

Abstract
The treatment of psoriasis has undergone a revolution with the advent of biologic therapies, including infliximab, etanercept, adalimumab, efalizumab, and alefacept. These medications are designed to target specific components of the immune system and are a major technological advancement over traditional immunosuppressive medications. These usually being well tolerated are being found useful in a growing number of immune-mediated diseases, psoriasis being just one example. The newest biologic, ustekinumab, is directed against the p40 subunit of the IL-12 and IL-23 cytokines. It has provided a new avenue of therapy for an array of T-cell-mediated diseases. Biologics are generally safe; however, there has been concern over the risk of lymphoma with use of these agents. All anti-TNF-α agents have been associated with a variety of serious and “routine” opportunistic infections.
Keywords: Adverse effects, biologics, psoriasis, therapy
Introduction
Psoriasis is a life-long chronic inflammatory skin condition affecting approximately 2% of the general population.[1,2] There are many clinical variants of psoriasis. Most patients have plaques with silver-white scale and an erythematous base. Some patients have joint involvement. There is strong evidence in favor of psoriasis being an immune-mediated disease with T-cells playing a central role.[3,4] However, the pathogenesis of psoriasis is complex and likely includes mediators of both the innate and adaptive immune systems. In support of an immune etiology, psoriasis can either develop or go into remission following a bone marrow transplantation.[5,6] To date, there is no consensus as to the antigens involved in the autoreactive immune response that is responsible for psoriasis. However, the cytokine secretion profile of the T-cells has been well characterized and both Th1 and Th17 cells have been found to play a role in the pathogenesis of psoriasis.[7] Th1 differentiation is mediated by IL-12. In contrast, Th17 cells develop in the presence of IL-1, IL-6, and TGF-α. Once differentiated, IL-23 is then required for their maintanance. Th1 cells release mediators such as TNF-α and IFN-α that lead to vasodilation, leukocyte migration and activation of keratinocytes.[4] This in turn leads to further activation of dendritic cells, creating a cycle of inflammation. Th-17 cells also stimulate keratinocyte activation and proliferation through secretion of IL-17 and IL-22.[8–10] A schematic of the activation process is shown in Figure 1.
Figure 1.
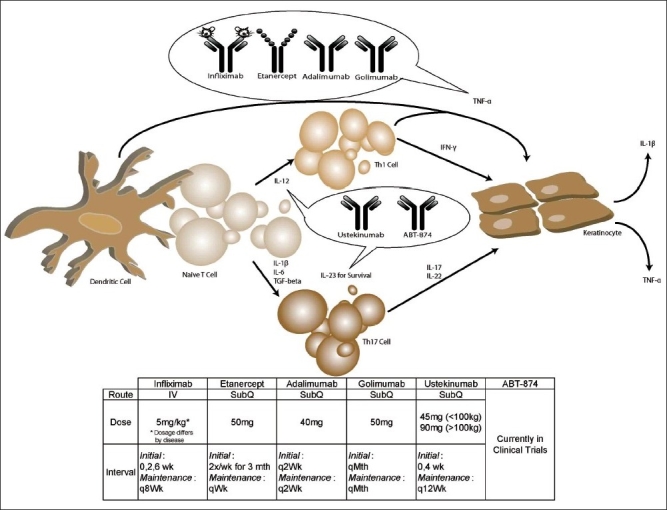
Biologics in psoriasis and their possible mechanisms. TNF- α secreted by antigen-presenting cells; Th-1 cells and keratinocytes can be neutralized by the anti-TNF biologics infliximab, etanercept, adalimumab, and golimumab. Adalimumab and golimumab are fully human antibodies directed against TNF-α. Infliximab was developed from a mouse anti-TNF antibody that was then partially humanized. Etanercept is a molecullarly engineered molecule formed by linking the TNF-α receptor to the Fc portion of an antibody. Ustekinumab and ABT-874 are directed against the p40 subunit of IL-12 and IL-23. IL-12 is needed for differentiation of naive cells into Th-1 cells and IL-23 is needed for the maintenance of IL-17-secreting Th17 cells. IFN-α secreted by Th-1 cells and IL-17 and IL-22 secreted by Th-17 cells activate keratinocytes, which in turn proliferate and secrete IL-12 and TNF-α.
