

Abstract
One of the hallmarks of both sickle cell disease (SCD) and thalassemia major (TM) is accelerated oxidative damage. Decreased antioxidant levels and increased oxidant stress biomarkers are found in both diseases. Although isolated vitamin deficiencies have been reported in TM and nontransfused SCD patients, a comprehensive evaluation of vitamin and trace mineral levels has never been performed in chronically transfused SCD or TM patients. As vitamins and trace minerals may be consumed as a result of chronic oxidative stress; we hypothesized that levels of these compounds would correlate with surrogates of iron overload, hemolysis, and inflammation in chronically transfused patients. Using a convenience sample of our group of chronically transfused patients we studied 43 patients with SCD (17 male, 26 female) and 24 patients with TM (13 male and 11 female). The age range for our patients varied from 1.5 to 31.4 years. Levels of vitamins A, thiamin, B6, B12, C, D, E as well as selenium, zinc, copper, and ceruloplasmin were measured. We found that 40–75% of the patients were deficient in A, C, D and selenium and 28–38% of the patients had low levels of B vitamins and folate. There was little association with iron overload, hemolysis, or inflammation. Although the precise mechanism of these deficiencies is unclear, they may contribute to the morbidity of chronically transfused hemoglobinopathy patients.
Introduction
Hemoglobinopathies are characterized by oxidant damage due to increased resting oxygen consumption and circulating prooxidative free hemoglobin (Hgb). In sickle cell disease (SCD), Hgb S is unstable and generates free radicals which damage cellular enzymes and membrane lipids [1]. Previous studies have shown high levels of malondialdehyde (MDA) and other oxidative biomarkers in the plasma of nontransfused SCD patients [2]. A similar finding has been shown in nontransfused thalassemia patients, due to the toxic effect of unpaired globin chains on the membrane [3]. Oxidative stress biomarkers are also elevated in chronically transfused SCD and thalassemia major (TM) patients and correlate most strongly with nontransferrin bound iron (NTBI) levels [4].
Vitamins and trace minerals represent key buffers against oxidative damage. However, chronic demands on oxidative buffering capacity may produce conditional deficiencies in key amino acids and enzymatic cofactors. In SCD, production of reactive oxygen species and hyperhemolysis have been postulated to be the dominant mechanisms for the consumption of these compounds [5,6]. Non transfused patients with SCD have been demonstrated to have reduced levels of zinc, selenium, and glutathione as well as vitamins A, C, riboflavin, D, and E [7-9]. Chronic transfusions, which decrease hemolysis by suppressing the production of abnormal red cells, could potentially improve the nutrient profile in SCD and TM patients. However, the production of free radicals induced by iron overload in transfused patients may override this effect. In addition, chronic inflammation, a hallmark of SCD, has been demonstrated to reduce levels of some of these nutrients [10,11]. The goal of the present study was to determine the prevalence of vitamin and mineral deficiencies in chronically transfused patients with SCD and TM and their relationship to age, iron-burden, hemolytic rate, and inflammatory markers.
Results
Table I summarizes our patient demographics. The patient populations were similar in age and gender, with a mean age of 14.5 years. In terms of chelation, the majority of the patients were on Deferasirox alone (51). Only 6 patients were on either Defiriprone (1) or Desferoxamine (5). Three patients were on Deferasirox and Desferal together, seven patients were not on chelators. All patients had normal renal function as measured by BUN and creatinine. Some patients had mild proteinuria which was due either to use of Deferasirox or SCD.
TABLE I. Demographics and Pertinent Lab Values.
| Reference range | Sickle cell N= 43 |
Thalassemia N= 24 |
P-value | |
|---|---|---|---|---|
| Male:female | – | 17:26 | 13:11 | NS |
| Age (yr) | – | 14.4 ± 6.6 (1.5–31.4) |
14.7 ± 7.6 (1.9–25.8) |
NS |
| HICa (mg/g dry wt) |
<1.6 mg/g dry wt | 19.5 ± 13.8 (2.2–70.2) |
13.7 ± 11.4 (2–39.5) |
<0.03 |
| Ferritin (ng/mL) | 5 months to 15 years: 10–140 Males > 15 years: 30–300 Females > 15 years: 10–150 |
3874 ± 4451 (81–27200) |
2089 ± 1920 (246–8230) |
<0.02 |
| % Saturation (Fe) | 15–50% | 68 ± 24 (11–103) |
84 ± 18 (36–106) |
<0.02 |
| LDH | 315–618 U/l | 1263 ± 717 (288–4209) |
445 ± 136 (292–717) |
<0.0001 |
| Plasma hemoglobin |
0–10 mg/dl | 50 ± 60 (3–330) |
16 ± 10 (3.5–49) |
<0.005 |
| hs CRP | <1.0 mg/l | 5.8 ± 8 (0.2–28.2) |
1.2 ± 3.4 (0.1–16.4) |
<0.004 |
Demographic and other lab values of the patients studied. Laboratory results are expressed as mean ± SD. P-values are indicated as shown.
