

Abstract
Acute pancreatitis (AP) is a common disease, which usually exists in its mild form. However, in a fifth of cases, the disease is severe, with local pancreatic complications or systemic organ dysfunction or both. Because the development of organ failure is the major cause of death in AP, early identification of patients likely to develop organ failure is important. AP is initiated by intracellular activation of pancreatic proenzymes and autodigestion of the pancreas. Destruction of the pancreatic parenchyma first induces an inflammatory reaction locally, but may lead to overwhelming systemic production of inflammatory mediators and early organ failure. Concomitantly, anti-inflammatory cytokines and specific cytokine inhibitors are produced. This anti-inflammatory reaction may overcompensate and inhibit the immune response, rendering the host at risk of systemic infection. At present, there is no specific treatment for AP. Increased understanding of the pathogenesis of systemic inflammation and development of organ dysfunction may provide us with drugs to ameliorate physiological disturbances.
Keywords: Acute pancreatitis, Organ failure, Inflammatory response, Immunosuppression, Coagulation
INTRODUCTION
Acute pancreatitis (AP) is a common disease though its incidence varies considerably in different countries from 5 to more than 100 per 100 000 inhabitants/year[1-5]. AP is a disease with wide clinical variation. Most of the patients suffer from mild disease but in about 20% of the cases the patient develops a severe form of the disease, with a mortality of 7%-15%[1,6,7]. The major cause of death is organ failure which complicates severe AP in 20%-80% of the cases[8,9].
AP has many distinct etiologies, though approximately 80% of all cases are caused by either gallstones or alcohol[10]. The frequency of different etiologies varies markedly in different countries. In Finland, alcohol is the causative factor in about 70% of the cases and the incidence of AP correlates with the rate of alcohol consumption[11].
Prognosis of severe AP has improved mainly because of efficacious conservative treatment in intensive care units. Also the correct timing of surgery is important but is indicated only for infected organized pancreatic necrosis when percutaneous drainage techniques are ineffective[12]. In recent years, many minimally invasive techniques, for example endoscopic and laparoscopic approaches, have been introduced to avoid the need for open necrosectomy[13]. However, in severe AP the incidence of intra-abdominal hypertension is 60%-80% and in some cases necessitates surgical decompression in the early phase of the disease[14]. The use of antimicrobial drug therapy does not protect against infected necrosis or reduce mortality in severe AP[15]. A factor which may affect the outcome is early endoscopic retrograde cholangiopancreatography (ERCP) in biliary AP with biliary obstruction[16]. Thus, despite extensive research during the recent decades, no mode of treatment has been developed that could dramatically improve the outcome of severe AP.
INFLAMMATORY RESPONSE IS SIMILAR IN AP AND SEPSIS
Previously, AP was considered to be a disease of the pancreas. Nowadays, however, there is strong evidence for systemic effects of the disease. The mechanisms of systemic inflammation resulting in organ failures are much alike in severe AP and in, for example, severe burn injury, multitrauma or sepsis[17] (Figure 1). Thus, a considerable amount of research data gained, for example, in sepsis studies could be applicable also in severe AP.
Figure 1.
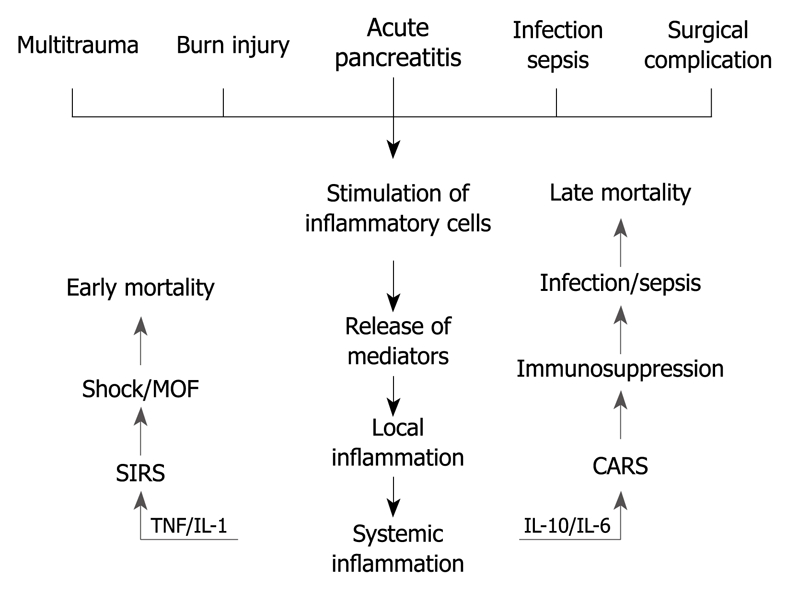
Two-phase hypothesis of development of multiorgan failure. MOF: Multiple organ failure; SIRS: Systemic inflammatory response syndrome; TNF: Tumor necrosis factor; IL: Interleukin; CARS: Compensatory anti-inflammatory response syndrome.