Biological therapy is the use of agents that can specifically target an immune or genetic mediator of a pathophysiological process. The introduction of biological-based therapies has greatly improved treatment of psoriasis. Several biological therapies have emerged over the past decade for psoriasis alone [Table 1]. Earlier agents disrupted activation and migration of T-cells and these include alefacept and efalizumab. Later agents have targeted TNF-α and these include infliximab, etanercept, and adalimumab. Recently, agents that target the p40 subunit shared by both IL-12 and IL-23 have been developed and these include ustekinumab and ABT-874. The sites of action of the anti-TNF and the anti-IL12/IL23 agents are indicated in Figure 1. Clinical trials that have investigated the role of biologics in psoriasis therapy are reviewed in Table 2a and 2b.
Table 1.
Biologics in treatment of psoriasis
| Biologic | Immunological construct | Mechanism of action | Manufacturer | Route |
|---|---|---|---|---|
| Alefacept | Human fusion protein of the first extracellular domain of LFA-3 fused Fc portion of human IgG1 | LFA-3 portion binds to CD2 on memory T-cells to block their activation. Fc portion binds to CD 16 on natural killer cells to induce apoptosis of memory T-cells | Astellas Pharma USA, Inc. | IV |
| Infliximab | Chimeric (murine-human) antibody against TNF-α | Binds TNF to neutralize its effects | Centocor Ortho Biotech Inc. | IV |
| Etanercept | Human fusion protein of the TNF receptor to Fc portion of IgG1 | Binds TNF to neutralize its effects | Amgen® and Wyeth® | SC |
| Adalimumab | Human monoclonal antibody against TNF | Binds TNF to neutralize its effects | Abbot Laboratories | SC |
| Golimumab | Human monoclonal antibody against TNF | Binds TNF to neutralize its effects | Centocor Ortho Biotech Inc. | SC |
| Ustekinumab | Human monoclonal antibody against the p40 subunit of IL-12 and IL-23 from human immunoglobulin transgenic mice | Blocks the actions of IL-12 and IL-23 | Centocor Ortho Biotech Inc. | SC |
| ABT-874 | Human monoclonal antibody against the p40 subunit of IL-12 and IL-23 isolated from human anti body phage display library | Blocks the actions of IL-12 and IL-23 | Abbot Laboratories | SC |
Table 2a.
Clinical trials of biologics for psoriasis
| Biologic | Study | Study design | Duration of study | Dosing | Antibody formation against Bologic |
|---|---|---|---|---|---|
| Alefacept |
|
|
|
|
|
| Infliximab |
|
|
|
|
|
| Etanercept |
|
|
|
|
|
| Adalimumab |
|
|
|
|
|
| Ustekinumab |
|
|
|
|
|
| ABT-874 |
|
|
|
|
|
DB = double-blind, PC = placebo controlled, PG = parallel group, CPP = chronic plaque psoriasis, IV = intravenous, PASI = psoriasis area and severity index
Table 2b.
Efficacies of biologics in clinical trials for psoriasis
| Biologic | Efficacy at primary endpoint | Notes |
|---|---|---|
| Alefacept |
|
|
| Infliximab |
|
|
| Etanercept |
|
|
| Adalimumab |
|
|
| Ustekinumab |
|
|
| ABT-874 |
|
|
Non-cytokine Biologics
Alefacept
Alefacept was designed to block the CD2/LFA-3 interaction important for T-cell function. Clinical trials with either IV[11,12] or IM[13] alefacept have found it to be effective in the treatment of psoriasis. All of these trials were 12 weeks in length, and tested for improvement at 2 weeks as their primary end point. Efficacy was maintained at 12 weeks. In one of the studies, alefacept was found to reduce the amount of memory effector T-cells without affecting the naïve T-cell population.[11] Another study included three different cohorts with a placebo crossover and placebo withdrawal. This trial found that the cohort that received two courses of alefacept had improved treatment efficiencies compared to the placebo crossover or withdrawal cohorts.[12]
A meta-analysis showed that alefacept treated patients had an overall 9% increased risk of adverse events.[14] The most common adverse effects noted in these studies were dizziness,[11] nausea,[11] infusion-related chills,[11,12] pharygitis,[12,13] headache,[13] and pruritus.[13] A meta-analysis of the safety of alefacept showed coronary artery disease in four subjects, cellulitis in three subjects, and myocardial infarction in three subjects, while none of these serious adverse events were noted in the placebo groups. IV dosing was noted to increase the incidence of serious adverse effects over IM dosing.[14]
Anti-alefacept antibodies were noted to develop in all three studies with up to 4% in the study in which alefacept was administered IM. These antibodies were found to be non-neutralizing.[13] No adverse events were correlated with the presence of the antibody.
