appear to be more important for termination than for binding. Because all arginine-guanine interactions appear to be essentially equivalent, this suggests that, beyond the importance of the interaction itself, some or all of these residues might also be essential to enable the conformation observed in the wt structure. Additional studies will be needed to establish their individual roles.
A model for DNA binding
Based on our results, we can recapitulate the events leading to specific MTERF1 binding and propose a model for how this protein promotes transcriptional termination. We have shown that MTERF1 is able to interact with DNA in a sequence-independent manner, suggesting that the protein probes the mitochondrial DNA randomly in search of its recognition sequence. Several of our observations suggest that unspecific DNA binding is likely to be structurally different from specific binding. Our data demonstrate that base-flipping determines the higher binding affinity observed for the specific termination sequence. This strongly suggests that base-flipping does not take place when binding to an unspecific sequence. Moreover, the stacking interactions that stabilize base-flipping do not appear to be responsible to discriminate against unspecific sequences. Thus the wt protein would be likely to promote base-flipping regardless of sequence if the DNA duplex was destabilized as in the wt structure. This would imply that sequence-unspecific binding is unlikely to lead to the DNA conformation observed in the wt structure, suggesting a need for MTERF1 to adopt different conformations. Finally, our termination results suggest that the five arginine residues involved in sequence recongition might play an important role in determining these events. We therefore propose a model for MTERF1 binding (Figure 5E) wherein binding to the correct sequence results in the six key arginine-guanine interactions being established (perhaps sequentially), which leads to a concurrent protein conformational change that bends and unwinds the DNA duplex. This in turn promotes the unstacking of three nucleotides, which are then stabilized in an extrahelical conformation by three stacking interactions. Our results indicate that base-flipping is essential for stable binding, suggesting that MTERF1 takes advantage of this mechanism to increase the stability of its interaction with DNA. Since we have shown that base-flipping is essential to promote transcriptional termination, this suggests that this activity is simply related to the ability of MTERF1 to successfully prevent the RNA polymerase from displacing it from the termination sequence. Similarly, acting as a temporary roadblock might be the mechanism by which MTERF1 participates in controlling mitochondrial replication.
Pathogenic mutations in the leu-tRNA
Our structures allow us to precisely determine which nucleotides of the mitochondrial DNA are contacting MTERF1 while binding to its termination sequence. In addition, the mechanism by which MTERF1 recognizes its binding sequence implies that the identity of bases far away from the center of the target sequence are critical for sequence recognition. Based on these observations, we surveyed a collection of pathogenic mitochondrial DNA mutations that occur in the MTERF1 binding sequence of the leu-tRNA and examined their potential to interfere with binding and transcriptional termination. Nine mutations (two of them on the same nucleotide; Figure 6A) fall in the sequence recognized by MTERF1. It is important to note that the pathogenic effects of these mutations might be simply due to an alteration on the leu-tRNA structure, and this appears indeed to be the case for the A3243G mutation (Sasarman et al., 2008). A3243G is frequently identified in MELAS patients and has been previously shown to slightly reduce MTERF1 binding (Hess et al., 1991) and proposed to interfere in vitro with transcriptional termination (Hess et al., 1991; Chomyn et al., 1992). We can understand these effects based on the crystal structure. A3243 is one of the three everted nucleotides (Figure 3A) and a transition mutation would maintain the basic structure of the purine ring, likely allowing the same conformation observed with the wt termination sequence. However, the mutation would substitute an A:T base pair by a stronger G:C base pair, which might explain the slight decrease in binding. In addition, the O6 of the guanine base would be located in close proximity of a phosphate oxygen (O2P in A3242), perhaps contributing to destabilization of the wt conformation. Our measurements confirm that A3243G results in slightly decreased binding (Figure 6A and 3SE). A similar binding affinity was observed for the A3243T mutation (Figure 6A). In this case the A:T base pair is preserved, but the larger purine ring is now located in the heavy strand, which would likely lead to a steric clash with Asn199 (Figure 3A). All remaining mutations would not be expected to severely conflict with MTERF1 sequence recognition, except for G3242A and G3249A. G3242 interacts in the structure with Arg251, while G3249 forms a double hydrogen bond with Arg387 (Figure 6B). Both of these mutations would eliminate a guanine-arginine interaction: the hydrogen bond between O6 of the guanine and the amino group of the arginine could not be formed in the mutant DNA. Consistent with the mild decrease in binding observed in the R251A mutant, G3242A resulted in weak or no effect on binding. However, the G3249A mutation appears to completely eliminate specific DNA binding (Figure 6A and 3SE), consistent with the effect of the R387A mutation.
