

Abstract
The early region of BK virus (BKV) is known to encode two well-characterized tumour (T) antigens, large T antigen (TAg) and small T antigen (tAg). In this study, we provide evidence of a third early BKV mRNA that codes for an additional early region product with an apparent molecular mass of 17–20 kDa. This truncated form of TAg (truncTAg) is expressed from an alternatively spliced mRNA that is derived from the excision of a second intron from the mRNA encoding TAg. The first 133 aa of truncTAg are identical to those of TAg but the additional splice results in translation from a different reading frame, adding three new amino acids before reaching a stop codon. TruncTAg is expressed in both BKV-transformed and lytically infected cells and it is found to be primarily localized to the nucleus. The function of BKV truncTAg is likely to be relevant to transformation, similar to the additional T antigens of simian virus 40, JC virus and mouse polyomavirus.
INTRODUCTION
BK virus (BKV) was first isolated in 1971 from the urine of an immunocompromised renal transplant patient (Gardner et al., 1971). It is a member of the polyomavirus family and is ubiquitous in the human population (Knowles et al., 2003). Following primary infection in early childhood, BKV persists in the kidneys and, upon immunosuppression of the human host, causes significant morbidity, particularly in bone marrow and renal transplant patients (Acott & Hirsch, 2007; Bohl & Brennan, 2007; Dropulic & Jones, 2008; Pavlakis et al., 2006). Polyomavirus nephropathy due to BKV reactivation is an emerging challenge in renal transplant recipients, in whom lytic replication can lead to destruction of the transplanted organ. BKV also has tumorigenic capabilities, which have been well demonstrated in experimental models (Tognon et al., 2003). It transforms rodent cells in culture, induces kidney tumours in transgenic mice and causes the immortalization of human cells either alone or in the presence of additional oncogenes such as c-Ha-ras or adenovirus E1A (reviewed by Imperiale & Major, 2007). Evidence in support of BKV as a potential cofactor in human cancers has been reported and others have observed the presence of BKV in cancerous prostates (Balis et al., 2007; Das et al., 2004, 2008; Fioriti et al., 2007; Lau et al., 2007; Zambrano et al., 2002). Understanding the complete life cycle of the virus and how it is regulated in these different clinical situations will be critical in the development of possible therapeutic interventions.
The BKV genome is divided into the early region, the late region and the non-coding control region, which has the origin of DNA replication as well as elements for transcriptional regulation of both the early and the late genes (Imperiale & Major, 2007). The early region encodes the large tumour (T) antigen (TAg) and the small tumour antigen (tAg), which are products of two alternatively spliced mRNAs and are the first proteins to be expressed during infection. The late region encodes the capsid proteins VP1, VP2 and VP3 and the agnoprotein, which are expressed after the onset of viral DNA replication. When BKV infects a cell, there are two possible outcomes. In a permissive host cell, early gene expression leads to DNA replication, which is followed by late gene expression, production of progeny viral particles and cell death. In a non-permissive cell, BKV lytic replication is blocked and abortive infection may result in oncogenic transformation through the uninterrupted expression of T antigens.
The ability of BKV to transform cells is a byproduct of its ability to deregulate cell growth control as a means to drive resting cells into S phase, in which viral DNA replication is maximized. The major transformation domains in TAg have been well defined for simian virus 40 (SV40) and, where they have been analysed, have also been found in BKV (Ahuja et al., 2005). These are: the N-terminal J domain, through which TAg interacts with the heat shock cognate 70 (Hsc70) co-chaperone protein (Campbell et al., 1997; Kelley & Georgopoulos, 1997); the N-terminal pRb binding domain, which contains a conserved LXCXE motif through which TAg associates with pRb and its family members, p107 and p130 (DeCaprio et al., 1988; Harris et al., 1996); and the C-terminal bipartite p53 binding region (Cavender et al., 1995; Kierstead & Tevethia, 1993; Li et al., 2003). Through its N terminus, TAg also interacts with two other proteins that are involved in cell growth control, the E3 ubiquitin ligase, CUL7 and the mitotic spindle checkpoint protein, Bub1 (Ali et al., 2004; Cotsiki et al., 2004; Kohrman & Imperiale, 1992).