Efalizumab
Efalizumab has been voluntarily withdrawn from the market in the USA partly due to the risks of progressive multifocal leukoencephalopathy. This antibody was manufactured by Genentech and was specific to the CD11a subunit of LFA-1.
Cytokine Biologics
Anti-TNF agents
Infliximab
Clinical trials with IV infliximab have shown it to have efficacy of reaching a PASI 75 at 10 weeks at 75.5-88% in those treated with 5 mg/kg when compared to 1.9-6% in the placebo group.[15–17] Two of the studies showed that an intermediate dose of 3 mg/kg was also effective in achieving PASI 75 at 10 weeks for 70.3-72% of those treated.[16,17] Efficacies were maintained over placebo for 46-50 weeks, a loss in response was noted in those subjects that developed anti-infliximab antibodies.[15,16] Although not a primary end point, one study noted 26% improvement and a 6% worsening in nail psoriasis of the infliximab and placebo treatment groups, respectively. The most common adverse effects were rhinitis, transaminitis, sinusitis, and headache.[15,16]
Etanercept
Phase 2 and phase 3 trials with etanercept delivered subcutaneously report that it is superior to placebo in achieving PASI 75.[18–22] At 12 weeks after treatment, the PASI 75 for biweekly subcutaneous injections of 25 mg or 50 mg were reported at 34% and 49%, respectively, while the placebo group had a 12 week PASI 75 of only 3-4%.[19,21] A dose response was noted from low to high dosing[19,21] and the efficacy continued to increase at 24 weeks.[21] A phase 3 study of a pediatric population revealed that after 12 weeks, subcutaneous dosing at 0.8 mg/kg resulted in 57% of patients receiving a PASI 75 while placebo dosing only achieved a PASI 75 in 11%.[20] Antibody formation against etanercept ranged from 1.1 to 18.3%.[18,19] A loss of response was correlated with the duration of therapy but the formed antibodies were not found to be neutralizing.
The most common side effects noted in adults were upper respiratory tract infections,[22] sinusitis,[22] headaches,[22] and injection site reactions[18,19,21,22] Injection site rections tended to occur more frequently during the first 12 weeks of therapy and approached placebo level frequencies afterward. The most common side effects noted in one pediatric study was an increased incidence of streptoccal pharyngitis and skin papillomas.[20]
Adalimumab
Subcutaneously injected adalimumab was found to have superior efficacy of achieving PASI 75 in comparison to placebo in several phase 2 and phase 3 trials. In one phase 2 study, increasing doses of adalimumab were compared against placebo and a dose response was observed. After 12 weeks of therapy it was found that placebo, 40 mg every other week and 40 mg weekly achieved a PASI 75 in 4%, 53%, and 80% of subjects, respectively.[23] Two other phase 3 studies found that 71-79.6% of subjects treated with 40 mg every other week achieved a PASI 75 in comparison to 7-18.9% of those treated with placebo after 16 weeks of treatment.[24,25] The higher rate of efficacy in achieving PASI 75 in the placebo groups of the phase 3 studies may have been related to the 16 week course of treatment in comparison to the 12 week treatment course in the phase 2 study. Antibodies against adalimumab developed in 8.8% of patients at some point during their treatment course and the presence of antibodies was correlated with a loss of response.[25]
One the phase 3 studies compared adalimumab treatment against methotrexate. At 16 weeks, the PASI 75 achieved by subjects in the methotrexate and the adalimumab treatment groups were 35.5% and 79.6%, respectively.[24] Because the methotrexate was started low and increased over time, the 16 weeks observation may have been too short to appropriately assess the methotrexate response.