Figure 6. Pathogenic DNA mutations in the mitochondrial termination sequence.

A. Scheme indicating the location of the mutations in the termination sequence and binding constants for titrations of a 22 bp oligonucleotide containing the leu-tRNA MTERF1 binding sequence carrying each of the pathogenic mutations into wt MTERF1. The DNA residues involved in arginine guanine interactions with MTERF1 are indicated by an asterisk. B. Interaction of R387 with G3249. Hydrogen-bonding distances are shown in orange. A simulated annealing fo-fc electron density map is shown (blue) contoured at 4σ. C. In vitro termination activity of wt MTERF1 on substrates containing each of the nine pathogenic mutations. FL, full-length run-off transcription. T, termination product. C, control lane without MTERF1. D. Quantification of termination activity on the different mutant sequences. The bar graph shows the percent termination observed in in vitro termination experiments with the termination sequence in the reverse orientation. Values correspond to the mean ± SD of at least three independent experiments.
To further analyze the functional consequences of these mitochondrial DNA mutations, we carried out termination assays using both the forward and reverse orientations of the termination sequence. The results (Figure 6C/D) are mostly consistent with the DNA binding data. As expected, only residual termination is observed in both orientations with the G3249A mutation. In addition, consistent with what was observed with the R251A mutant, the G3242A mutation, while not substantially altering binding, results nevertheless in a strong decrease in termination. The A3243G mutation moderately reduces termination, while termination on A3243T is only marginally weaker than for the wt sequence. The reduction in termination observed for A3243G is consistent with what was previously reported (Hess et al., 1991). Unexpectedly, one additional mutation appears to moderately affect termination: G3244A, while not affecting DNA binding, results in a moderate but reproducible effect on termination. Interestingly, no specific interaction is observed in the crystal structure with any of the nucleotides in this base pair. However, it is adjacent to the extrahelical A:T base pair and displays a high base pair buckle (see Experimental Procedures). It is possible that the strength of the G:C base pair in the wt sequence is important to maintain this conformation and that the G3244A mutation thus leads to a slight destabilization of the bound MTERF1 that is sufficient to make it more readily displaced by the RNA polymerase.
Conclusions
We have reported the structure of human MTERF1. Our structure suggests that the mterf proteins constitute a family of dedicated DNA binding proteins, as can be readily concluded from inspection of the protein fold and the relatively high conservation between family members (Figure 1S). In addition, our results suggest a unique mechanism of sequence recognition and binding where sequence-independent binding is followed by sequence recognition, resulting in a conformational change leading to unwinding of the DNA double-helix and base flipping. Base flipping appears to be critical to confer on MTERF1 the ability to stably bind to a specific site in DNA. Stable DNA binding, in turn, is necessary for the ability of this protein to promote transcriptional termination. Since base flipping does not affect the conformation of the DNA duplex, our results suggest that MTERF1 promotes termination by interfering with the elongation machinery. In this respect it is interesting to stress that, as was previously observed (Asin-Cayuela et al., 2005), MTERF1 appears to be much more efficient at promoting termination from the LSP than from the HSP. This might reflect the need to prevent transcription from the LSP from interfering with rRNA synthesis, and is consistent with the stronger affinity for the light strand (Nam and Kang, 2005) and the large number of interactions established with this strand. While our structure does not address if or how MTERF1 can form the MtDNA loop implicated in rRNA synthesis, the extensive protein-DNA interaction surface observed in the structure indicates that MTERF1 would not be able to simultaneously associate with two DNA duplexes. This therefore implies that it is unlikely that the loop can be mediated by a single MTERF1 molecule. On the other hand, the partial duplex melting observed in the structure, together with the preferential binding to the light strand and relatively low number of interactions with the heavy strand, suggest a mechanism by which MTERF1 or other mterf proteins could facilitate transcriptional initiation by contributing to initial duplex melting. In this respect it is important to note that Phe243 appears to be conserved in MTERF2 and MTERF3 (Linder et al., 2005; Figure 1S), and that MTERF2 has been suggested to positively regulate transcriptional initiation (Wenz et al., 2009). Finally, it is easy to imagine how this binding fold can be utilized to recognize different sequences given that only five guanine residues appear to be essential for sequence recognition. An entirely different specificity could perhaps be obtained by alteration of the handful of residues responsible for sequence recognition. In this respect, it is interesting to note that both the modular architecture of MTERF1 and its sequence recognition mechanism are highly reminiscent of the PUF family of RNA binding proteins, where recognition of individual bases can be modulated by mutation of residues at key positions (Lu et al., 2009). These similarities also perhaps suggest that the mterf fold is not necessarily restricted to binding double stranded DNA.