In addition to expressing TAg and tAg, some polyomaviruses, including mouse polyomavirus (MPyV), SV40 and the human polyomavirus, JC virus (JCV), encode additional early mRNAs that are produced by secondary RNA splicing events on the TAg mRNA. MPyV produces two additional mRNAs. The first encodes middle T antigen, a membrane-associated protein that is identical to tAg at its N terminus and which stimulates cellular proliferation (Ichaso & Dilworth, 2001), and the second encodes tiny T antigen, which shares its N terminus with TAg but has six unique amino acids at its C terminus (Riley et al., 1997). Tiny T antigen is found in both the cytoplasm and the nucleus and it enhances the ATPase activity of Hsc70 through its J domain. SV40 encodes a protein called 17KT, in which the first 131 of its 135 aa are the same as the N-terminal 131 aa of TAg (Zerrahn et al., 1993). 17KT is reported to be a nuclear protein and is expressed at higher levels in transformed cells than in lytically infected cells. It has been demonstrated to have transforming activity in both rodent and human cells (Boyapati et al., 2003; Zerrahn et al., 1993). JCV encodes proteins known as T′135, T′136 and T′165, which are produced from three alternatively spliced early mRNAs (Trowbridge & Frisque, 1995). All three JCV T′ proteins share their N-terminal 132 aa with the N terminus of TAg. The 33 C-terminal aa of T′165 are also found in TAg but the other two T′ proteins have unique C-termini due to the use of a different open reading frame (ORF) after excision of the intron. The JCV T′ proteins are reported to be nuclear proteins and to have transformation potential (Bollag et al., 2000, 2006). The detection of a third early protein of BKV has also been reported, but it was assumed to be a proteolytic degradation product of TAg and therefore was not previously characterized (Bollag et al., 1989).
In various experiments in our laboratory, we noticed a faster migrating band in Western blot analyses of BKV TAg. In this study, we show that BKV does indeed encode a third early protein by alternative splicing of the BKV early pre-mRNA. We demonstrate that this protein, which we call truncated TAg (truncTAg), is expressed in both BKV-transformed and lytically infected cells. Similar to the shortened forms of TAg expressed by the other polyomaviruses, BKV truncTAg is found primarily in the nucleus. The expression of this BKV early protein and how it may relate to lytic infection and transformation are discussed in this report.
METHODS
Cells and virus.
BSC and BSC-BKT cells [BSC cells stably transfected with the early region of BKV (Dunlop strain)], were maintained as described previously (Harris et al., 1996). Vero cells (ATCC CCL-81) were maintained in Dulbecco's modified Eagle's medium (Gibco) containing 10 % fetal bovine serum (Hyclone), 100 U penicillin ml−1 and 100 μg streptomycin ml−1 (Cambrex). Primary human prostate epithelial cells (PrECs; Lonza) were maintained for up to five passages in prostate epithelial cell basal medium (Lonza) supplemented with bovine pituitary extract, hydrocortisone, human epidermal growth factor, epinephrine, transferrin, insulin, retinoic acid, triiodothyronine and GA-1000, obtained as PrEGM SingleQuots (Lonza). All cells were grown at 37 °C in a 5 % CO2 humidified incubator. For viral infection, PrECs were grown in 12-well plates to 50 % confluence and infected with BKV (TU strain) at an m.o.i. of 5 infectious units per cell. Cells were harvested 2 days post-infection (p.i.) and lysed in ice-cold Tris lysis buffer (50 mM Tris, pH 8.0, 120 mM NaCl, 0.5 % Igepal, 1 mM EGTA, 100 μg phenylmethylsulfonylfluoride ml−1, 50 μg aprotinin ml−1, 50 μg leupeptin ml−1 and 1 mM sodium orthovanadate). Primary human renal proximal tubule epithelial cells (RPTECs) were maintained and infected with BKV; protein lysates were prepared at 2 days p.i., as described previously (Abend et al., 2007).
Western blotting.
Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane using overnight wet transfer at 60 V at 4 °C. The membrane was subsequently blocked at room temperature for 1 h or overnight at 4 °C in 5 % non-fat dried milk in PBS containing 0.1 % Tween 20 (PBS-T). After blocking, the membrane was incubated with primary antibody (PAb416; 1 : 3000) in 5 % non-fat dried milk in PBS-T overnight at 4 °C. The membrane was then washed with PBS-T and incubated with horseradish peroxidase-conjugated sheep anti-mouse IgG antibody (Amersham) in 5 % non-fat dried milk in PBS-T for 1 h at room temperature. The membrane was again washed with PBS-T and developed using either the ECL Plus reagent (Amersham) or Luminol (Millipore).
RT-PCR.
Total cellular RNA was isolated from BSC-BKT cells using an RNeasy kit (Qiagen) according to the manufacturer's instructions. Reverse transcription was performed using the iScript cDNA Synthesis kit (Bio-Rad) with 1 μg RNA according to the manufacturer's instructions or the Sensiscript RT kit (Qiagen) with 50 ng of RNA and 1 μM random hexamer primers (BD Biosciences) according to the manufacturer's instructions. Primers A and F (Supplementary Table S1, available in JGV Online) were used to amplify full-length mRNAs and subsequently clone truncTAg. PCR preparations (50 μl total volume) contained 3 μl cDNA template, 1 μM each primer, 1 mM dNTPs, 2 mM MgSO4 and 1 U Platinum Taq High Fidelity polymerase (Invitrogen). The PCR programme consisted of initial denaturation for 2 min at 95 °C followed by 31 cycles of denaturation at 95 °C for 45 s, annealing at 55 °C for 1 min and elongation at 68 °C for 2 min 40 s, followed by a final elongation step at 68 °C for 7 min.