The most common side effects were upper respiratory infections,[25] nasopharyngitis,[24] headache,[24] and cellulitis.[25]
Anti-p40 (IL-12/IL-23)
Ustekinumab
Ustekinumab is the first of a new class of biological drugs that prevent the actions of IL-12 and IL-23 by binding to their mutual subunit p40. Two phase 3 studies show that subcutaneously injected ustekinumab has superior efficacy in comparison to placebo. Both studies utilized a 12 week placebo controlled period during which ustekinumab had an efficacy of achieving PASI 75 in 66.7-67.1% and 66.4-75.7% in those treated with 45 mg or 90 mg, respectively.[26,27] In the placebo group, 3.1-3.7% achieved PASI 75. Both of these trials included a placebo crossover group that attained similar treatment efficacies as the ustekinumab treatment group. The dosing of ustekinumab is more spaced out than previous biologics with subcutaneous injections given at week 0, week 4, and then at 12 week intervals, making treatment more convenient. The development of antibodies against ustekinumab has been shown to have clinical implications as the antibodies were found to be neutralizing.[26] This study showed that subjects could be split into full responders and partial responders, the latter defined as those subjects that achieved PASI 50 but not PASI 75 by 28 weeks. Partial responders had increased the prevalence of antibodies against ustekinumab.[26]
The most common side effects were injection site reactions.[26] As this is a newly introduced drug, there is little long-term usage studies and post-market surveillance will be important in understanding long-term side effects.
ABT-874
ABT-874 is another antibody generated against the p40 subunit and designed to block the actions of IL-12 and IL-23. One phase 2 trial investigated the use of ABT-874 with progressively increasing doses, showing a dose response relationship.[28] While 3% of subjects in the placebo achieved PASI 75 at 12 weeks, 90% of those treated with 200 mg every 4 weeks achieved PASI 75 at 12 weeks. It was found that increasing the dosing beyond 200 mg every 4 weeks did not provide any increase in achieving PASI 75. The most common adverse events were injection site reactions and nasopharyngitis. The development of antibodies against ABT-874 were not reported.
Discussion
With the growth in development of biological therapies, there are several effective options for the treatment of chronic plaque psoriasis, which is the most prevalent form of psoriasis. Several generalizations can be made from review of the clinical trial literature. It is interesting to note that in most studies, the placebo group had a larger dropout rate than the treatment group [Table 2b], and this may alter the actual differences between the treatment and placebo group. All of the studies compared treatment against placebo, but only one study compared the biological therapy against methotrexate.[24] The formation of antibodies against the biological drug is not uncommon and can affect the long-term efficacy of the biologic. Studies in the use of biological therapies and immunosuppresants for rheumatoid arthritis and Crohn's disease show that combined dosing of a biological agent with another immunosppressive agent, such as methotrexate, decreases the formation of antibodies against the biological agent.[29,30] Although resistance to one biological agent does not imply resistance to another agent, it would be inconvenient to keep switching agents given the chronic nature of psoriasis. A better solution may be to concomitantly treat patients with both a biological agent and another immunospressant, such as methotrexate. Case reports describe the utility in combining methotrexate with a biological agent.[31] However, no studies have investigated the combined therapy of biological agents and methotrexate for psoriasis and currently there are little data on the efficacy or the side effects of combined therapy.