The initial phase of AP involves triggering events that lead to premature activation of pancreatic proteases as a result of intracellular co-localization of the digestive and lysosomal enzymes. This leads to disruption of the acinar cells[18-20]. Then, activated enzymes escape into the interstitium of the pancreas and cause pancreatic autodigestion[21,22]. Furthermore, pancreatic damage is followed by activation of local inflammatory cells and various inflammatory mediators (cytokines) (Figure 1). Localized inflammation is the body’s initial physiologic protective response, which is generally strictly controlled at the site of injury. Loss of the local control results in excessive uncontrolled activation of inflammatory cells and mediators. This response is defined as systemic inflammatory response syndrome[23]. Proinflammatory cytokines, tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) are released via portal vein and lymph fluid drainage to the circulation[24]. In addition, the production of cytokines enhances proinflammatory signals. IL-6 stimulates the synthesis of acute phase proteins (i.e. C-reactive protein and procalcitonin) in the liver[25]. Proinflammatory cytokines activate the vascular endothelium in the whole body, which leads to enhanced leakage of the capillary veins and triggers migration of leukocytes into tissues. promoting activation of coagulation cascades. Circulating neutrophils and monocytes become activated, as evidenced by the increased expression of adhesion molecules (e.g. CD11b), and release their proteolytic enzymes and oxygen radicals, which damage vascular endothelial cells and organ parenchymal cells[26]. Enhanced amounts of tissue fluid together with impaired microcirculation lead to lack of oxygen, which results in dysfunction of vital organs and, ultimately, in organ failure[27,28]. While lungs and kidneys have a wide capillary bed, they are especially susceptible to phlogistic mediators inducing end organ injury.
ANTI-INFLAMMATORY RESPONSE
Systemic inflammation in AP is concomitantly associated with rapidly-strengthening compensatory anti-inflammatory response syndrome (CARS)[29,30]. An anti-inflammatory response may be sufficient to control the systemic inflammatory reaction, a sign of good prognosis. However, CARS may be excessive leading to immune deficiency or suppression, which renders the host susceptible to secondary infections. In AP, an increased number of infections has been observed in a later stage of the disease[31], i.e. at the phase of immune suppression.
In immunosuppression, monocytes are characterized by a markedly reduced human leukocyte antigen-DR (HLA-DR) expression, and a profound reduction of their ability to produce proinflammatory cytokines, for example TNF-α[32,33]. Monocytes with low HLA-DR density show impaired antigen presentation capacity[34,35]. IL-10, the most potent anti-inflammatory cytokine, is responsible for the decreased monocyte HLA-DR expression[36]. IL-1 receptor antagonist (IL-1ra) and IL-6 are also important anti-inflammatory cytokines. IL-1ra binds competitively to the IL-1 receptor and blocks IL-1 mediated responses[37]. Previously, IL-6 was regarded as a proinflammatory cytokine. However, some years ago it was shown to act predominantly as an anti-inflammatory cytokine and, for example prevents synthesis of IL-1β and TNF-α[38].
Altered cellular immune function in AP was first reported by García-Sabrido et al[39] in 1989 when they showed a correlation between poor outcome and anergy to delayed-type hypersensitivity testing. In addition to proinflammatory mediators[40,41], the IL-10 plasma level has also been shown to be increased in severe AP[42] and predicts development of organ failure in the very early phase of AP[43]. In AP patients, decreased monocyte HLA-DR expression is associated with development of secondary infections[30]. Decreased monocyte HLA-DR expression has also been shown to predict development of organ failure[44] and a fatal outcome[45]. In AP patients, high plasma levels of IL-10 and IL-6 correlate with decreased monocyte HLA-DR expression[30]. IL-10 and IL-6 can be measured by a chemiluminescent immunoassay that could be available on a 24-h basis at the hospital emergency unit. Flow cytometric measurement of monocyte HLA-DR expression can be used as a routine clinical test and provides results within 30-60 min of sampling. In addition to monocyte functions, there are changes also in lymphocyte count and proportion of T-helper cells that can be measured by flow cytometry in severe AP[46].
DEVELOPMENT OF ORGAN FAILURE WORSENS PROGNOSIS
The classification of AP severity is based on a consensus conference held in Atlanta in 1992[47]. According to the Atlanta classification, AP is classified as severe if systemic and/or local complications (for example pancreatic necrosis, pseudocyst or abscess) are present. Otherwise, AP is classified as mild. Organ failure occurs in half of patients with pancreatic necrosis[48].