HIC, hepatic iron concentration (as determined by MRI); Fe, iron; LDH, lactate dehydrogenase; hs-CRP, high sensitivity C reactive protein.
HIC values were calculated by MRI R2 and R2* methods.
Somatic iron burden was significantly higher in SCD patients whether assessed by hepatic iron concentration (HIC, 42%) or ferritin (85%). Despite having greater total body iron, transferrin saturation was nearly 23% lower in SCD patients. SCD patients had higher surrogate values for hemolysis and inflammation. LDH and cell free Hgb were nearly three times larger in SCD patients, indicating that transfusions were incompletely effective in suppressing endogenous RBC production. High sensitivity C-reactive protein (hs-CRP) was also almost 5-fold larger in SCD patients, consistent with greater chronic inflammation [12]. Table II summarizes the mean values and percent of abnormal nutrient values for SCD and TM patients. No gender differences were noted. The magnitude and spectrum of deficiencies was nearly identical for the two groups. Over half of the SCD and TM patients were deficient in vitamins A, C, and D25-OH. Thiamin, B6 and folate were deficient in roughly 1/3 of patients. B12 levels were within population norms. Alpha-tocopherol was abnormal in 29.2% of TM patients but only 10.5% of the SCD patients (P = 0.09).
TABLE II. Nutrient Levels in Chronically Transfused Sickle Cell Patients (Top) and Thalassemia Patients (Bottom).
| Nutrient | Reference range |
Sickle cell patients |
Thalassemia patients |
||
|---|---|---|---|---|---|
| Mean ± S.D. | %abn | Mean ± S.D. | %abn | ||
| A (μ/dl) | 38–98 | 34. 4 ± 8.5 | 73.70% | 34.6 ± 12.2 | 52.40% |
| Thiamin (μ/l) | 2.4–11.7 | 3.3 ± 21.6 | 38.50% | 4.1 ± 4.0 | 37.50% |
| B6 (ng/ml) | 3.3–26 | 8.1 ± 27.7 | 34.20% | 7.0 ± 5.9 | 34.80% |
| B12 (pg/ml) | 200–1100 | 664.4 ± 399.2 | 4.70% | 528 ± 152.0 | 0 |
| Folate (ng/ml) | >8 | 13.5 ± 6.0 | 28.60% | 11.8 ± 7.7 | 37.50% |
| C (mg/dl) | 0.2–1.9 | 0.28 ± 0.29 | 56.70% | 0.32 ± 0.40 | 66.70% |
| D25 (pg/ml) | 20–100 | 16.3 ± 6.2 | 74.40% | 17.1 ± 8.5 | 50.00% |
| D1-25 (pg/ml) | 15–60 | 61.1 ± 18.2 | 48.60% | 59.9 ± 19.5 | 39.10% |
| E-alpha (mg/dl) | 5.7–19.9 | 8.7 ± 4.7 | 10.50% | 7.5 ± 7.5 | 29.20% |
| E-gamma (mg/dl) | <4.3 | 2.3 ± 1.3 | 7.90% | 3.0 ± 5.0 | 4.20% |
| Selenium (μ/l) | 110–160 | 107.1 ± 13.3 | 67.50% | 99.5 ± 20.7 | 75.00% |
| Zinc (μ/dl) | 65–124 | 81.9 ± 17.6 | 24.30% | 83.0 ± 15.6 | 8.30% |
| Copper (μ/l) | 590–1180 | 1092.1 ± 221.1* | 34.20% | 851 ± 155.0* | 0 |
| Ceruloplasmin (mg/dl) | 24–71 | 33.7 ± 7.5* | 4.70% | 26.2 ± 4.8* | 16.70% |
The vitamins and trace minerals measured are listed with their normal ranges. The patients’ mean values plus or minus the standard deviation are in the second column. The percent of patients whose measured values fell outside the normal range are listed as percent abnormal (third column). All abnormal values were indicative of deficiencies with the exception of D1, 25-OH, and Cu which were elevated in SCD. The values where there is a difference between SCD and TM patients are indicated with a “*” symbol. Blood samples were drawn while patients were fasting overnight and their usual iron chelator was held for 24 hr.
