

Abstract
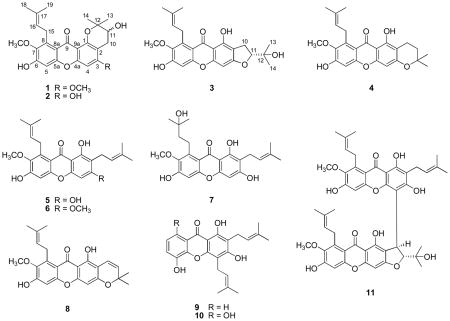
Bioassay-guided fractionation of a chloroform-soluble extract of Garcinia mangostana stem bark using the HT-29 human colon cancer cell line and an enzyme-based ELISA NF-κB assay, led to the isolation of a new xanthone, 11-hydroxy-3-O-methyl-1-isomangostin (1). The structure of 1 was elucidated by spectroscopic data analysis. In addition, ten other known compounds, 11-hydroxy-1-isomangostin (2), 11α-mangostanin (3), 3-isomangostin (4), α-mangostin (5), β-mangostin (6), garcinone D (7), 9-hydroxycalabaxanthone (8), 8-deoxygartanin (9), gartanin (10), and cratoxyxanthone (11), were isolated. Compounds 4–8 exhibited cytotoxicity against the HT-29 cell line with ED50 values of 4.9, 1.7, 1.7, 2.3, and 9.1 μM, respectively. In an ELISA NF-κB assay, compounds 5–7, 9, and 10 inhibited p65 activation with IC50 values of 15.9, 12.1, 3.2, 11.3, and 19.0 μM, respectively, and 6 showed p50 inhibitory activity with an IC50 value of 7.5 μM. α-Mangostin (5) was further tested in an in vivo hollow fiber assay, using HT-29, LNCaP, and MCF-7 cells, but it was found to be inactive at the highest dose tested (20 mg/kg).
Garcinia mangostana L. (Clusiaceae) is well-known in southeastern Asia for its pleasant-tasting fruits, commonly known as mangosteen, which is now used widely as a botanical dietary supplement in several countries.1 Xanthones are the most characteristic secondary metabolite constituents of G. mangostana and over 80 compounds of this type have been isolated and characterized from the various parts of this plant.1,2 The biological effects of the mangosteen xanthones are diverse, and include antioxidant, antibacterial, antifungal, antimalarial, anti-inflammatory, cytotoxic, and HIV-1 inhibitory activities.1,2 Recent phytochemical investigations on the fruits of G. mangostana at The Ohio State University have resulted in the isolation of xanthones with antioxidant,3 aromatase inhibitory,4 and quinone reductase-inducing activities.5
As part of a collaborative project directed towards the discovery of novel natural product anticancer agents,6 a CHCl3-soluble extract of the stem bark of G. mangostana collected in Indonesia showed cytotoxic activity against a “gatekeeper” HT-29 human colon cancer cell line with an ED50 value of 1.6 μg/mL. This extract also inhibited p50 and p65 activation with 57% and 67% inhibition at 50 μg/mL, respectively, in an ELISA NF-κB (nuclear factor-kappaB) assay. Therefore, it was subjected to bioactivity-guided fractionation, leading to the isolation of twelve xanthones, including a new compound (1). The structure elucidation of 1 and the biological evaluation of all compounds isolated are described herein.