The larger studies reviewed here have focused on the therapy of plaque psoriasis and it is unclear how effective the biological therapies will be in treatment of other forms of psoriasis. Smaller studies have suggested that some of the biologics may be useful for other forms of psoriasis.[32–34]
Unlike the TNF blockers that have been studied for a longer duration of time[35,36] and used extensively in rheumatology, the IL-12/23 blockers are new treatment options and the long-term effects are still largely unknown. Because these biological agents act earlier in the immune response chain, in comparison to the TNF-α blockers, they are potentially more immunosuppressive and thus infection is a concern. In particular, Th-17 cells, whose actions are antagonized by IL-12/23 blockers, are important in protection against bacteria and fungi.[37]
Biologics are generally safe and well tolerated. However, like all medications, they have adverse effects. Importantly, these medications can predispose patients to infections and increased their risk of developing a malignancy.[38–41] All anti-TNF-α agents have been associated with a variety of serious and “routine” opportunistic infections.[38] From a public health standpoint, the development of active tuberculosis in some patients who receive TNF-α inhibitor therapy is a matter of serious concern.[38,39] There is also an increased risk for a variety of malignant conditions such as lymphoma, leukemia, and melanomas.[40,41]
As the use of TNF-α antagonists becomes widespread, further cases of tuberculosis associated with TNF-α blockade can be expected, especially in developing countries with high incidences of tuberculosis.[38] To prevent the reactivation of latent tuberculosis, appropriate screening of patients with Mantoux test and chest X-ray should be performed before initiating anti-TNF therapy, and begin treatment if latent infection is found. The screening strategies employed in Europe and North America have reduced the occurrence of TNF-α inhibitor-associated tuberculosis. Tuberculosis in patients treated with anti-TNF agents may present with extrapulmonary or disseminated disease. Thus, clinicians should be vigilant in monitoring for tuberculosis in their patients treated with TNF-α inhibitors. The role of screening in the prevention of other opportunistic infections is far less certain. No official guidelines currently exist for many of these opportunistic infections, but various authors have made recommendations regarding screening options, as summarized in Table 3.[38,42–44] Because of systemic immune suppression, a variant clinical presentation is expected; atypical signs and symptoms as well as atypical pathogens should be considered. Patients receiving TNF-α inhibitor treatment should be closely monitored for serious infections and should be educated about how to avoid infectious complications.[38] Although rare, clinicians need to closely monitor for malignancy, and induction of autoimmune diseases (psoriasis, lupus) in patients receiving anti-TNF agents.
Table 3.
Suggested screening tests for certain infections before initiating anti-TNF therapy (39,43,44,45)
| Infection | Recommended screening |
|---|---|
| Tuberculosis | PPD, chest X-ray at baseline and PPD every 12 months. |
| Histoplasmosis | Consider chest radiograph and urine histoplasmin antigen testing at baseline and every 3 – 4 months for patients who live or have lived in endemic areas. |
| Coccidioidomycosis | Chest radiograph and serologic testing with IgM and IgG tests at baseline. Consider follow-up testing every 3 – 4 months for patients who live or have lived in endemic areas. |
The development of biological therapies has revolutionized psoriasis treatment. Despite the growing number of biological therapies that are entering the clinical arena, many more biological remain on the horizon, including the targeting of IL-21[45] or IL-22.[10] With time, long-term side effects and efficacies will become clearer and help determine which ones are the most suitable for long-term care of psoriasis.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Raychaudhuri SP, Gross J. A comparative study of pediatric onset psoriasis with adult onset psoriasis. Pediatr Dermatol. 2000;17:174–8. doi: 10.1046/j.1525-1470.2000.01746.x. [DOI] [PubMed] [Google Scholar]