For amplifications using primers B, C, D and E, PCR preparations contained 200 nM each primer, 200 μM dNTPs and 1 U Titanium Taq DNA polymerase (Clontech). The PCR programme consisted of initial denaturation for 5 min at 94 °C followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s and elongation at 72 °C for 1 min, followed by a final elongation step at 72 °C for 7 min.
Cloning of RT-PCR products and sequence analysis.
The pGEM-T Easy cloning kit (Promega) was used to clone the full-length RT-PCR products. Plasmid DNA was purified using the QIAprep Spin Miniprep kit (Qiagen). Clones were screened by digestion with either NotI or EcoRI (NEB) and plasmids containing inserts were sequenced by the DNA Sequencing Core at the University of Michigan. Sequences were analysed using lasergene software from dnastar. For expression studies, the truncTAg cDNA was excised from the pGEM-T clone using EcoRI (NEB) and cloned into the EcoRI site of pcDNA3.1(+), resulting in plasmid pcDNA3.1(+)-truncTAg. Proper orientation of the insert was confirmed by restriction mapping.
To visualize the location of truncTAg in the presence of TAg, an N-terminal FLAG-tagged version of truncTAg was created. The full-length truncTAg cDNA was excised from pcDNA3.1(+) by digestion with NheI and BamHI, followed by BfaI to obtain a 56 bp fragment containing the 5′ UTR and a 772 bp fragment containing the ORF and 3′ UTR. The FLAG tag was inserted after the fourth codon of the truncTAg cDNA using the 772 bp BfaI/BamHI fragment as a template in a PCR consisting of 1 μM primers FLAGT and F (Supplementary Table S1), 1 mM dNTPs, 2 mM MgSO4 and 1 U Platinum Taq High Fidelity polymerase. The PCR programme consisted of initial denaturation for 2 min at 95 °C followed by 30 cycles of denaturation at 95 °C for 45 s, annealing at 58 °C for 1 min and elongation at 68 °C for 1 min. The PCR product was purified using the QIAquick PCR Purification kit (Qiagen) and then used as template along with the 56 bp 5′ UTR fragment from the NheI/BfaI digestion in an overlapping PCR, consisting of 1 μM primers A and F (Supplementary Table S1), 1 mM dNTPs, 2 mM MgSO4 and 1 U Platinum Taq High Fidelity polymerase. The PCR programme consisted of initial denaturation for 2 min at 95 °C followed by 30 cycles of denaturation at 95 °C for 45 s, annealing at 53 °C for 1 min and elongation at 68 °C for 1 min. The product was first ligated into the pGEM-T Easy plasmid and then moved to pcDNA3.1(+) as described above, resulting in plasmid pcDNA3.1(+)-FLAGtruncTAg. Sequence and orientation were confirmed by the DNA Sequencing Core at the University of Michigan.
Transfection.
Transfections were performed with BSC, BSC-BKT and Vero cells using Effectene reagents (Qiagen). Transfection complexes were prepared according to the manufacturer's instructions with a DNA to Effectene reagent ratio of 1 : 25 (BSC), 1 : 15 (BSC-BKT) or 1 : 20 (Vero). BSC cells were seeded into a 12-well plate from a 70 % confluent 10 cm dish immediately prior to transfection and each well was transfected with 0.6 μg pcDNA3.1(+)-truncTAg, pcDNA3.1(+)-FLAGtruncTAg or pcDNA3.1(+)-TAg, which contains the cDNA encoding BKV TAg. Whole-cell lysates were prepared 2 days post-transfection (p.t.) for Western blot analysis. BSC-BKT and Vero cells were transfected with 0.6 μg pcDNA3.1(+)-FLAGtruncTAg in two-well chamber slides (Nunc-LabTek) and analysed for localization of truncTAg by an immunfluorescence assay at 2 days p.t.
Immunofluorescence assay.
Transfected cells were fixed 2 days p.t. with 4 % paraformaldehyde in PBS for 20 min and then washed three times with PBS. Antigen retrieval was carried out using 0.1 % Triton X-100 in PBS for 5 min, followed by two washes with PBS. Slides were blocked in PBS containing 5 % goat serum for 30 min at room temperature and then incubated with 1 : 1000 monoclonal anti-FLAG M2 antibody (Sigma, catalogue no. F1804) in PBS containing 5 % goat serum overnight at room temperature in a humidified box. Slides were then washed twice in PBS and incubated with 1 : 500 fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG antibody (Sigma) in PBS containing 5 % goat serum. Slides were washed twice in PBS, air-dried for 15 min and mounted with Vectashield with DAPI (Vector Laboratories). Representative pictures were taken using an Olympus BX41 microscope with a Plan ×20/0.65 objective.