Compound 1 was obtained as a yellow amorphous powder and produced a molecular ion peak at m/z 463.1729 [M+Na]+ in the HRESITOFMS, corresponding to the sodiated elemental formula, C25H28O7Na. The IR spectrum showed absorption bands at 3350 cm−1 for one or more hydroxy groups and at 1614 and 1456 cm−1 for aromatic groups.7 The UV spectrum of 1 exhibited absorption maxima at 242, 254, and 303 nm, indicating the presence of a xanthone system.7 The 1H and 13C NMR spectra of 1 were similar to those of the known compound, 11-hydroxy-1-isomangostin (2),8 except for the presence of signals for a second methoxy group at δH 3.83 (3H, s, OCH3-3) and δC 56.0 (CH3, OCH3-3). The positions of two methoxy groups were assigned C-3 and C-7 by the 1H-13C HMBC correlations between signals at δH 3.83 (3H, s, OCH3) to δC 162.0 (C, C-3) and δH 3.78 (3H, s, OCH3) to δC 142.9 (C, C-7), respectively. Further detailed analysis of the 1H-1H COSY, 1H-13C HSQC, and 1H-13C HMBC NMR data (Figure 1) allowed unambiguous assignments for all of the 1H and 13C NMR signals of 1. The Mosher ester method was utilized in an attempt to determine an absolute configuration of 1.9 However, the reaction between the secondary hydroxy group at C-11 and the R- and S-MTPA-Cl reagents yielded a mixture of S- and R-MTPA esters and a mixture of R- and S-MTPA esters of 1, respectively, indicating the presence of a racemic mixture, but not in a 1:1 mixtures due to the slightly positive specific rotation observed. Thus, the structure of the new compound 1 was elucidated as 11-hydroxy-3-O-methyl-1-isomangostin.
Figure 1.
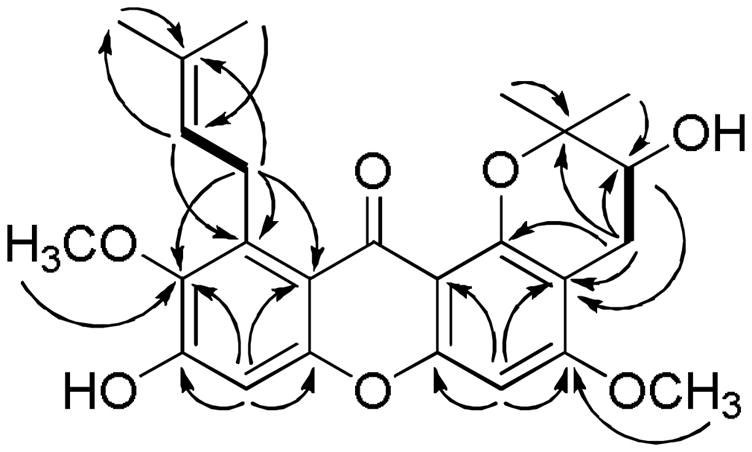
Important 1H-1H COSY ( ) and 1H-13C HMBC (→) correlations of compound 1.
) and 1H-13C HMBC (→) correlations of compound 1.
Compounds 2 and 3 exhibited NMR spectroscopic data identical to those of 11-hydroxy-1-isomangostin8 and mangostanin.10 To attempt to determine the absolute configuration of 2, the Mosher ester method was performed,9 but these reactions again produced evidence for the presence of a racemic mixture. As observed for 1, the slightly positive specific rotation value of 2 could be also caused by an unequal ratio of 11R and 11S isomers present. Compound 3 was isolated previously from the fruits of G. mangostana, but its relative configuration has not been reported thus far. The energy-minimized stereostructure of 3 showed a dihedral angle of 26 between H-11 and H-10β, which corresponded to the coupling constant value with 3JHH = 8~9 Hz, from the Karplus correlation equation (3JHH = A + B cos Φ + C cos 2Φ; A = 7, B = −1, C = 5, Φ = dihedral angle).7,11 The dihedral angle between H-11 and H-10α was calculated as 151° in the energy-minimized stereostructure of 3, with the expected calculated coupling constant value 3JHH = 9~10 Hz.7,11 These computational calculations were compared to the actual coupling constants observed in the 1H NMR spectrum for 3 at δH 4.83 (1H, dd, J = 9.5, 8.2 Hz, H-11), 3.18 (1H, dd, J = 15.8, 8.2 Hz, H-10β), and 3.12 (1H, dd, J = 15.8, 9.5 Hz, H-10α). Based on these observations, the hydroxy group at C-11 was established as α (Figure 2). Therefore, compound 3 was determined as 11α-mangostanin.