The most common organ failure in severe AP is respiratory failure. Also other organs may fail, e.g. renal, hepatic, cardiovascular, digestive, neurologic, coagulation[17] or endocrine or immunologic systems[49]. Failure of different organs has different effects on the prognosis of the disease. Mortality rates in severe AP patients with respiratory, renal, and hepatic failure are 43%, 63%, and 83%, respectively[50]. Lately, the Atlanta classification has been criticized because it does not separate organ failure from local complications[51]. Furthermore, the definition of organ failure in the Atlanta classification is so wide that it includes many patients with a good prognosis. To overcome these shortcomings we have categorized the patients into 3 groups: patients who develop vital organ failure, patients who meet the Atlanta criteria for severe AP but do not develop organ failure, and patients who neither meet the Atlanta criteria nor develop organ failure[41,43,44,52,53]. Indeed, in terms of mortality and morbidity, the organ failure group is the critical one[8,9] regardless of the presence or absence of necrosis of the pancreas[54]. Organ failure often develops early in the course of AP. As many as 37%-76% of AP patients who will develop organ failure may have it already at admission[55,56] or develop it during the first 24 h[44]. Also a difference between transient (< 48 h) and persistent (> 48 h) organ failure has been noted[57,58]. Patients with transient organ failure have an uneventful recovery but persistent organ failure predicts death and local complications.
The mortality of AP is biphasic. Half of the mortality takes place during the first week of the disease and is related to severe multiorgan failure[6]. If the patient survives the first week, he/she may get infectious complications, such as sepsis, with worsening organ dysfunction. These events increase mortality during the second phase of the disease[6]. Knowing the biphasic nature of the mortality is highly relevant to treatment and research of novel treatment modalities in severe AP because the first peak occurs during on-going pro- and/or anti-inflammatory responses whereas the second peak occurs at the phase of immune suppression.
IMMUNE-MODULATION THERAPY
Current treatment of AP remains unspecific and supportive and is mainly targeted to aggressively prevent systemic complications by intensive care. Delayed admission to the intensive care unit results in a 4-fold excess in mortality[59]. Therefore, early identification of patients who develop a severe AP with organ failure is essential for improved prognosis by earlier intervention with appropriate resuscitation in specialized hospitals.
At present, no specific medical therapy for AP exists. Several experimental studies and clinical trials have attempted to discover medical treatment to inhibit the systemic inflammatory reaction and, thus, the development of multiple organ dysfunction. In a rat model of AP, in contrast to expectations, anti-TNF antibody was found to be harmful[60]. However, when given prophylactically anti-TNF antibodies decreased the severity of AP[61]. In experimental AP, block of the cytokine cascade at the level of the IL-1 receptor with a specific antagonist (IL-1ra) decreased pancreatic damage[62]. Furthermore, anti-inflammatory therapy with an IL-10 agonist[63,64] and an anti-IL-8 antibody[65] have shown beneficial effects. In addition, anti-adhesion therapy with anti-intercellular adhesion molecule-1 antibody ameliorates lung injury in mice with severe AP[66]. Platelet-activating factor antagonist lexipafant was promising in the first clinical trials[67,68] but in the final trial the results were disappointing[55]. In the complex network of inflammatory responses, a combination therapy to inhibit several proinflammatory agents could be useful.
In clinical AP, patients are typically admitted to the emergency unit in a fairly late stage of the disease when organ failure is about to develop or may already be present. Then the patient has CARS or probably even immunosuppression, initiation of anti-inflammatory therapies could worsen the prognosis. Thus, the therapeutic window for anti-inflammatory therapies may be too narrow in clinical practice[55]. The problem of early-occurring organ dysfunction in clinical trials is highlighted by the lexipafant study[55], in which 44% of the patients met the criteria of organ failure prior to receiving the first dose of the study drug, an inhibitor of platelet activating factor, or placebo.
In anergic septic patients immunostimulation with interferon-γ has proven to be beneficial[33]. In a clinical study of septic patients, granulocyte macrophage stimulating factor (GM-CSF) treatment was well tolerated and increased monocyte HLA-DR expression and TNF-α production capacity[69]. In severe AP, there is a defect in monocyte function. However, priming of monocytes in vitro by interferon-γ and GM-CSF increased HLA-DR expression and restored lipopolysaccharide-induced TNF-α secretion[70]. Yet, it is of note, that immunosuppression may be confined to the peripheral blood while the lungs and the other end organs may still be in the pro-inflammatory stage. Therefore, immunostimulatory treatment must be used cautiously and clinicians should have the means to monitor the patients’ immunoinflammatory state. Currently, the best method is measurement of monocyte HLA expression by flow cytometry. It would be optimal to depress the pro-inflammatory reaction in the patients at risk of excessive immune suppression so that inappropriate CARS would be prevented. An interesting question is whether the patients in whom a deep immune suppression develops, benefit from antimicrobial prophylaxis, along with immunostimulation.
COAGULATION SYSTEM IS RELATED TO INFLAMMATION
In systemic inflammation, several proinflammatory cytokines activate the coagulation process in the body[71] and activated endothelial cells seem to have an important contributing role in that process. Systemic coagulation activation results in thrombosis in small and middle-sized vessels in many organs, resulting in disseminated intravascular coagulation (DIC), that leads to decreased blood circulation of organs and organ failure. In DIC, consumption of platelets is increased resulting in thrombocytopenia which is a common sign of severe AP[72]. The D-dimer level has been shown to be high in severe AP and serves as a marker of DIC[73]. In experimental severe AP, it was demonstrated that microcirculatory disorders affecting capillary blood flow, capillary permeability and leukocyte endothelial interaction are present not only in the pancreas but also in the colon, liver, and lungs[28].