RESULTS
Expression of truncTAg in BKV-transformed BSC cells and lytically infected primary human epithelial cells
We have observed bands that migrated at an apparent molecular mass of 17–20 kDa in Western blots of protein lysates from BKV-transformed and infected cells that were probed with PAb416, a mouse monoclonal antibody that was originally raised against SV40 TAg (Harlow et al., 1981). This antibody recognizes an epitope contained within aa 82–130 of SV40 TAg (Arthur et al., 1988) and cross-reacts with BKV TAg (Harris et al., 1996). A representative Western blot of whole-cell lysates from BKV-transformed and BKV lytically infected cells probed with PAb416 is shown in Fig. 1. In addition to TAg, we detected two proteins in BSC-BKT cells with apparent molecular masses of 17–20 kDa. When lysates from primary PrEC and RPTE cells infected with BKV were analysed, similar proteins were observed, along with an additional, more slowly migrating polypeptide. Since these polypeptides were present only in cells expressing the BKV early region and were recognized by the anti-TAg antibody, we are referring to them collectively as truncTAg.
Fig. 1.

Detection of truncTAg in whole-cell lysates. Aliquots of total cell protein (30 μg) from BSC and BSC-BKT, mock- or BKV-infected PrECs, or mock- or BKV-infected RPTECs were separated on an 8 % SDS-PAGE gel, transferred to a membrane by Western blot and probed with PAb416. Protein size markers (Magic Mark XP, Invitrogen) are indicated (kDa).
Expression of a third BKV early mRNA
There are two possible explanations for the appearance of truncTAg: proteolytic degradation of TAg or alternative splicing of early BKV pre-mRNA. Given the precedent of the existence of alternatively spliced mRNAs in other polyomaviruses, we hypothesized that BKV truncTAg is also a product of such an alternative splicing event. Total cell RNA was extracted from BSC-BKT cells or BKV-infected RPTE cells and RT-PCR was performed with several sets of primers (Supplementary Table S1) designed to span the splice junction sites of TAg and tAg and to survey the BKV early region. The RT-PCR products of primers A and F, using BSC-BKT RNA as a template, are shown in Fig. 2, and similar results were obtained by using RNA from lytically infected RPTE cells (data not shown). In addition to the predicted amplification products for tAg mRNA (2537 bp) and TAg mRNA (2259 bp), two prominent products of approximately 825 and 1200 bp were observed. The greater intensities of these two bands are likely to be the result of more efficient amplification due to the small size of the amplicons, rather than being indicative of the abundance of these transcripts. When primers B, C, D and E were used to amplify cDNA from BSC-BKT cells, products were obtained that were consistent with the existence of alternatively spliced mRNAs (Table 1). The RT-PCR products from three of the reactions were cloned and multiple clones were sequenced.
Fig. 2.
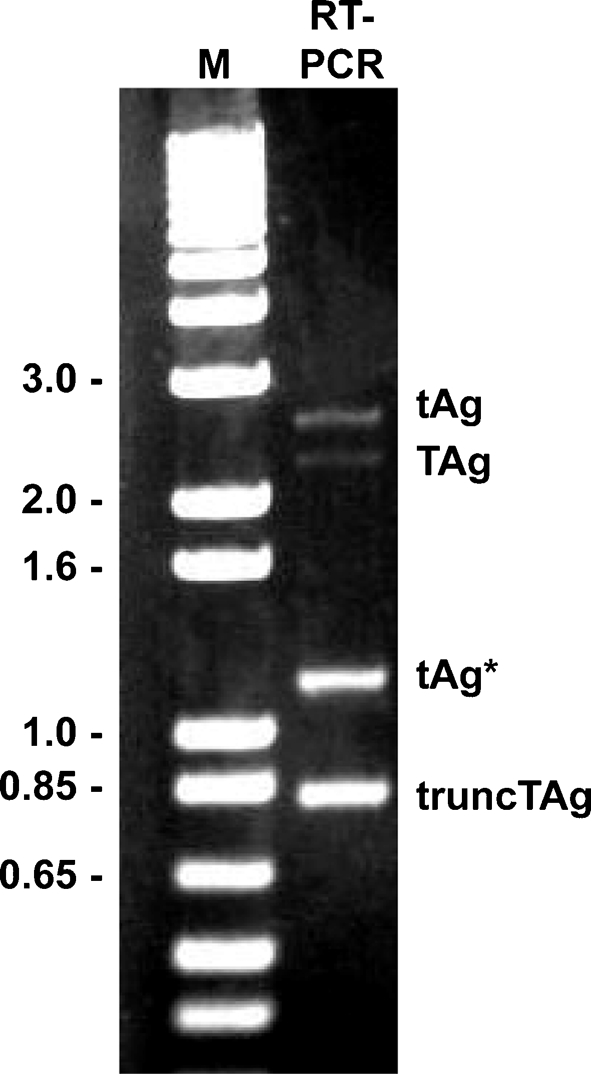
RT-PCR analysis of BKV early region RNA. RNA was isolated from BSC-BKT cells and used as template in RT-PCR analysis with primers A and F (Supplementary Table S1). The products were analysed on a 0.8 % agarose gel. M, 1 kb Plus Ladder marker (kb; Invitrogen).
Table 1.