Figure 2.

Dihedral angles in the energy-minimized stereostructure of compound 3.
Other known compounds were identified in the present investigation as 3-isomangostin (4),9 α-mangostin (5),12 β-mangostin (6),13 garcinone D (7),14 9-hydroxycalabaxanthone (8),15 8-deoxygartanin (9),16 gartanin (10),17 cratoxyxanthone (11),8 and mangostanol,18 respectively, by comparison of their physical and spectroscopic data with those reported previously. Compounds 2–4, 9, and 10 were isolated as constituents of the stem bark part of this plant for the first time. In addition, cratoxyxanthone (11) has been isolated only from the bark of Cratoxylum cochinchinense (Lour.) Bl. (Clusiaceae) previously,8 so this is the first report of its isolation from a plant of the genus Garcinia.
All compounds isolated in the present investigation were tested in vitro for their cytotoxic activity against the HT-29 human colon cancer cell line (Table 1). The major active compounds, α-mangostin (5) and β-mangostin (6), have been found cytotoxic against various human cancer cells,19–22 including DLD-1 human colon cancer cells,23 and compounds 4, 7, and 8 have been also reported for their cytotoxicity against epidermoid carcinoma (KB), breast cancer (BC-1), or small cell lung cancer (NCI-H187).19 However, this is the first report of the evaluation of xanthones from G. mangostana for their cytotoxicity against the HT-29 colon cancer cell line. In an enzyme-based ELISA NF-κB assay, all compounds except for 3 and 4 and mangostanol were tested for their p50 (NF-κB1) and p65 (RelA) inhibitory activities, with β-mangostin (6) and garcinone D (7), respectively, being the most active substances found (Table 1). The major cytotoxic isolate, α-mangostin (5), was chosen for evaluation in an in vivo hollow fiber assay, which is used as a secondary bioassay in our drug discovery program to prioritize leads for subsequent analysis in traditional xenograft models.24 However, compound 5 was found to be inactive against HT-29 and LNCaP (hormone-dependent human prostate cancer) cells implanted at the intraperitoneal (i.p.) site at doses of 2.5, 5, 10, and 20 mg/kg (Figure S1, Supporting Information). Therefore, on the basis of these results in the hollow fiber assay, α-mangostin (5) does not seem to be promising as a potential anticancer agent.
Table 1.
In Vitro Activity of Compounds 4–10 in Cytotoxicity (HT-29 Cell Line) and ELISA NF-κB (p65 and p50) Assays.
| compound | 4 | 5 | 6 | 7 | 8 | 9 | 10 | camptothecind | rocaglamided |
|---|---|---|---|---|---|---|---|---|---|
| cytotoxicitya | 4.9 | 1.7 | 1.7 | 2.3 | 9.1 | >10 | >10 | 0.06 | - |
| NF-κB (p65)b | NTc | 15.9 | 12.1 | 3.2 | >20 | 11.3 | 19.0 | - | 0.08 |
| NF-κB (p50)b | NTc | >20 | 7.5 | >20 | >20 | >20 | >20 | - | 2.0 |
Compounds 1–3 and 11 were inactive (ED50 >10 μM).
Compounds 1, 2, and 11 were inactive (IC50 >20 μM).
Compounds 3 and 4 were not tested in the NF-κB assays because of the limited amounts available.
Positive control substances.