Base flipping is a key feature of the interaction of MTERF1 with DNA. Base-flipping was identified as a mechanism used by HhaI methyltransferase to access its substrate (Kimasauskas et al., 1994). Since then, a large number of base-flipping enzymes have been identified that take advantage of this mechanism (Reinisch et al., 1995; Roberts and Cheng, 1998; Huffman et al., 2005). It is commonly employed by different DNA repair and replication proteins that require access to a specific base in the DNA duplex (Huffman et al., 2005). Because nucleotide eversion is not energetically favorable, in these proteins, as in MTERF1, the everted base is usually stabilized by π-stacking interactions. Base-flipping is usually utilized either to help recognize specific features of the DNA molecule or to assist in the catalytic mechanism, usually to gain access to a substrate that is otherwise buried in the DNA double-helix. In this case, base-flipping appears to be employed solely to stabilize the protein on DNA and make it more difficult to be displaced. The fact that this mechanism can be successfully utilized for a completely different purpose further illustrates the frequent recycling of structural motifs and solutions that has occurred throughout evolution.
An important conclusion derived from the crystal structure is that two pathogenic mutations that interfere with arginine-guanine interactions involved in sequence recognition by MTERF1 result in a strong impairment in the ability of MTERF1 to terminate transcription. G3242A does not substantially affect binding, but very strongly reduces the ability of MTERF1 to terminate transcription, while G3249A appears to completely eliminate specific binding by MTERF1 and consequently severely impairs termination. Their strong effect on termination suggests that the pathogenic effects of the G3249A and G3242A MtDNA mutations might be at least partially mediated by interfering with the activity of MTERF1 at the leu-tRNA site. The G3249A mutation leads to the development of a variant of Kearns-Sayre syndrome (Seneca et al., 2001), a mitochondrial myopathy, while the G3242A mutation has been associated with an uncharacterized mitochondrial disorder (Mimaki et al., 2009). It has been previously suggested that transcriptional deregulation might be one of the mechanisms leading to mitochondrial dysfunction. This is consistent with the fact that mice deficient in MTERF2 develop features of mitochondrial myopathies (Wenz et al., 2009). Further experiments will be needed to address whether the G3249A and G3242A mutations result in transcriptional alterations in vivo and if this effect is sufficient to explain their clinical phenotype.
Experimental Procedures
Protein expression and purification
WT MTERF1 (residues 57–399), TFB2M and POLRMT (a generous gift of C. Gustafsson) were cloned into pTEV-HMBP3, allowing expression of a fusion with his-tagged maltose binding protein (MBP) cleavable by TEV protease. The triple R162A-F243A-Y288A mutant and each of the five arginine mutants were constructed by QuikChange mutagenesis (Stratagene). His-tagged TFAM was cloned in pET-22. Proteins were overexpressed in Arctic Xpress E.coli (DE3) cells (Stratagene) at 16°C for 20 hr. WT and mutant MTERF1 proteins and TFB2M were purified using ProBond Resin (Invitrogen), followed by overnight TEV protease cleavage, Heparin and Mono S chromatography. POLRMT was purified by Heparin chromatography, overnight TEV protease cleavage, second Heparin and Mono S chromatography. TFAM was purified using ProBond Resin, Heparin and Mono S chromatography. Proteins were concentrated using a 10,000 MWCO Amicon Ultra-15 device. Concentrated proteins were stored in 20 mM HEPES (pH 8.0), 200 mM KCl, 5% glycerol, and 1mM DTT.
Crystallization and structure determination
Natural and 5Br-dC 22-mer oligonucleotides were synthesized, annealed and combined with the protein in a 2:1 ratio. Crystals grew at room temperature in 100 mM Bis-Tris, pH 5.0–6.0, 18–21% PEG 3350, 200 mM potassium sodium tartrate. Data collection was performed at beamlines X25 and X29 at the National Synchrotron Light Source (BNL). Both datasets were processed using HKL2000 (Otwinowski and Minor, 1997; Table I). The wt structure was determined by Multiwavelength Anomalous Diffraction (Hendrickson and Ogata, 1997) using three datasets. Heavy atom sites were located with Shelx (Sheldrick, 1990). Subsequent refinement and phase calculation to 3.6 Å were performed using SHARP (Fortelle and Bricogne, 1997). Density modification and phase extension were performed using DM (Cowtan, 1994), and model building with Coot (Emsley and Cowtan, 2004). The resulting model was refined against the native data set using Phenix (Terwilliger, 2002). The model includes all nucleotides and residues 73–396 of MTERF1. Model quality was assessed using MOLPROBITY (Davis et al., 2007). The mutant structure was solved by molecular replacement using molrep (Vagin A, 1997). Analyses of the helical parameters of the DNA molecule were carried out using Curves+ (Lavery et al., 2009).