RT-PCR products of BKV early region RNA
| Primers | Products (bp) | |||||
|---|---|---|---|---|---|---|
| Dunlop* | TU† | |||||
| TAg | tAg | truncTAg | TAg | tAg | truncTAg | |
| A and F | 2259 | 2537 | 810 | 2266 | 2543 | 818 |
| B and E | 1965 | 2243 | 516 | |||
| C and D | 394 | 672 | NP‡ | |||
| C and E | 1793 | 2071 | 344 | |||
The results of the sequence analysis suggest that the smallest band represents an alternatively spliced BKV early mRNA that is 810 (Dunlop) or 818 (TU) nt in length. The transcript contains a single ORF that would encode a protein with a theoretical molecular mass of 16 098 Da, which is consistent with the electrophoretic mobility of truncTAg. This mRNA is generated by the excision of two introns, one of which is also excised to produce the TAg mRNA and the second of which utilizes additional splice donor and acceptor sites (Fig. 3). The sequences flanking these donor and acceptor sites (nt 4413–4405 and 2977–2962, respectively) fit the consensus sequences for splice sites (Burset et al., 2001; Mount, 1982). The protein encoded from this alternatively spliced transcript is predicted to be identical to BKV TAg up to a lysine residue at position 133. By analogy to SV40 TAg, a presumed nuclear localization signal (NLS; PKKKRKV; Kalderon et al., 1984) is also present in this region of BKV TAg. Thus, truncTAg, like SV40 17KT, contains the entire NLS except for the final valine residue (Fig. 3b). Excision of the second intron causes translation in a different ORF after the splice, resulting in three additional unique codons followed by a stop codon (Fig. 3b). The additional 1200 bp band (tAg*) is found to be derived by splicing of only this C-terminal intron and is therefore predicted to encode tAg (Fig. 3a).
Fig. 3.

BKV early mRNA structures and truncTAg intron analysis. (a) Diagram of the four early region mRNAs produced by BKV. Boxes indicate exons and lines indicate introns. The unspliced transcript is shown, with the locations of the annealing sites for the primers (Supplementary Table S1) indicated by horizontal arrows. Vertical arrows indicate the positions of the start and stop codons for each transcript. (b) Diagram showing consensus splice donor and acceptor sequences, the DNA sequences of the splice junctions for the truncTAg second intron and the predicted codons adjoining the splice sites. The predicted codons for SV40 17KT are included for comparison.
To prove that this alternatively spliced mRNA does indeed encode the protein we detected in the Western blots, we cloned a full-length cDNA of the proposed truncTAg into pcDNA3.1(+), transfected it into BSC cells and analysed the protein lysates (Fig. 4). The protein species expressed from the cDNA migrated identically to truncTAg produced during BKV infection of RPTE cells (RPTE-BKV) and from a cDNA expression vector for TAg; the TAg mRNA can be further spliced to remove the second intron and produce the truncTAg transcript. We were, however, only able to detect the two fastest migrating bands after transfection of truncTAg cDNA, whereas three distinct truncTAg species were produced in the presence of TAg. Similarly, transfection of the FLAG-tagged truncTAg expression plasmid also produced only two truncTAg bands which, as expected, migrated more slowly than the untagged truncTAg species.
Fig. 4.

Expression of truncTAg from the cDNA clone. BSC cells were transfected with pcDNA3.1(+)-TAg, pcDNA3.1(+)-truncTAg or pcDNA3.1(+)-FLAGtruncTAg, and whole-cell lysates were prepared at 2 days p.t. Aliquots of total cell protein (30 μg) were separated on a 10 % SDS-PAGE gel and analysed by Western blot, probing with PAb416. *, A non-specific band of approximately 70 kDa; No Tfxn, untransfected cells; RPTE-BKV, BKV-infected RPTE whole-cell lysates.
Subcellular localization of truncTAg
Given the proximity of the NLS to the C terminus of truncTAg, we wished to determine its subcellular localization. Subcellular fractionation was not useful for this analysis, because manipulations during fractionation can cause leakage of small nuclear proteins into the cytoplasm. We instead used an immunofluorescence assay to directly observe the location of truncTAg in transfected cells. Due to the ability of PAb416 to recognize both TAg and truncTAg, it was necessary to tag truncTAg in order to observe the behaviour of the protein both in the absence and presence of other early viral proteins. We cloned the sequence encoding the FLAG epitope after the fourth codon of the truncTAg cDNA and obtained a plasmid expressing an N-terminal FLAG-tagged truncTAg. Transfection of Vero cells with pcDNA3.1(+)-FLAGtruncTAg (Fig. 5) or pcDNA3.1(+)-truncTAg (data not shown) resulted in identical nuclear localization of truncTAg. To determine whether the presence of TAg or tAg would affect the localization of truncTAg, we transfected BSC-BKT cells, which express the entire early region of BKV, with the FLAG-tagged truncTAg (Fig. 5). TruncTAg remained completely localized to the nucleus, suggesting that it has a functional NLS and that its location is not altered in the presence of other viral proteins.
Fig. 5.