Experimental Section
General Experimental Procedures
The melting point was measured on a Fisher-Johns 12-144 melting point apparatus with a 12-142T thermometer (Fisher Scientific, Pittsburgh, PA), and is uncorrected. Optical rotations were measured with a Perkin-Elmer 343 automatic polarimeter. UV and IR spectra were obtained with a Shimadzu UV 160U spectrophotometer and Thermo Scientific Nicolet™ 6700 FT-IR spectrometer, respectively. 1D and 2D NMR experiments were performed on Bruker Avance DPX-300 and DRX-400 spectrometers with tetramethylsilane (TMS) as internal standard. Electrospray ionization (ESI) mass spectrometric analyses were performed with a 3-Tesla Finnigan FTMS-2000 Fourier Transform mass spectrometer. Silica gel (65–250 and 230–400 mesh, Sorbent Technologies, Atlanta, GA) and Sephadex LH-20 (Supelco, Bellefonte, PA) were used for column chromatography. Thin-layer chromatographic (TLC) analysis was performed on Silica G (silica gel, 0.2 mm layer thickness, Sorbent Technologies, Atlanta, GA) and RP-18 F254s (Merck, Germany) TLC plates, with visualization under UV light (254 and 365 nm) and 10% (v/v) sulfuric acid spray followed by heating (120 °C, 2 min). A Sunfire PrepC18 column (150 mm × 19 mm i.d., Waters, Milford, MA) and a Sunfire guard column (5 μm, 10 mm × 19 mm i.d., Waters, Milford, MA) were used for preparative HPLC, along with a Waters system composed of a 600 controller, a 717 Plus autosampler, and a 2487 dual wavelength absorbance detector.
Plant Material
The stem bark of G. mangostana (400 g) was collected at Pangradin village, Jasinga, West Java, Indonesia, by S. R. in August, 2005, who also identified this plant. A voucher specimen (acquisition number 2285414) has been deposited in the John G. Searle Herbarium of the Field Museum of Natural History, Chicago, Illinois.
Extraction and Isolation
The dried stem bark of G. mangostana (400 g) was extracted with MeOH (3 × 1 L) overnight at room temperature. The solvent was evaporated in vacuo to afford a MeOH extract (75 g), which was then suspended in MeOH-H2O (9:1, 1 L), and partitioned with hexane (3 × 1 L). To the defatted residue, which was dried in vacuo, was added 10% MeOH in H2O (1 L), and then this was partitioned with CHCl3 (3 × 1 L). The CHCl3-soluble layer was washed with 1% aqueous NaCl (3 × 1 L) to provide a partially detannified CHCl3 extract (14 g). This CHCl3 extract was subjected to silica gel column chromatography (CC; φ 3.5 cm; 60–250 mesh, 200 g), using gradient mixtures of MeOH in CHCl3 (0→1%) as mobile phases, affording nine fractions (FI - FIX). These fractions were evaluated against the HT-29 cell line, and, of these fractions, FI - FV were found to be active (ED50 <10 μg/mL). Compounds 6 (300 mg, 0.075% w/w) and 5 (2.5 g, 0.625% w/w) were isolated from fraction FI and FII, respectively, by precipitation in MeOH. The residual portion of fraction FI (464 mg), eluted with 100% CH2Cl2 from the first separation, was subjected to silica gel CC (φ 2.5 cm; 230–400 mesh, 50 g), with hexanes-EtOAc (4:1→1:1) as solvent system, yielding 8 (8.0 mg, 0.002% w/w). Fraction FII (3.3 g, after partial removal of α-mangostin), also eluted with 100% CH2Cl2 from the first purification step, was separated by silica gel CC (φ 3 cm; 230–400 mesh, 100 g), using gradient mixtures of hexanes-EtOAc-MeOH (20:10:1→10:10:1) for elution, affording seven sub-fractions. Sub-fraction FII-1 (100 