Protein C is a natural anticoagulant in blood and seems to have an essential role in the regulation of the coagulation cascade in inflammation. Protein C is activated by the thrombin-thrombomodulin complex at the endothelial surface[74]. Activated protein C (APC) inactivates factor V and factor VIII and inhibits thrombin generation. In experimental studies, APC has shown also to have anti-inflammatory effects[75,76]. Septic patients have a decreased protein C level in blood and it correlates with poor prognosis[77]. In meningococcemia patients with DIC, treatment with APC has prevented development of organ failure, and decreased mortality[78]. In patients with severe sepsis, treatment with APC was safe and resulted in decreased mortality[79,80]. In severe AP, protein C pathway defects have been shown to be associated with development of organ failure[81]. In a rat model of severe AP, APC treatment reduced inflammation in the pancreas and lungs and improved survival[82]. These findings encouraged us to perform a placebo-controlled clinical trial of APC in the treatment of severe AP at the Helsinki University Central Hospital.
FUTURE DIRECTIONS
Deeper understanding of the cell biology and physiology of AP is necessary to permit the design of effective interventions concerning the inflammatory response process. Improved methods to accurately identify those patients with severe AP in need of monitoring and treatment in intensive care units are urgently needed. A means to monitor the state of the immune response (monocyte HLA-DR expression) during hospitalization of severe AP patients is important. Whether monitoring signaling pathways of circulating leukocytes, such as nuclear factor κB, signal transducers and activators of transcription and members of the mitogen activated protein kinase family helps us to find the patients at risk for secondary infections and, thus, late organ failure is at present under research[83]. Future treatment strategies will probably focus on combination therapy aimed at suppressing excessive activation of the inflammatory response and preventing concomitantly overcompensating CARS and eventually immunosuppression. Because AP shares many features of sepsis, an urgent question is whether APC provides a means to alleviate severe AP, as it does in patients with life-threatening sepsis. Finally, the analysis of signaling patterns of leukocytes may reveal novel therapeutic targets in severe AP.
Footnotes
Peer reviewers: Naoaki Sakata, MD, PhD, Division of Hepato-Biliary Pancreatic Surgery, Tohoku University Graduate School of Medicine, 1-1 Seiryo-machi, Aoba-ku, Sendai, Miyagi 980-8574, Japan; Udayakumar Navaneethan, MD, Department of Internal Medicine, University of Cincinnati College of Medicine, 231 Albert Sabin Way, Cincinnati, OH 45267, United States
S- Editor Wang JL L- Editor Cant MR E- Editor Lin YP
References
- 1.Eland IA, Sturkenboom MJ, Wilson JH, Stricker BH. Incidence and mortality of acute pancreatitis between 1985 and 1995. Scand J Gastroenterol. 2000;35:1110–1116. doi: 10.1080/003655200451261. [DOI] [PubMed] [Google Scholar]
- 2.Banks PA. Epidemiology, natural history, and predictors of disease outcome in acute and chronic pancreatitis. Gastrointest Endosc. 2002;56:S226–S230. doi: 10.1067/mge.2002.129022. [DOI] [PubMed] [Google Scholar]
- 3.Goldacre MJ, Roberts SE. Hospital admission for acute pancreatitis in an English population, 1963-98: database study of incidence and mortality. BMJ. 2004;328:1466–1469. doi: 10.1136/bmj.328.7454.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersson R, Andersson B, Haraldsen P, Drewsen G, Eckerwall G. Incidence, management and recurrence rate of acute pancreatitis. Scand J Gastroenterol. 2004;39:891–894. doi: 10.1080/00365520410007061. [DOI] [PubMed] [Google Scholar]
- 5.Fagenholz PJ, Castillo CF, Harris NS, Pelletier AJ, Camargo CA Jr. Increasing United States hospital admissions for acute pancreatitis, 1988-2003. Ann Epidemiol. 2007;17:491–497. doi: 10.1016/j.annepidem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 6.McKay CJ, Evans S, Sinclair M, Carter CR, Imrie CW. High early mortality rate from acute pancreatitis in Scotland, 1984-1995. Br J Surg. 1999;86:1302–1305. doi: 10.1046/j.1365-2168.1999.01246.x. [DOI] [PubMed] [Google Scholar]