All abnormal results, with the exception of copper, represent deficiencies.
Trace metal deficiencies were also common. Selenium deficiency was ubiquitous in both populations (67.5% SCD and 75% TM). Mean zinc levels were comparable in SCD and TM, and deficiencies were present in 24.3% and 8.3%, respectively (P = 0.31). SCD patients demonstrated mean copper and ceruloplasmin that were at the upper limits of normal and 1/3 of patients had increased copper levels. Both copper and ceruloplasmin levels were higher in SCD than TM. No TM or SCD patient had decreased serum copper although circulating ceruloplasmin levels were decreased in 1/6 of TM patients.
Table III summarizes the predictors of nutrient deficiencies; only statistically significant comparisons are included. Age was the strongest predictor of lower nutrient levels in both SCD and TM patients. Folate, D25-OH, copper and ceruloplasmin levels decreased with increasing age in both groups. Levels of γ-tocopherol increased in SCD patients as they aged. HIC had little association with nutrient levels; although there were weak negative correlations with lower Cu levels in SCD patients and lower D25-OH levels in TM patients. Higher gamma tocopherol levels correlated with higher HIC in TM patients. hs-CRP was positively correlated with copper and ceruloplasmin levels (r2 = 0.37) and negatively correlated with vitamin A levels (r2 = 0.28) in SCD patients (see Fig. 1).
TABLE III. Significant Relationships among Vitamin/Mineral Levels, Age, Hepatic Iron Concentration, and High Sensitivity CRP.
| Age |
HIC |
Transf % |
hs-CRP |
|||||
|---|---|---|---|---|---|---|---|---|
| SCD | TM | SCD | TM | SCD | TM | SCD | TM | |
| r 2 | r 2 | r 2 | r 2 | – | – | r 2 | r 2 | |
| Vitamin A | – | – | – | – | – | – | ↓ 0.28 | – |
| Folate | ↓ 0.37 | ↓ 0.21 | – | – | ↓ 0.30 | – | – | – |
| D25-OH | ↓ 0.23 | ↓ 0.45 | – | ↓ 0.16 | – | – | – | – |
| D1-25 | ↓ 0.37 | – | – | – | – | – | – | – |
| γ tocopherol | ↑ 0.30 | – | – | ↑ 0.22 | – | – | – | – |
| Copper | ↓ 0.25 | ↓ 0.25 | ↓ 0.15 | – | – | – | ↑ 0.35 | – |
| Ceruloplasmin | ↓ 0.15 | ↓ 0.37 | – | – | – | – | ↑ 0.25 | – |
This table is restricted to vitamin/mineral levels demonstrating physiologically relevant correlations (r2 > 0.15). These include Vitamin A, Folate, Vitamin D25-OH, Vitamin D 1-25, γ-tocopherol, Copper, and Ceruloplasmin. Negative and positive correlations are indicated by respective arrows as shown beneath the table. Multivariate model was limited to the independent variables analyzed in TABLE III.
indicates positive correlation.
indicates negative correlation.
All P < 0.0125. Bold type indicates retained significance <0.05 in multivariate analysis.
Figure 1.
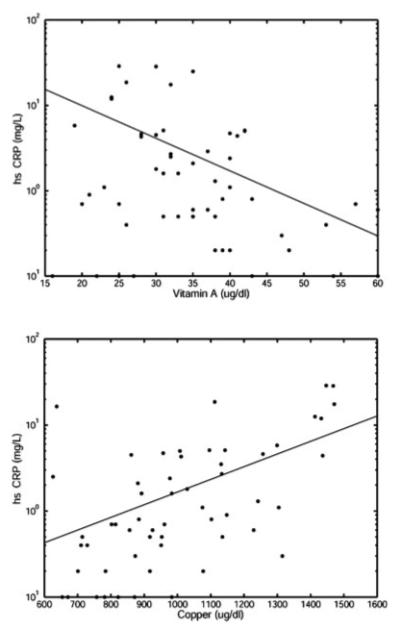
Graphs of the relationship between hs-CRP and vitamin A (top) (r2= 0.37, P < 0.0027, and negatively correlated), and hs CRP and copper (bottom) (r2 = 0.28, P < 0.0014, and positively correlated). Vitamin A and copper are on a linear scale, whereas hs CRP is on a log scale. The appropriate fits are shown.
hs-CRP levels were not elevated in TM patients and were not associated with any nutritional deficiencies. Hemolysis, as measured by LDH and cell-free Hgb, did not predict nutritional deficiencies in either group.