mg), eluted with hexanes-EtOAc-MeOH (20:10:1), was further purified by preparative HPLC, using an isocratic mixture of MeOH-H2O (8:2, 8 mL/min) as solvent system, to afford 9 (tR 12.1 min, 3.1 mg, 0.00077% w/w) and 10 (tR 19.5 min, 2.2 mg, 0.00055% w/w). Fraction FIII (987 mg), eluted with 0.1% MeOH in CH2Cl2 from the first separation, was subjected again to silica gel CC (φ 2.5 cm; 230–400 mesh, 50 g), using gradient mixtures of MeOH in CHCl3 (1→10%) as mobile phases, providing ten sub-fractions. Passage over Sephadex LH-20 of the third fraction (210 mg) from this column, which was eluted with 1% MeOH in CHCl3, using 100% MeOH, afforded mangostanol (2.2 mg, 0.00055% w/w). Fraction FIV (1.1 g), eluted with 0.15% MeOH in CH2Cl2 from the first separation, was separated by silica gel CC (φ 3 cm; 230–400 mesh, 70 g), using a gradient solvent system of CH2Cl2-acetone (99:1→9:1), providing 16 sub-fractions. Sub-fraction 10 (100 mg), eluted with CH2Cl2-acetone (95:5), was subjected to Sephadex LH-20 chromatography using 100% MeOH as solvent, yielding 11 (15.5 mg, 0.0038% w/w). The fourth fraction (10 mg) from this Sephadex LH-20 column was purified by preparative HPLC, using an isocratic mixture of CH3CN-H2O (9:1, 8 mL/min) as solvent system, to afford 3 (tR 18.5 min, 3.4 mg, 0.00085% w/w). The combined sub-fractions 11 and 12 (97 mg), eluted with CH2Cl2-acetone (95:5), were subjected to passage over Sephadex LH-20 (100% MeOH), providing four sub-fractions. The first sub-fraction (20.1 mg) from this separation was purified by preparative HPLC using an isocratic mixture of MeOH-H2O (85:15, 5 mL/min) as solvent system, to obtain 1 (tR 11.8 min, 5.1 mg, 0.00127%). Combined sub-fractions 13 and 14 (162 mg), eluted with CH2Cl2-acetone (95:5), were also separated by Sephadex LH-20 CC (100% MeOH), affording 7 (28.2 mg, 0.007% w/w) and 4 (3.2 mg, 0.0008% w/w). Fraction FV (242 mg), eluted with 0.2% MeOH in CH2Cl2 from the first separation, was subjected to Sephadex LH-20 CC, using 100% MeOH as solvent system, and then purified by preparative HPLC using an isocratic mixture of CH3CN-H2O (9:1, 8 mL/min) as solvent system, furnishing 2 (tR 9.2 min, 3.9 mg, 0.00097% w/w).
11-Hydroxy-3-O-methyl-1-isomangostin (1)
yellow amorphous powder; mp 130–132 °C; [α]D 25 +9.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 242 (4.43), 252 (4.39), 303 (4.18) nm; IR (film) νmax 3350, 2931, 1614, 1456, 1374, 1272, 1208, 1181, 1138, 1115 cm−1; 1H NMR (CDCl3, 400 MHz) δ 6.76 (1H, s, H-5), 6.29 (1H, s, H-4), 5.33 (1H, t, J = 6.4 Hz, H-16), 4.07 (2H, t, J = 6.4 Hz, H-15), 3.85 (1H, t, J = 4.9 Hz, H-11), 3.83 (3H, s, OCH3-3), 3.78 (3H, s, OCH3-7), 2.84 (1H, dd, J = 17.3, 4.9 Hz, H-10β), 2.69 (1H, dd, J = 17.3, 4.9 Hz, H-10α), 1.80 (3H, s, H-19), 1.65 (3H, s, H-18), 1.48 (3H, s, H-14), 1.36 (3H, s, H-13); 13C NMR (CDCl3, 100 MHz) δ 176.9 (C, C-9), 162.0 (C, C-3), 157.4 (C, C-4a), 154.6 (C, C-6), 154.3 (C, C-5a), 153.7 (C, C-1), 142.9 (C, C-7), 137.2 (C, C-8), 131.4 (C, C-17), 124.3 (CH, C-16), 115.3 (C, C-8a), 108.0 (C, C-9a), 104.0 (C, C-2), 101.4 (CH, C-5), 90.7 (CH, C-4), 78.2 (C, C-12), 68.8 (CH, C-11), 61.9 (CH3, OCH3-7), 56.0 (CH3, OCH3-3), 26.6 (CH2, C-15), 26.5 (CH2, C-10), 26.1 (CH3, C-18), 24.7 (CH3, C-13), 22.6 (CH3, C-14), 18.5 (CH3, C-19); ESIMS (positive mode) m/z 463.17 [M+Na]+ (100), 322.80 (10), 172.89 (40); HRESITOFMS (positive mode) m/z 463.1729 [M+Na]+ (calcd for C25H28O7, 463.1733).