- 7.Floyd A, Pedersen L, Nielsen GL, Thorladcius-Ussing O, Sorensen HT. Secular trends in incidence and 30-day case fatality of acute pancreatitis in North Jutland County, Denmark: a register-based study from 1981-2000. Scand J Gastroenterol. 2002;37:1461–1465. doi: 10.1080/003655202762671369. [DOI] [PubMed] [Google Scholar]
- 8.de Beaux AC, Palmer KR, Carter DC. Factors influencing morbidity and mortality in acute pancreatitis; an analysis of 279 cases. Gut. 1995;37:121–126. doi: 10.1136/gut.37.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenner S, Sica G, Hughes M, Noordhoek E, Feng S, Zinner M, Banks PA. Relationship of necrosis to organ failure in severe acute pancreatitis. Gastroenterology. 1997;113:899–903. doi: 10.1016/s0016-5085(97)70185-9. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg W, Tenner S. Acute pancreatitis. N Engl J Med. 1994;330:1198–1210. doi: 10.1056/NEJM199404283301706. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola M, Nordback I. Pancreatitis in Finland between 1970 and 1989. Gut. 1993;34:1255–1260. doi: 10.1136/gut.34.9.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puolakkainen P, Kemppainen E, Leppäniemi A, Sainio V, Hietaranta A, Haapiainen R. Current principles of treatment in acute pancreatitis. Ann Chir Gynaecol. 1998;87:200–203. [PubMed] [Google Scholar]
- 13.Navaneethan U, Vege SS, Chari ST, Baron TH. Minimally invasive techniques in pancreatic necrosis. Pancreas. 2009;38:867–875. doi: 10.1097/MPA.0b013e3181b3b237. [DOI] [PubMed] [Google Scholar]
- 14.De Waele JJ, Leppäniemi AK. Intra-abdominal hypertension in acute pancreatitis. World J Surg. 2009;33:1128–1133. doi: 10.1007/s00268-009-9994-5. [DOI] [PubMed] [Google Scholar]
- 15.Jafri NS, Mahid SS, Idstein SR, Hornung CA, Galandiuk S. Antibiotic prophylaxis is not protective in severe acute pancreatitis: a systematic review and meta-analysis. Am J Surg. 2009;197:806–813. doi: 10.1016/j.amjsurg.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Nowak A, Marek TA, Nowakowska-Duława E, Rybicka J, Kaczor R. Biliary pancreatitis needs endoscopic retrograde cholangiopancreatography with endoscopic sphincterotomy for cure. Endoscopy. 1998;30:A256–A259. doi: 10.1055/s-2007-1001451. [DOI] [PubMed] [Google Scholar]
- 17.Deitch EA. Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg. 1992;216:117–134. doi: 10.1097/00000658-199208000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steer ML, Meldolesi J. Pathogenesis of acute pancreatitis. Annu Rev Med. 1988;39:95–105. doi: 10.1146/annurev.me.39.020188.000523. [DOI] [PubMed] [Google Scholar]
- 19.Otani T, Chepilko SM, Grendell JH, Gorelick FS. Codistribution of TAP and the granule membrane protein GRAMP-92 in rat caerulein-induced pancreatitis. Am J Physiol. 1998;275:G999–G1009. doi: 10.1152/ajpgi.1998.275.5.G999. [DOI] [PubMed] [Google Scholar]
- 20.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest. 2001;108:1387–1395. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warshaw AL. Damage prevention versus damage control in acute pancreatitis. Gastroenterology. 1993;104:1216–1219. doi: 10.1016/0016-5085(93)90299-r. [DOI] [PubMed] [Google Scholar]
- 22.Steer ML. Frank Brooks memorial Lecture: The early intraacinar cell events which occur during acute pancreatitis. Pancreas. 1998;17:31–37. doi: 10.1159/000026152. [DOI] [PubMed] [Google Scholar]
- 23.Bone RC. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med. 1996;24:163–172. doi: 10.1097/00003246-199601000-00026. [DOI] [PubMed] [Google Scholar]
- 24.Montravers P, Chollet-Martin S, Marmuse JP, Gougerot-Pocidalo MA, Desmonts JM. Lymphatic release of cytokines during acute lung injury complicating severe pancreatitis. Am J Respir Crit Care Med. 1995;152:1527–1533. doi: 10.1164/ajrccm.152.5.7582288. [DOI] [PubMed] [Google Scholar]
- 25.Castell JV, Gómez-Lechón MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242:237–239. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]
- 26.Osman MO, Jensen SL. Acute pancreatitis: the pathophysiological role of cytokines and integrins. New trends for treatment? Dig Surg. 1999;16:347–362. doi: 10.1159/000018746. [DOI] [PubMed] [Google Scholar]
- 27.Menger MD, Plusczyk T, Vollmar B. Microcirculatory derangements in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2001;8:187–194. doi: 10.1007/s005340170015. [DOI] [PubMed] [Google Scholar]
- 28.Foitzik T, Eibl G, Hotz B, Hotz H, Kahrau S, Kasten C, Schneider P, Buhr HJ. Persistent multiple organ microcirculatory disorders in severe acute pancreatitis: experimental findings and clinical implications. Dig Dis Sci. 2002;47:130–138. doi: 10.1023/a:1013284008219. [DOI] [PubMed] [Google Scholar]