Discussion
These results demonstrate that chronically transfused patients with TM or SCD have significant deficiencies in both fat and water soluble nutrients which are potentially clinically relevant. For example, the levels of Vitamin A found in these patients have been shown to be related to poorer health outcomes in children with and without SCD [10,13]. In addition, Vitamin D levels lower than 20 ng/ml have been associated with multiple problems in the general population [14] and with iron cardiomyopathy in TM [15].
Potential contributors to observed deficiencies are illustrated in Fig. 2, including hemolysis, iron toxicity, ineffective erythropoiesis, inflammation, anemia, diet, and absorption. SCD patients exhibit greater hemolysis and inflammation than TM patients, while TM patients have greater iron toxicity and ineffective erythropoiesis. Despite these phenotypic differences, the spectrum of nutritional deficiencies was remarkably similar between the two diseases (Table II). Intravascular hemolysis, although a potent predictor of vascular disease in SCD, was not correlated with any nutritional deficiencies in both SCD and TM. Inflammation is important to the pathophysiology of SCD [16,17] and markers such as Vitamin A levels decrease in the presence of inflammation while copper levels rise [10,18]. We found that hs-CRP was positively correlated with copper and negatively correlated with Vitamin A in our study (see Fig. 1). Despite these findings, we were unable to demonstrate a correlation between other nutrient deficiencies and inflammation in our patients.
Figure 2.
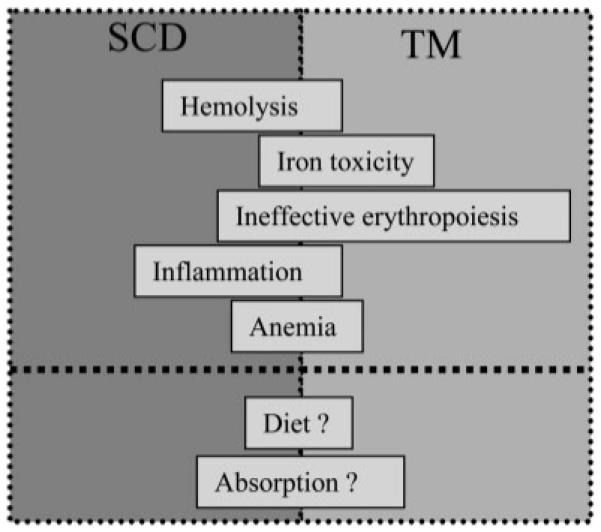
Schematic representing potential contributors to vitamin and mineral deficiencies in chronically transfused TM and SCD patients, including hemolysis, iron toxicity, ineffective erythropoiesis, inflammation, chronic anemia, diet, and malabsorption. Left–right position of the bar signifies the relative importance in a particular disease state. Little is known regarding the actors below the bold dotted line, thus, they are indicated by question marks and are centered with respect to the disease states.
HIC and ferritin values were greater in the SCD than the TM patients, suggesting decreased chelation compliance in this population. Chronically transfused SCD patients have a lower prevalence of iron-mediated endocrine and cardiac mediated dysfunction [19]. Regardless, iron overload offered little overall predictive value for antioxidant status other than mild elevations in γ-tocopherol. The negative relationship between HIC and vitamin D25-OH levels has been previously described in hereditary hemochromatosis, and is thought to reflect inhibition of hepatic 25-hydroxylation by excess iron [20].
Despite their many pathophysiologic differences, SCD and TM share some common phenotypes. All patients in this study were chronically anemic. Chronically, anemic patients maintain increased cardiac output to maintain oxygen delivery [21]. This produces a mildly hypercatabolic state, increased resting energy expenditure, and chronic oxidative stress [22-25]. These findings could contribute to increased consumption of nutrients. Malabsorption could also play a role in our findings. Both SCD and TM patients develop siderosis of the exocrine pancreas. In TM pancreatic iron negatively correlates with circulating pancreatic trypsin levels [26] and TM patients have been documented to have significant decreases in stool elastase [27]. While these observations could clearly contribute to malabsorption of fat soluble vitamins, it does not explain the low levels of water soluble vitamins found in these patients.