TruncTAg is localized to the nucleus. Vero (top panels) or BSC-BKT (bottom panels) cells were transfected with pcDNA3.1(+)-FLAGtruncTAg and analysed for localization of FLAG-tagged truncTAg by immunofluorescence, probing with an anti-FLAG epitope antibody as described in Methods. Representative fields of cells are shown at ×200 magnification. Anti-FLAG panels (left) are pseudocoloured in red; areas of colocalization with DAPI appear pink in the merged images (right).
DISCUSSION
In this study, we present evidence for the expression of a third BKV early protein, truncTAg, in both BKV-transformed and lytically infected cells. The existence of this form of BKV TAg was first reported by Bollag et al. (1989) but was presumed to be a proteolytic degradation product of TAg. We show that truncTAg is encoded by the alternative splicing of the BKV early pre-mRNA. It is produced by the removal of two introns, one of which is also removed to generate the mRNA for TAg and the second of which uses downstream splice donor and acceptor sites that are well-conserved among BKV strains. Sequence analysis of RT-PCR products from three different sets of primers confirmed the excision of this second intron; the fact that this intron is also removed to produce a second tAg-expressing mRNA (tAg*) supports the conclusion that it is authentic. Additionally, we demonstrate that a full-length cloned cDNA encodes a protein identical to that observed in BKV-transformed and BKV-infected lysates. Finally, we show that the protein localizes to the nucleus of cells both in the absence and presence of TAg and tAg.
BKV truncTAg consists of 136 aa and is collinear with the first 133 aa of the N terminus of TAg. This part of TAg contains the J domain and the binding region for pRb family proteins. By homology to SV40, BKV truncTAg also contains the binding domains for the E3 ubiquitin ligase, CUL7, and the mitotic spindle checkpoint protein, Bub1 (Ali et al., 2004; Cotsiki et al., 2004). We therefore predict that truncTAg would be able to deregulate cell growth during infection. Additionally, the N terminus of BKV TAg contains potential phosphorylation sites that are also present in truncTAg. The occurrence of these phosphorylation sites in the N terminus of JCV and SV40 TAg and their involvement in multiple regulatory functions has been demonstrated (Deppert et al., 1991; Scheidtmann et al., 1991; Swenson & Frisque, 1995). Phosphorylation may account for the multiple truncTAg bands we have detected in both transformed and infected lysates. It is possible that expression of TAg is required for post-translational modification of truncTAg, since expression of truncTAg alone results in only the two faster migrating bands, while all three bands are produced in BSC-BKT cells, after transfection with a TAg cDNA and in a viral infection.
Other polyomaviruses also express shortened early proteins similar to truncTAg. In MPyV, a truncated TAg called tiny T antigen is known to stimulate the ATPase activity of Hsc70 through its DnaJ motif (Riley et al., 1997). In SV40, the smaller form of TAg is known as the 17KT protein (Zerrahn et al., 1993). Expression of 17KT in rat F111 fibroblasts induces transformation of these cells (Zerrahn et al., 1993) and 17KT can complement J domain mutations in SV40 TAg to promote transformation of human fibroblasts, reduce p130 levels and stimulate entry into S phase by inducing the release of E2F (Boyapati et al., 2003). The SV40 17KT can also induce premature senescence through its ability to bind Bub1 (Gjoerup et al., 2007). Of the three JCV truncated TAg proteins, T′135, T′136, and T′165, T′136 is the closest in structure to BKV truncTAg (Bollag et al., 2000; Trowbridge & Frisque, 1995). T′136 is the major T′ product in JCV transformed cells, binds to both p107 and p130, and alters the phosphorylation status of both proteins (Bollag et al., 2006). Additionally, both 17KT and the JCV T′ proteins promote the degradation of pRb family proteins and have the ability to cooperate with a mutant Ras protein to immortalize rat embryo fibroblasts (Bollag et al., 2006; Boyapati et al., 2003). The JCV T′ proteins have also been shown to be involved in efficient viral DNA replication (Prins & Frisque, 2001). We predict that BKV truncTAg will have functions relating to transformation that are comparable to those of the other shortened T antigens, since they have similar protein domains. In addition, we are uniquely positioned to study the role of truncTAg during lytic infection in a primary cell culture system, which would allow us to gain insight into the role of these proteins in pathogenesis. Studies involving characterization of mutant viruses that do not express truncTAg are in progress. The transformation potential of truncTAg in primary PrECs will also be of interest.
In conclusion, we have provided evidence that BKV encodes a third early mRNA that is translated to produce a truncated form of TAg that localizes to the nucleus. The function of this protein during lytic infection and transformation remains to be determined. Expression of truncated T antigen proteins is conserved over the polyomavirus family, thus suggesting that these proteins play an important role in pathogenesis.
Supplementary Material
Acknowledgments
We would like to thank the members of the Imperiale lab for assistance and critical reading of the manuscript and Angela Hopkinson for technical assistance. This work was supported by a postdoctoral fellowship from the US Army Medical Research and Materiel Command, Department of Defense to D. D. (W81xWH-06-1-0132), in part through the University of Michigan's Cancer Center Support Grant (P30 CA46592) and by research grants to M. J. I. from the NIH (CA118970 and AI060584). J. R. A. was supported in part by the Frederick G. Novy Fellowship.