- 2.Raychaudhuri SP, Farber EM. The prevalence of psoriasis in the world. J Eur Acad Dermatol Venereol. 2001;15:16–7. doi: 10.1046/j.1468-3083.2001.00192.x. [DOI] [PubMed] [Google Scholar]
- 3.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 4.Raychaudhuri SP, Kundu-Raychaudhuri S, Tamura K, Masunaga T, Kubo K, Hanaoka K, et al. FR255734, a humanized, Fc-Silent, Anti-CD28 antibody, improves psoriasis in the SCID mouse-psoriasis xenograft model. J Invest Dermatol. 2008;128:1969–76. doi: 10.1038/jid.2008.38. [DOI] [PubMed] [Google Scholar]
- 5.Gardembas-Pain M, Ifrah N, Foussard C, Boasson M, Saint Andre JP, Verret JL. Psoriasis after allogeneic bone marrow transplantation. Arch Dermatol. 1990;126:1523. doi: 10.1001/archderm.1990.01670350139033. [DOI] [PubMed] [Google Scholar]
- 6.Eedy DJ, Burrows D, Bridges JM, Jones FG. Clearance of severe psoriasis after allogenic bone marrow transplantation. BMJ. 1990;300:908. doi: 10.1136/bmj.300.6729.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–94. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolk K, Witte E, Warszawska K, Schulze-Tanzil G, Witte K, Philipp S, et al. The Th17 cytokine IL-22 induces IL-20 production in keratinocytes: A novel immunological cascade with potential relevance in psoriasis. Eur J Immunol. 2009;39:3570–81. doi: 10.1002/eji.200939687. [DOI] [PubMed] [Google Scholar]
- 9.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis CN, Krueger GG. Alefacept Clinical Study Group. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med. 2001;345:248–55. doi: 10.1056/NEJM200107263450403. [DOI] [PubMed] [Google Scholar]
- 12.Krueger GG, Papp KA, Stough DB, Loven KH, Gulliver WP, Ellis CN, et al. A randomized, double-blind, placebo-controlled phase III study evaluating efficacy and tolerability of 2 courses of alefacept in patients with chronic plaque psoriasis. J Am Acad Dermatol. 2002;47:821–33. doi: 10.1067/mjd.2002.127247. [DOI] [PubMed] [Google Scholar]
- 13.Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. 2003;139:719–27. doi: 10.1001/archderm.139.6.719. [DOI] [PubMed] [Google Scholar]
- 14.Brimhall AK, King LN, Licciardone JC, Jacobe H, Menter A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Br J Dermatol. 2008;159:274–85. doi: 10.1111/j.1365-2133.2008.08673.x. [DOI] [PubMed] [Google Scholar]
- 15.Reich K, Nestle FO, Papp K, Ortonne JP, Evans R, Guzzo C, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367–74. doi: 10.1016/S0140-6736(05)67566-6. [DOI] [PubMed] [Google Scholar]
- 16.Menter A, Feldman SR, Weinstein GD, Papp K, Evans R, Guzzo C, et al. A randomized comparison of continuous vs.intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2007;56:31. doi: 10.1016/j.jaad.2006.07.017. e-15. [DOI] [PubMed] [Google Scholar]
- 17.Gottlieb AB, Evans R, Li S, Dooley LT, Guzzo CA, Baker D, et al. Infliximab induction therapy for patients with severe plaque-type psoriasis: a randomized, double-blind, placebo-controlled trial. J Am Acad Dermatol. 2004;51:534–42. doi: 10.1016/j.jaad.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 18.Tyring S, Gordon KB, Poulin Y, Langley RG, Gottlieb AB, Dunn M, et al. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol. 2007;143:719–26. doi: 10.1001/archderm.143.6.719. [DOI] [PubMed] [Google Scholar]
- 19.Papp KA, Tyring S, Lahfa M, Prinz J, Griffiths CE, Nakanishi AM, et al. A global phase III randomized controlled trial of etanercept in psoriasis: safety, efficacy, and effect of dose reduction. Br J Dermatol. 2005;152:1304–12. doi: 10.1111/j.1365-2133.2005.06688.x. [DOI] [PubMed] [Google Scholar]
- 20.Paller AS, Siegfried EC, Langley RG, Gottlieb AB, Pariser D, Landells I, et al. Etanercept treatment for children and adolescents with plaque psoriasis. N Engl J Med. 2008;358:241–51. doi: 10.1056/NEJMoa066886. [DOI] [PubMed] [Google Scholar]
- 21.Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014–22. doi: 10.1056/NEJMoa030409. [DOI] [PubMed] [Google Scholar]
- 22.Gottlieb AB, Matheson RT, Lowe N, Krueger GG, Kang S, Goffe BS, et al. A randomized trial of etanercept as monotherapy for psoriasis. Arch Dermatol. 2003;139:1627–32. doi: 10.1001/archderm.139.12.1627. [DOI] [PubMed] [Google Scholar]