- 29.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Mentula P, Kylänpää ML, Kemppainen E, Jansson SE, Sarna S, Puolakkainen P, Haapiainen R, Repo H. Plasma anti-inflammatory cytokines and monocyte human leucocyte antigen-DR expression in patients with acute pancreatitis. Scand J Gastroenterol. 2004;39:178–187. doi: 10.1080/00365520310008278. [DOI] [PubMed] [Google Scholar]
- 31.Beger HG, Bittner R, Block S, Büchler M. Bacterial contamination of pancreatic necrosis. A prospective clinical study. Gastroenterology. 1986;91:433–438. doi: 10.1016/0016-5085(86)90579-2. [DOI] [PubMed] [Google Scholar]
- 32.Hershman MJ, Appel SH, Wellhausen SR, Sonnenfeld G, Polk HC Jr. Interferon-gamma treatment increases HLA-DR expression on monocytes in severely injured patients. Clin Exp Immunol. 1989;77:67–70. [PMC free article] [PubMed] [Google Scholar]
- 33.Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 34.Tonegawa S. Antibody and T-cell receptors. JAMA. 1988;259:1845–1847. [PubMed] [Google Scholar]
- 35.Wolk K, Döcke WD, von Baehr V, Volk HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 2000;96:218–223. [PubMed] [Google Scholar]
- 36.Fumeaux T, Pugin J. Role of interleukin-10 in the intracellular sequestration of human leukocyte antigen-DR in monocytes during septic shock. Am J Respir Crit Care Med. 2002;166:1475–1482. doi: 10.1164/rccm.200203-217OC. [DOI] [PubMed] [Google Scholar]
- 37.Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- 38.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117:1162–1172. doi: 10.1378/chest.117.4.1162. [DOI] [PubMed] [Google Scholar]
- 39.García-Sabrido JL, Valdecantos E, Bastida E, Tellado JM. The anergic state as a predictor of pancreatic sepsis. Zentralbl Chir. 1989;114:114–120. [PubMed] [Google Scholar]
- 40.Kylänpää-Bäck ML, Takala A, Kemppainen E, Puolakkainen P, Kautiainen H, Jansson SE, Haapiainen R, Repo H. Cellular markers of systemic inflammation and immune suppression in patients with organ failure due to severe acute pancreatitis. Scand J Gastroenterol. 2001;36:1100–1107. doi: 10.1080/003655201750422738. [DOI] [PubMed] [Google Scholar]
- 41.Kylänpää-Bäck ML, Takala A, Kemppainen EA, Puolakkainen PA, Leppäniemi AK, Karonen SL, Orpana A, Haapiainen RK, Repo H. Procalcitonin, soluble interleukin-2 receptor, and soluble E-selectin in predicting the severity of acute pancreatitis. Crit Care Med. 2001;29:63–69. doi: 10.1097/00003246-200101000-00016. [DOI] [PubMed] [Google Scholar]
- 42.Chen CC, Wang SS, Lu RH, Chang FY, Lee SD. Serum interleukin 10 and interleukin 11 in patients with acute pancreatitis. Gut. 1999;45:895–899. doi: 10.1136/gut.45.6.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mentula P, Kylänpää ML, Kemppainen E, Jansson SE, Sarna S, Puolakkainen P, Haapiainen R, Repo H. Early prediction of organ failure by combined markers in patients with acute pancreatitis. Br J Surg. 2005;92:68–75. doi: 10.1002/bjs.4786. [DOI] [PubMed] [Google Scholar]
- 44.Mentula P, Kylänpää-Bäck ML, Kemppainen E, Takala A, Jansson SE, Kautiainen H, Puolakkainen P, Haapiainen R, Repo H. Decreased HLA (human leucocyte antigen)-DR expression on peripheral blood monocytes predicts the development of organ failure in patients with acute pancreatitis. Clin Sci (Lond) 2003;105:409–417. doi: 10.1042/CS20030058. [DOI] [PubMed] [Google Scholar]
- 45.Richter A, Nebe T, Wendl K, Schuster K, Klaebisch G, Quintel M, Lorenz D, Post S, Trede M. HLA-DR expression in acute pancreatitis. Eur J Surg. 1999;165:947–951. doi: 10.1080/110241599750008053. [DOI] [PubMed] [Google Scholar]
- 46.Curley PJ, McMahon MJ, Lancaster F, Banks RE, Barclay GR, Shefta J, Boylston AW, Whicher JT. Reduction in circulating levels of CD4-positive lymphocytes in acute pancreatitis: relationship to endotoxin, interleukin 6 and disease severity. Br J Surg. 1993;80:1312–1315. doi: 10.1002/bjs.1800801031. [DOI] [PubMed] [Google Scholar]
- 47.Bradley EL 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, Ga, September 11 through 13, 1992. Arch Surg. 1993;128:586–590. doi: 10.1001/archsurg.1993.01420170122019. [DOI] [PubMed] [Google Scholar]
- 48.Lankisch PG, Pflichthofer D, Lehnick D. No strict correlation between necrosis and organ failure in acute pancreatitis. Pancreas. 2000;20:319–322. doi: 10.1097/00006676-200004000-00015. [DOI] [PubMed] [Google Scholar]