Footnotes
A supplementary table of primers used for RT-PCR is available with the online version of this paper.
References
- Abend, J. R., Low, J. A. & Imperiale, M. J. (2007). Inhibitory effect of gamma interferon on BK virus gene expression and replication. J Virol 81, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acott, P. D. & Hirsch, H. H. (2007). BK virus infection, replication and diseases in pediatric kidney transplantation. Pediatr Nephrol 22, 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja, D., Sáenz-Robles, M. T. & Pipas, J. M. (2005). SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24, 7729–7745. [DOI] [PubMed] [Google Scholar]
- Ali, S. H., Kasper, J. S., Arai, T. & DeCaprio, J. A. (2004). Cul7/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J Virol 78, 2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur, A. K., Hoss, A. & Fanning, E. (1988). Expression of simian virus 40 T antigen in Escherichia coli: localization of T-antigen origin DNA-binding domain to within 129 amino acids. J Virol 62, 1999–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balis, V., Sourvinos, G., Soulitzis, N., Giannikaki, E., Sofras, F. & Spandidos, D. A. (2007). Prevalence of BK virus and human papillomavirus in human prostate cancer. Int J Biol Markers 22, 245–251. [DOI] [PubMed] [Google Scholar]
- Bohl, D. L. & Brennan, D. C. (2007). BK virus nephropathy and kidney transplantation. Clin J Am Soc Nephrol 2 (Suppl 1), S36–S46. [DOI] [PubMed] [Google Scholar]
- Bollag, B., Chuke, W. F. & Frisque, R. J. (1989). Hybrid genomes of the polyomaviruses JC virus, BK virus and simian virus 40: identification of sequences important for efficient transformation. J Virol 63, 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag, B., Prins, C., Snyder, E. L. & Frisque, R. J. (2000). Purified JC virus T and T′ proteins differentially interact with the retinoblastoma family of tumor suppressor proteins. Virology 274, 165–178. [DOI] [PubMed] [Google Scholar]
- Bollag, B., Kilpatrick, L. H., Tyagarajan, S. K., Tevethia, M. J. & Frisque, R. J. (2006). JC virus T'135, T'136 and T′165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol 12, 428–442. [DOI] [PubMed] [Google Scholar]
- Boyapati, A., Wilson, M., Yu, J. & Rundell, K. (2003). SV40 17KT antigen complements DnaJ Mutations in large T antigen to restore transformation of primary human fibroblasts. Virology 315, 148–158. [DOI] [PubMed] [Google Scholar]
- Burset, M., Seledtsov, I. A. & Solovyev, V. V. (2001). Splice DB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res 29, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, K. S., Mullane, K. P., Aksoy, I. A., Stubdal, H., Zalvide, J., Pipas, J. M., Silver, P. A., Roberts, T. M., Schaffhausen, B. S. & DeCaprio, J. A. (1997). DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev 11, 1098–1110. [DOI] [PubMed] [Google Scholar]
- Cavender, J. F., Conn, A., Epler, M., Lacko, H. & Tevethia, M. J. (1995). Simian virus 40 large T antigen contains two independent activities that cooperate with a ras oncogene to transform rat embryo fibroblasts. J Virol 69, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsiki, M., Lock, R. L., Cheng, Y., Williams, G. L., Zhao, J., Perera, D., Freire, R., Entwistle, A., Golemis, E. A. & other authors (2004). Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci U S A 101, 947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, D., Shah, R. B. & Imperiale, M. J. (2004). Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene 23, 7031–7046. [DOI] [PubMed] [Google Scholar]
- Das, D., Wojno, K. & Imperiale, M. J. (2008). BK virus as a cofactor in the etiology of prostate cancer in its early stages. J Virol 82, 2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio, J. A., Ludlow, J. W., Figge, J., Shew, J. Y., Huang, C. M., Lee, W. H., Marsilio, E., Paucha, E. & Livingston, D. M. (1988). SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54, 275–283. [DOI] [PubMed] [Google Scholar]
- Deppert, W., Kurth, M., Graessmann, M., Graessmann, A. & Knippschild, U. (1991). Altered phosphorylation at specific sites confers a mutant phenotype to SV40 wild-type large T antigen in a flat revertant of SV40-transformed cells. Oncogene 6, 1931–1938. [PubMed] [Google Scholar]
- Dropulic, L. K. & Jones, R. J. (2008). Polyomavirus BK infection in blood and marrow transplant recipients. Bone Marrow Transplant 41, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioriti, D., Russo, G., Mischitelli, M., Anzivino, E., Bellizzi, A., Di Monaco, F., Di Silverio, F., Giordano, A., Chiarini, F. & Pietropaolo, V. (2007). A case of human polyomavirus BK infection in a patient affected by late stage prostate cancer: could viral infection be correlated with cancer progression? Int J Immunopathol Pharmacol 20, 405–411. [DOI] [PubMed] [Google Scholar]
- Gardner, S. D., Field, A. M., Coleman, D. V. & Hulme, B. (1971). New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1, 1253–1257. [DOI] [PubMed] [Google Scholar]
- Gjoerup, O. V., Wu, J., Chandler-Militello, D., Williams, G. L., Zhao, J., Schaffhausen, B., Jat, P. S. & Roberts, T. M. (2007). Surveillance mechanism linking Bub1 loss to the p53 pathway. Proc Natl Acad Sci U S A 104, 8334–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., Crawford, L. V., Pim, D. C. & Williamson, N. M. (1981). Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol 39, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, K. F., Christensen, J. B. & Imperiale, M. J. (1996). BK virus large T antigen: interactions with the retinoblastoma family of tumor suppressor proteins and effects on cellular growth control. J Virol 70, 2378–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichaso, N. & Dilworth, S. M. (2001). Cell transformation by the middle T-antigen of polyoma virus. Oncogene 20, 7908–7916. [DOI] [PubMed] [Google Scholar]
- Imperiale, M. J. & Major, E. O. (2007). Polyomaviruses. In Fields Virology, 5th edn, pp. 2263–2298. Edited by D. M. Knipe & P. M. Howley. Philadelphia: Lippincott, Williams & Wilkins.