- 23.Gordon KB, Langley RG, Leonardi C, Toth D, Menter MA, Kang S, et al. Clinical response to adalimumab treatment in patients with moderate to severe psoriasis: double-blind, randomized controlled trial and open-label extension study. J Am Acad Dermatol. 2006;55:598–606. doi: 10.1016/j.jaad.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Saurat JH, Stingl G, Dubertret L, Papp K, Langley RG, Ortonne JP, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION) Br J Dermatol. 2008;158:558–66. doi: 10.1111/j.1365-2133.2007.08315.x. [DOI] [PubMed] [Google Scholar]
- 25.Menter A, Tyring SK, Gordon K, Kimball AB, Leonardi CL, Langley RG, et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial. J Am Acad Dermatol. 2008;58:106–15. doi: 10.1016/j.jaad.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371:1675–84. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 27.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–74. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 28.Kimball AB, Gordon KB, Langley RG, Menter A, Chartash EK, Valdes J, et al. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: Results of a randomized, placebo-controlled, phase 2 trial. Arch Dermatol. 2008;144:200–7. doi: 10.1001/archdermatol.2007.63. [DOI] [PubMed] [Google Scholar]
- 29.Vermeire S, Noman M, van Assche G, Baert F, D'Haens G, Rutgeerts P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn's disease. Gut. 2007;56:1226–31. doi: 10.1136/gut.2006.099978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66:921–6. doi: 10.1136/ard.2006.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirby B, Marsland AM, Carmichael AJ, Griffiths CE. Successful treatment of severe recalcitrant psoriasis with combination infliximab and methotrexate. Clin Exp Dermatol. 2001;26:27–9. doi: 10.1046/j.1365-2230.2001.00753.x. [DOI] [PubMed] [Google Scholar]
- 32.Myers W, Christiansen L, Gottlieb AB. Treatment of palmoplantar psoriasis with intramuscular alefacept. J Am Acad Dermatol. 2005;53:S127–9. doi: 10.1016/j.jaad.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Prossick TA, Belsito DV. Alefacept in the treatment of recalcitrant palmoplantar and erythrodermic psoriasis. Cutis. 2006;78:178–80. [PubMed] [Google Scholar]
- 34.Carr D, Tusa MG, Carroll CL, Pearce DJ, Camacho F, Kaur M, et al. Open label trial of alefacept in palmoplantar pustular psoriasis. J Dermatolog Treat. 2008;19:97–100. doi: 10.1080/09546630701364776. [DOI] [PubMed] [Google Scholar]
- 35.Castelo-Soccio L, van Voorhees AS. Long-term efficacy of biologics in dermatology. Dermatol Ther. 2009;22:22–33. doi: 10.1111/j.1529-8019.2008.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brimhall AK, King LN, Licciardone JC, Jacobe H, Menter A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: A meta-analysis of randomized controlled trials. Br J Dermatol. 2008;159:274–85. doi: 10.1111/j.1365-2133.2008.08673.x. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z, O'Shea JJ. Th17 cells: A new fate for differentiating helper T-cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 38.Raychaudhuri SP, Nguyen CT, Raychaudhuri SK, Gershwin ME. Incidence and nature of infectious disease in patients treated with anti-TNF agents. Autoimmun Rev. 2009;9:67–81. doi: 10.1016/j.autrev.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 40.Day R. Adverse reactions to TNF-alpha inhibitors in rheumatoid arthritis. Lancet. 2002;359:540–1. doi: 10.1016/S0140-6736(02)07718-8. [DOI] [PubMed] [Google Scholar]
- 41.Pallavicini FB, Caporali R, Sarzi-Puttini P, Atzeni F, Bazzani C, Gorla R, et al. Tumour necrosis factor antagonist therapy and cancer development: Analysis of the LORHEN registry. Autoimmun Rev. 2010;9:175–80. doi: 10.1016/j.autrev.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005;84:291–302. doi: 10.1097/01.md.0000180044.19285.9a. [DOI] [PubMed] [Google Scholar]
- 43.Giles JT, Bathon JM. Serious infections associated with anticytokine therapies in the rheumatic diseases. J Intensive Care Med. 2004;19:320–34. doi: 10.1177/0885066604267854. [DOI] [PubMed] [Google Scholar]
- 44.Strangfeld A, Listing J. Infection and musculoskeletal conditions: Bacterial and opportunistic infections during anti-TNF therapy. Best Pract Res Clin Rheumatol. 2006;20:1181–95. doi: 10.1016/j.berh.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 45.Caruso R, Botti E, Sarra M, Esposito M, Stolfi C, Diluvio L, et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat Med. 2009;15:1013–5. doi: 10.1038/nm.1995. [DOI] [PubMed] [Google Scholar]