- 49.Kox WJ, Volk T, Kox SN, Volk HD. Immunomodulatory therapies in sepsis. Intensive Care Med. 2000;26 Suppl 1:S124–S128. doi: 10.1007/s001340051129. [DOI] [PubMed] [Google Scholar]
- 50.Halonen KI, Pettilä V, Leppäniemi AK, Kemppainen EA, Puolakkainen PA, Haapiainen RK. Multiple organ dysfunction associated with severe acute pancreatitis. Crit Care Med. 2002;30:1274–1279. doi: 10.1097/00003246-200206000-00019. [DOI] [PubMed] [Google Scholar]
- 51.Bollen TL, van Santvoort HC, Besselink MG, van Leeuwen MS, Horvath KD, Freeny PC, Gooszen HG. The Atlanta Classification of acute pancreatitis revisited. Br J Surg. 2008;95:6–21. doi: 10.1002/bjs.6010. [DOI] [PubMed] [Google Scholar]
- 52.Lindström O, Jarva H, Meri S, Mentula P, Puolakkainen P, Kemppainen E, Haapiainen R, Repo H, Kylänpää L. Elevated levels of the complement regulator protein CD59 in severe acute pancreatitis. Scand J Gastroenterol. 2008;43:350–355. doi: 10.1080/00365520701763209. [DOI] [PubMed] [Google Scholar]
- 53.Lindström O, Tukiainen E, Kylänpää L, Mentula P, Rouhiainen A, Puolakkainen P, Rauvala H, Repo H. Circulating levels of a soluble form of receptor for advanced glycation end products and high-mobility group box chromosomal protein 1 in patients with acute pancreatitis. Pancreas. 2009;38:e215–e220. doi: 10.1097/MPA.0b013e3181bb59a7. [DOI] [PubMed] [Google Scholar]
- 54.Remes-Troche JM, Uscanga LF, Peláez-Luna M, Duarte-Rojo A, González-Balboa P, Teliz MA, Chan-Nunez C, Campuzano M, Robles-Díaz G. When should we be concerned about pancreatic necrosis? Analysis from a single institution in Mexico City. World J Surg. 2006;30:2227–2233; discussion 2234-2235. doi: 10.1007/s00268-006-0148-8. [DOI] [PubMed] [Google Scholar]
- 55.Johnson CD, Kingsnorth AN, Imrie CW, McMahon MJ, Neoptolemos JP, McKay C, Toh SK, Skaife P, Leeder PC, Wilson P, et al. Double blind, randomised, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut. 2001;48:62–69. doi: 10.1136/gut.48.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Isenmann R, Rau B, Beger HG. Early severe acute pancreatitis: characteristics of a new subgroup. Pancreas. 2001;22:274–278. doi: 10.1097/00006676-200104000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Buter A, Imrie CW, Carter CR, Evans S, McKay CJ. Dynamic nature of early organ dysfunction determines outcome in acute pancreatitis. Br J Surg. 2002;89:298–302. doi: 10.1046/j.0007-1323.2001.02025.x. [DOI] [PubMed] [Google Scholar]
- 58.Johnson CD, Abu-Hilal M. Persistent organ failure during the first week as a marker of fatal outcome in acute pancreatitis. Gut. 2004;53:1340–1344. doi: 10.1136/gut.2004.039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brivet FG, Emilie D, Galanaud P. Pro- and anti-inflammatory cytokines during acute severe pancreatitis: an early and sustained response, although unpredictable of death. Parisian Study Group on Acute Pancreatitis. Crit Care Med. 1999;27:749–755. doi: 10.1097/00003246-199904000-00029. [DOI] [PubMed] [Google Scholar]
- 60.Guice KS, Oldham KT, Remick DG, Kunkel SL, Ward PA. Anti-tumor necrosis factor antibody augments edema formation in caerulein-induced acute pancreatitis. J Surg Res. 1991;51:495–499. doi: 10.1016/0022-4804(91)90171-h. [DOI] [PubMed] [Google Scholar]
- 61.Grewal HP, Mohey el Din A, Gaber L, Kotb M, Gaber AO. Amelioration of the physiologic and biochemical changes of acute pancreatitis using an anti-TNF-alpha polyclonal antibody. Am J Surg. 1994;167:214–218; discussion 218-219. doi: 10.1016/0002-9610(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 62.Norman J, Franz M, Messina J, Riker A, Fabri PJ, Rosemurgy AS, Gower WR Jr. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery. 1995;117:648–655. doi: 10.1016/s0039-6060(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 63.Van Laethem JL, Marchant A, Delvaux A, Goldman M, Robberecht P, Velu T, Devière J. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology. 1995;108:1917–1922. doi: 10.1016/0016-5085(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 64.Osman MO, Jacobsen NO, Kristensen JU, Deleuran B, Gesser B, Larsen CG, Jensen SL. IT 9302, a synthetic interleukin-10 agonist, diminishes acute lung injury in rabbits with acute necrotizing pancreatitis. Surgery. 1998;124:584–592. [PubMed] [Google Scholar]