- Kalderon, D., Roberts, B. L., Richardson, W. D. & Smith, A. E. (1984). A short amino acid sequence able to specify nuclear location. Cell 39, 499–509. [DOI] [PubMed] [Google Scholar]
- Kelley, W. L. & Georgopoulos, C. (1997). The T/t common exon of simian virus 40, JC, and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc Natl Acad Sci U S A 94, 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierstead, T. D. & Tevethia, M. J. (1993). Association of p53 binding and immortalization of primary C57BL/6 mouse embryo fibroblasts by using simian virus 40 T-antigen mutants bearing internal overlapping deletion mutations. J Virol 67, 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, W. A., Pipkin, P., Andrews, N., Vyse, A., Minor, P., Brown, D. W. & Miller, E. (2003). Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol 71, 115–123. [DOI] [PubMed] [Google Scholar]
- Kohrman, D. C. & Imperiale, M. J. (1992). Simian virus 40 large T antigen stably complexes with a 185-kilodalton host protein. J Virol 66, 1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K., Lacey, S. F., Chen, Y. Y., Chen, W. G. & Weiss, L. M. (2007). Low frequency of BK virus in prostatic adenocarcinomas. APMIS 115, 743–749. [DOI] [PubMed] [Google Scholar]
- Li, D., Zhao, R., Lilyestrom, W., Gai, D., Zhang, R., DeCaprio, J. A., Fanning, E., Jochimiak, A., Szakonyi, G. & Chen, X. S. (2003). Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 423, 512–518. [DOI] [PubMed] [Google Scholar]
- Mount, S. M. (1982). A catalogue of splice junction sequences. Nucleic Acids Res 10, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis, M., Haririan, A. & Klassen, D. K. (2006). BK virus infection after non-renal transplantation. Adv Exp Med Biol 577, 185–189. [DOI] [PubMed] [Google Scholar]
- Prins, C. & Frisque, R. J. (2001). JC virus T′ proteins encoded by alternatively spliced early mRNAs enhance T antigen-mediated viral DNA replication in human cells. J Neurovirol 7, 250–264. [DOI] [PubMed] [Google Scholar]
- Riley, M. I., Yoo, W., Mda, N. Y. & Folk, W. R. (1997). Tiny T antigen: an autonomous polyomavirus T antigen amino-terminal domain. J Virol 71, 6068–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidtmann, K. H., Buck, M., Schneider, J., Kalderon, D., Fanning, E. & Smith, A. E. (1991). Biochemical characterization of phosphorylation site mutants of simian virus 40 large T antigen: evidence for interaction between amino- and carboxy-terminal domains. J Virol 65, 1479–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson, J. J. & Frisque, R. J. (1995). Biochemical characterization and localization of JC virus large T antigen phosphorylation domains. Virology 212, 295–308. [DOI] [PubMed] [Google Scholar]
- Tognon, M., Corallini, A., Martini, F., Negrini, M. & Barbanti-Brodano, G. (2003). Oncogenic transformation by BK virus and association with human tumors. Oncogene 22, 5192–5200. [DOI] [PubMed] [Google Scholar]
- Trowbridge, P. W. & Frisque, R. J. (1995). Identification of three new JC virus proteins generated by alternative splicing of the early viral mRNA. J Neurovirol 1, 195–206. [DOI] [PubMed] [Google Scholar]
- Zambrano, A., Kalantari, M., Simoneau, A., Jensen, J. L. & Villarreal, L. P. (2002). Detection of human polyomaviruses and papillomaviruses in prostatic tissue reveals the prostate as a habitat for multiple viral infections. Prostate 53, 263–276. [DOI] [PubMed] [Google Scholar]
- Zerrahn, J., Knippschild, U., Winkler, T. & Deppert, W. (1993). Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J 12, 4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.