11-Hydroxy-1-isomangostin (2)
[α]D25 +11.4 (c 0.1, MeOH).
11α-Mangostanin (3)
[α]D25 −1.75 (c 0.1, MeOH).
Cratoxyxanthone (11)
[α]D25 +0.06 (c 0.13, MeOH).
Cytotoxicity Assay
Cytotoxic potential against HT-29 was determined using a established protocol.25 Camptothecin was employed as the positive control (ED50 = 0.06 μM).
Enzyme-based ELISA NF-κB Assay
The NF-κB p65 and p50 inhibitory activity assay was conducted according to a published protocol.26,27 Rocaglamide was used as the positive control and exhibited IC50 values of 0.08 and 2.0 μM in this assay.
Hollow Fiber Assay
α-Mangostin (5) was evaluated in the in vivo hollow fiber model, using HT-29, LNCaP, and MCF-7 cells, according to a procedure described in the literature.28,29 Paclitaxel was used as the positive control for this experiment at a dose of 20 mg/kg.
Supplementary Material
Acknowledgments
This work was supported by grants U19-CA52956 and P01-CA125066 funded by the National Cancer Institute, NIH, Bethesda, Maryland. A.-R. H. was supported by a Korea Research Foundation Grant (KRF-2007-357-E00035), funded by the Korean Government (MOEHRD), Seoul, South Korea.
Footnotes
Supporting Information Available: Data for in vivo hollow fiber evaluation of α-mangostin (5) against LNCaP and HT-29 cells, 1D and 2D NMR spectra for compound 1, and tables of NMR data of the known compounds 2–11. This material is available free of charge via the Internet at http://pubs.acs.org/jnp.
References and Notes
- 1.Chin YW, Kinghorn AD. Mini-Rev Org Chem. 2008;5:355–364. doi: 10.2174/157019308786242223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Obolskiy D, Pischel I, Siriwatanametanon N, Heinrich M. Phytother Res. 2009;23:1047–1065. doi: 10.1002/ptr.2730. [DOI] [PubMed] [Google Scholar]
- 3.Jung HA, Su BN, Keller WJ, Mehta RG, Kinghorn AD. J Agric Food Chem. 2006;54:2077–2082. doi: 10.1021/jf052649z. [DOI] [PubMed] [Google Scholar]
- 4.Balunas MJ, Su B, Brueggemeier RW, Kinghorn AD. J Nat Prod. 2008;71:1161–1166. doi: 10.1021/np8000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin YW, Jung HA, Chai H, Keller WJ, Kinghorn AD. Phytochemistry. 2008;69:754–758. doi: 10.1016/j.phytochem.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 6.Kinghorn AD, Carcache de Blanco EJ, Chai H-B, Orjala J, Farnsworth NR, Soejarto DD, Oberlies NH, Wani MC, Kroll DJ, Pearce CJ, Swanson SM, Kramer RA, Rose WC, Emanuel S, Vite GD, Jarjoura D, Cope FO. Pure Appl Chem. 2009;81:1051–1063. doi: 10.1351/PAC-CON-08-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavia DL, Lampman GM, Kriz GS. Introduction to Spectroscopy. Thomson Learning, Ltd; London: 2001. [Google Scholar]
- 8.Sia GL, Bennett GJ, Harrison LJ, Sim KY. Phytochemistry. 1995;38:1521–1528. doi: 10.1016/0031-9422(94)00314-j. [DOI] [PubMed] [Google Scholar]
- 9.(a) Dale JA, Mosher HS. J Am Chem Soc. 1972;95:512–519. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4902–4906. [Google Scholar]; (c) Su BN, Park EJ, Mbwambo ZH, Santarsiero BD, Mesecar AD, Fong HHS, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:1278–1282. doi: 10.1021/np0202475. [DOI] [PubMed] [Google Scholar]
- 10.Nilar, Harrison LJ. Phytochemistry. 2002;60:541–548. doi: 10.1016/s0031-9422(02)00142-5.. In this reference, the compound number of mangostanin should be 15 rather than the erroneous 13.