- 65.Osman MO, Kristensen JU, Jacobsen NO, Lausten SB, Deleuran B, Deleuran M, Gesser B, Matsushima K, Larsen CG, Jensen SL. A monoclonal anti-interleukin 8 antibody (WS-4) inhibits cytokine response and acute lung injury in experimental severe acute necrotising pancreatitis in rabbits. Gut. 1998;43:232–239. doi: 10.1136/gut.43.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lundberg AH, Fukatsu K, Gaber L, Callicutt S, Kotb M, Wilcox H, Kudsk K, Gaber AO. Blocking pulmonary ICAM-1 expression ameliorates lung injury in established diet-induced pancreatitis. Ann Surg. 2001;233:213–220. doi: 10.1097/00000658-200102000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kingsnorth AN, Galloway SW, Formela LJ. Randomized, double-blind phase II trial of Lexipafant, a platelet-activating factor antagonist, in human acute pancreatitis. Br J Surg. 1995;82:1414–1420. doi: 10.1002/bjs.1800821039. [DOI] [PubMed] [Google Scholar]
- 68.McKay CJ, Curran F, Sharples C, Baxter JN, Imrie CW. Prospective placebo-controlled randomized trial of lexipafant in predicted severe acute pancreatitis. Br J Surg. 1997;84:1239–1243. [PubMed] [Google Scholar]
- 69.Nierhaus A, Montag B, Timmler N, Frings DP, Gutensohn K, Jung R, Schneider CG, Pothmann W, Brassel AK, Schulte Am Esch J. Reversal of immunoparalysis by recombinant human granulocyte-macrophage colony-stimulating factor in patients with severe sepsis. Intensive Care Med. 2003;29:646–651. doi: 10.1007/s00134-003-1666-6. [DOI] [PubMed] [Google Scholar]
- 70.Kylanpaa ML, Mentula P, Kemppainen E, Puolakkainen P, Aittomaki S, Silvennoinen O, Haapiainen R, Repo H. Monocyte anergy is present in patients with severe acute pancreatitis and is significantly alleviated by granulocyte-macrophage colony-stimulating factor and interferon-gamma in vitro. Pancreas. 2005;31:23–27. doi: 10.1097/01.mpa.0000164449.23524.94. [DOI] [PubMed] [Google Scholar]
- 71.Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586–592. doi: 10.1056/NEJM199908193410807. [DOI] [PubMed] [Google Scholar]
- 72.Lasson A, Ohlsson K. Consumptive coagulopathy, fibrinolysis and protease-antiprotease interactions during acute human pancreatitis. Thromb Res. 1986;41:167–183. doi: 10.1016/0049-3848(86)90227-6. [DOI] [PubMed] [Google Scholar]
- 73.Salomone T, Tosi P, Palareti G, Tomassetti P, Migliori M, Guariento A, Saieva C, Raiti C, Romboli M, Gullo L. Coagulative disorders in human acute pancreatitis: role for the D-dimer. Pancreas. 2003;26:111–116. doi: 10.1097/00006676-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 74.Shen L, Dahlbäck B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J Biol Chem. 1994;269:18735–18738. [PubMed] [Google Scholar]
- 75.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K. Activated protein C prevents LPS-induced pulmonary vascular injury by inhibiting cytokine production. Am J Physiol. 1997;272:L197–L202. doi: 10.1152/ajplung.1997.272.2.L197. [DOI] [PubMed] [Google Scholar]
- 76.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 77.Fisher CJ Jr, Yan SB. Protein C levels as a prognostic indicator of outcome in sepsis and related diseases. Crit Care Med. 2000;28:S49–S56. doi: 10.1097/00003246-200009001-00011. [DOI] [PubMed] [Google Scholar]
- 78.White B, Livingstone W, Murphy C, Hodgson A, Rafferty M, Smith OP. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood. 2000;96:3719–3724. [PubMed] [Google Scholar]
- 79.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 80.Abraham E, Laterre PF, Garg R, Levy H, Talwar D, Trzaskoma BL, François B, Guy JS, Brückmann M, Rea-Neto A, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332–1341. doi: 10.1056/NEJMoa050935. [DOI] [PubMed] [Google Scholar]
- 81.Lindstrom O, Kylanpaa L, Mentula P, Puolakkainen P, Kemppainen E, Haapiainen R, Fernandez JA, Griffin JH, Repo H, Petaja J. Upregulated but insufficient generation of activated protein C is associated with development of multiorgan failure in severe acute pancreatitis. Crit Care. 2006;10:R16. doi: 10.1186/cc3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alsfasser G, Warshaw AL, Thayer SP, Antoniu B, Laposata M, Lewandrowski KB, Fernández-del Castillo C. Decreased inflammation and improved survival with recombinant human activated protein C treatment in experimental acute pancreatitis. Arch Surg. 2006;141:670–676; discussion 676-677. doi: 10.1001/archsurg.141.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Puolakkainen P, Repo H, Vainionpää S, Kylänpää L, Oiva J, Kemppainen E, Mustonen H. Defects in monocyte signaling in patients with acute pancreatitis. Pancreas. 2009;38:1039. [Google Scholar]