Binding measurements
ITC experiments were performed with a VP-ITC calorimeter (Microcal) at 4 °C. WT or mutant MTERF1 (20–25 μM) was titrated with 10-μl injections of 200–250 μM 22 bp oligonucleotide pairs. Samples were prepared by dialyzing all interacting components against a buffer containing 20 mM Hepes, pH 8.0, 200 mM KCl, 2.5% glycerol, 1 mM EDTA. Data were analyzed using the ORIGIN software.
Transcriptional termination
Assays were adapted from Asin-Cayuela et al., 2005. Nucleotides 491 to 790 of the human mitochondrial DNA (containing the HSP) were cloned between the NcoI and HindIII sites of pET-22 (Novagen), and the termination sequence was inserted 100 bp from the initiation site in both orientations by site directed mutagenesis. Additional mutations were carried out by site directed mutagenesis. The substrate was linearized using HindIII. Reactions were carried out in 20 μl and contained 30ng DNA, 20 mM Hepes, pH 8.0, 5% glycerol, 10 mM MgCl2, 150 mM KCL, 1 mM DTT, 100 μg/ml BSA, 0.4 mM ATP, 0.15 mM CTP and GTP, 0.01 mM UTP and 1 μl of [α-32P]UTP. 1.5, 3 or 6 pmol of WT or mutant MTERF1 was added and the mixture incubated for 30 min at RT. Transcription was initiated by addition of 400 fmol POLRMT, 500 fmol TFB2M and 2.5 pmol TFAM. Reactions were incubated for 30 min at 32°C and stopped by adding 100 μl of 1% SDS, 20 mM EDTA, 300 mM NaAc, 20 μg Calf Thymus DNA and 0.12 mg/ml glycogen. Products were phenol extracted, ethanol precipitated, resuspended in 20 μl of loading buffer and analyzed by PAGE.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. Bebenek, Bogenhagen, De los Santos, Demple, Garcia-Segura and Kunkel for critical reading of the manuscript and the NSLS Protein Crystallography group. NSLS beamlines x25 and x29 are mainly supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and the National Center for Research Resources of the National Institutes of Health. This work was supported by R00 ES015421 to MGD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Asin-Cayuela J, Schwend T, Farge G, Gustafsson CM. The human mitochondrial transcription termination factor (mTERF) is fully active in vitro in the non-phosphorylated form. J Biol Chem. 2005;280:25499–25505. doi: 10.1074/jbc.M501145200. [DOI] [PubMed] [Google Scholar]
- Asin-Cayuela J, Gustafsson CM. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem Sci. 2007;32:111–117. doi: 10.1016/j.tibs.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Chomyn A, Martinuzzi A, Yoneda M, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci USA. 1992;89:4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. Joint CCP4 and ESF-EACMB Newsletter on Protein. Crystallography. 1994;31:34–38. [Google Scholar]
- Davis I, Leaver-Fay A, Chen V, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Martinez-Azorin F, Micol V, et al. The human mitochondrial transcription termination factor (mTERF) is a multizipper protein but binds to DNA as a monomer, with evidence pointing to intramolecular leucine zipper interactions. EMBO J. 1997;16:1066–1079. doi: 10.1093/emboj/16.5.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortelle EDL, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods in Enzymology. 1997;276 doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- Gaspari M, Larsson N, Gustafsson CM. The transcription machinery in mammalian mitochondria. Biochim Biophys Acta. 2004;1659:148–152. doi: 10.1016/j.bbabio.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Groves MR, Barford D. Topological characteristics of helical repeat proteins. Curr Opin Struct Biol. 1999;9:383–389. doi: 10.1016/s0959-440x(99)80052-9. [DOI] [PubMed] [Google Scholar]
- Hendrickson WA, Ogata CM. Phase determination from multiwavelength anomalous diffraction measurements. Methods in Enzymology. 1997;276 doi: 10.1016/S0076-6879(97)76074-9. [DOI] [PubMed] [Google Scholar]
- Hess JF, Parisi MA, Bennett JL, et al. Impairment of mitochondrial transcription termination by a point mutation associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1991;351:236–239. doi: 10.1038/351236a0. [DOI] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Hyvärinen AK, Pohjoismäki JLO, Reyes A, et al. The mitochondrial transcription termination factor mTERF modulates replication pausing in human mitochondrial DNA. Nucleic Acids Res. 2007;35:6458–6474. doi: 10.1093/nar/gkm676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Detecting folding motifs and similarities in protein structures. Methods Enzymol. 1997;277:525–545. doi: 10.1016/s0076-6879(97)77029-0. [DOI] [PubMed] [Google Scholar]
- Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse B, Narasimhan N, Attardi G. Termination of transcription in human mitochondria: identification and purification of a DNA binding protein factor that promotes termination. Cell. 1989;58:391–397. doi: 10.1016/0092-8674(89)90853-2. [DOI] [PubMed] [Google Scholar]
- Lavery R, Moakher M, Maddocks JH, et al. Conformational analysis of nucleic acids revisited: Curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder T, Park CB, Asin-Cayuela J, et al. A family of putative transcription termination factors shared amongst metazoans and plants. Curr Genet. 2005;48:265–269. doi: 10.1007/s00294-005-0022-5. [DOI] [PubMed] [Google Scholar]
- Lu G, Dolgner SJ, Hall TMT. Understanding and engineering RNA sequence specificity of PUF proteins. Curr Opin Struct Biol. 2009;19:110–115. doi: 10.1016/j.sbi.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Cho J, Cesare AJ, et al. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- Metodiev MD, Lesko N, Park CB, et al. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Mimaki M, Hatakeyama H, Ichiyama T, et al. Different effects of novel mtDNA G3242A and G3244A base changes adjacent to a common A3243G mutation in patients with mitochondrial disorders. Mitochondrion. 2009;9:115–122. doi: 10.1016/j.mito.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Nam S, Kang C. DNA light-strand preferential recognition of human mitochondria transcription termination factor mTERF. J Biochem Mol Biol. 2005;38:690–694. doi: 10.5483/bmbrep.2005.38.6.690. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276 doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Park CB, Asin-Cayuela J, Camara Y, et al. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell. 2007;130:273–285. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Asin-Cayuela J, Erdjument-Bromage H, et al. MTERF2 is a nucleoid componenet in mammalian mitochondria. Biochim Biophys Acta. 2009;1787:296–302. doi: 10.1016/j.bbabio.2009.01.018. [DOI] [PubMed] [Google Scholar]
- Reinisch KM, Chen L, Verdine GL, et al. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell. 1995;82:143–153. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- Roberti M, Bruni F, Loguercio Polosa P, Manzari C, Gadaleta MN, Cantatore P. MTERF3, the most conserved member of the mTERF-family, is a modular factor involved in mitochondrial protein synthesis. Biochim Biophys Acta. 2006;1757:1199–1206. doi: 10.1016/j.bbabio.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Roberti M, Polosa PL, Bruni F, et al. The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim Biophys Acta. 2009;1787:303–311. doi: 10.1016/j.bbabio.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Cheng X. Base flipping. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially supressed by overexpression of EFTu and EFG2. Human Mol Genet. 2008;17:3697–3707. doi: 10.1093/hmg/ddn265. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Seneca S, Verhelst H, De Meirleir L, et al. A new mitochondrial point mutation in the transfer RNA(Leu) gene in a patient with a clinical phenotype resembling Kearns-Sayre syndrome. Arch Neurol. 2001;58:1113–1118. doi: 10.1001/archneur.58.7.1113. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. Phase annealing in SHELX-90: direct methods for larger structures. Acta Cryst A. 1990;46:467–473. [Google Scholar]
- Sibanda BL, Chirgadze DY, Blundell TL. Crystal Structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sologub M, Litonin D, Anikin M, et al. TFB2 is a transient component of the catalytic site of the human mitochondrial RNA polymerase. Cell. 2009;139:934–944. doi: 10.1016/j.cell.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. Automated structure solution, density modification and model building. Acta Cryst D. 2002;58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- Vagin ATA. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- Wenz T, Luca C, Torraco A, et al. mTERF2 regulates oxidative phosphorylation by modulating mtDNA transcription. Cell Metab. 2009;9:499–511. doi: 10.1016/j.cmet.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.