- 11.Crews P, Rodriguez J, Jaspars M. Organic Structure Analysis. Oxford University Press; New York: 1998. [Google Scholar]
- 12.Sen AK, Sarkar KK, Mazumder PC, Banerji N, Uusuori R, Hase TA. Phytochemistry. 1982;21:1747–1750. [Google Scholar]
- 13.Likhitwitayawuid K, Phadungcharoen T, Krungkrai J. Planta Med. 1998;64:70–72. doi: 10.1055/s-2006-957370. [DOI] [PubMed] [Google Scholar]
- 14.Bennett GJ, Harrison LJ, Sia GL, Sim KY. Phytochemistry. 1993;32:1245–1251. [Google Scholar]
- 15.Sen AK, Sarkar KK, Mazumder PC, Banerji N, Uusvuori R, Hase TA. Phytochemistry. 1980;19:2223–2225. [Google Scholar]
- 16.Nguyen LHD, Vo HT, Pham HD, Connolly JD, Harrison LJ. Phytochemistry. 2003;63:467–470. doi: 10.1016/s0031-9422(02)00433-8. [DOI] [PubMed] [Google Scholar]
- 17.Govindachari TR, Kalyanaraman PS, Muthukumaraswamy N, Pai BR. Tetrahedron. 1971;27:3919–3926. [Google Scholar]
- 18.Chairungsrilerd N, Takeuchi K, Ohizumi Y, Nozoe S, Ohta T. Phytochemistry. 1996;43:1099–1102. [Google Scholar]
- 19.Suksamrarn S, Komutiban O, Ratananukul P, Chimnoi N, Lartpornmatulee N, Suksamrarn A. Chem Pharm Bull. 2006;54:301–305. doi: 10.1248/cpb.54.301. [DOI] [PubMed] [Google Scholar]
- 20.Ee GCL, Daud S, Izzaddin SA, Rahmani M. J Asian Nat Prod Res. 2008;10:481–485. doi: 10.1080/10286020801948490. [DOI] [PubMed] [Google Scholar]
- 21.Laphookhieo S, Syers JK, Kiattansakul R, Chantrapromma K. Chem Pharm Bull. 2006;54:745–747. doi: 10.1248/cpb.54.745. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto K, Akao Y, Yi H, Ohguchi K, Ito T, Tanaka T, Kobayashi E, Iinuma M, Nozawa Y. Bioorg Med Chem. 2004;12:5799–5806. doi: 10.1016/j.bmc.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 23.Akao Y, Nakagawa Y, Iinuma M, Nozawa Y. Int J Mol Sci. 2008;9:355–370. doi: 10.3390/ijms9030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi Q, Pezzuto JM, Farnsworth NR, Wani MC, Kinghorn AD, Swanson SM. J Nat Prod. 2009;72:573–580. doi: 10.1021/np800767a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Likhitwitayawuid K, Angerhofer CK, Cordell GA, Pezzuto JM, Ruangrungsi N. J Nat Prod. 1993;56:30–38. doi: 10.1021/np50091a005. [DOI] [PubMed] [Google Scholar]
- 26.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Nucleic Acids Res. 2001;29:e21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salim AA, Pawlus AD, Chai HB, Farnsworth NR, Kinghorn AD, Carcache-Blanco EJ. Bioorg Med Chem Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mi Q, Cui B, Silva GL, Lantvit DD, Lim E, Chai H, Hollingshead MG, Mayo JG, Kinghorn AD, Pezzuto JM. Cancer Lett. 2002;184:13–20. doi: 10.1016/s0304-3835(02)00202-1. [DOI] [PubMed] [Google Scholar]
- 29.Hollingshead MG, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. Life Sci. 1995;57:131–41. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.