Most prior work in SCD has focused on nontransfused SCD patients, with deficiencies in both water and fat-soluble vitamins. Although thiamine deficiency has not been reported, deficiencies in riboflavin and pyridoxine have been noted in SCD [28] as well as decreases in serum pyridoxal 5-phosphate concentration [29]. Homocysteine levels are increased in SCD disease [30]. In addition deficiencies in vitamins A, D, and E have been reported in this group [31-34]. Vitamin D deficiency is associated with decreased bone density in SCD patients [35]. Vitamin A deficiency has been linked to increased hospitalization, poor growth and lower Hgb [10].
Dietary intake has been documented in several studies to be inadequate in SCD patients and to decline further as patients age [36,37]. This is particularly important for folate, which is a nutrient required by patients with increased red cell turnover and which is routinely supplemented in SCD patients despite supplementation in the US diet. As our patients have high red cell turnover, we measured serum folate. This value does not assess long term folate status. Nevertheless we also found nearly one third of our patients were folate deficient. Although we prescribe folic acid to our patients, we did not assess compliance. Thus, the low folate levels seen in this study may be due to non compliance in addition to other factors such as poor diet or malabsorption.
Only one study has examined antioxidant defenses in chronically transfused SCD patients. Alpha-tocopherol levels were lower in transfused than in nontransfused patients and were negatively correlated with number of units transfused [38]. MDA levels, a marker of cellular oxidant damage, was found to be increased 1.8-fold in TM patients compared to controls, but not in SCD patients. MDA levels were positively correlated with HIC, but not NTBI [4].
In contrast with SCD, prior work in thalassemia primarily reflects chronically transfused patients. TM patients have been shown to have low levels of Vitamins E and A as well as decreased RBC superoxide dismutase; these deficiencies were correlated with increased levels of lipid peroxidation [39]. Similar findings were reported in Italian patients where vitamin E and A deficiencies were inversely correlated with liver enzyme levels, suggesting that liver damage may play a role in the extent of depletion of these lipid soluble antioxidants [40]. Vitamin D deficiency has also been reported in several thalassemia cohorts [41].
There are a number of limitations to the present study. We did not have a control group, such as non affected siblings in the same household or non transfused SCD patients. We did not measure dietary intake which could play a role in these findings. In addition, because we do not have age-specific norms in our study, it is possible that our results are artificially skewed. We did not use trace metal free tubes when measuring trace elements such as Zn, Se, and Cu. Nevertheless, only Cu was elevated in this study and was well correlated with ceruloplasmin levels indicating it was not a spurious finding. Finally, because almost all of our patients were on Deferasirox for chelation, we could not meaningfully assess whether the type of chelation had an effect on the findings.
Nevertheless, the breadth and depth of these deficiencies are striking. For example, many authors now consider vitamin D25-OH levels less than 30 ng/ml insufficient for proper bone mineralization and muscle function [14]. Serum PTH and bone density data was incomplete in this study population and further work will be necessary to characterize their relationship to vitamin D stores. Thus, this study is also limited by a lack of measurable functional outcomes which prevent us from determining the degree of harm these deficiencies pose for the patients.
In summary, chronically transfused SCD and TM have broad spectrum nutritional deficiencies of both water and fat soluble nutrients. The precise mechanism of the abnormalities is unclear and is probably multifactorial. In contrast to our underlying hypothesis, iron overload, hemolysis, and inflammatory stress appear to play relative minor roles in these deficiencies. Careful nutritional studies will need to be performed to determine contributions of diet and malabsorption. Regardless of the underlying etiology, these results suggest that all patients with TM and SCD who are chronically transfused should have periodic nutritional evaluation and supplementation as necessary. Further studies of the consequences of these deficiencies are also warranted.
Methods
Vitamin and micronutrient panels were obtained as part of annual routine clinical screening. Permission for review of medical records and waiver of consent was obtained from the Committee on Clinical Investigation at Children’s Hospital Los Angeles. A convenience sample of 43 chronically transfused patients with SCD and 24 patients with TM were recruited from our population of chronically transfused hemoglobinopathy patients, and had nutrition panels drawn at the time of a regularly scheduled transfusion. All patients were requested to fast over night and hold their iron chelator for 24 hr prior to having blood drawn for the study. Levels of the following vitamins were obtained: A, thiamin, B6, B12, C, D 25OH, D1-25, and E (alpha and gamma tocopherol). Levels of folate, selenium, zinc, copper, and ceruloplasmin were also obtained. Trace element-free tubes were not used in this study. Samples were collected and processed according to clinical laboratory specifications, including rapid collection and freezing of samples for measurement of Vitamin C. (Quest Nichols Institute, San Juan Capistrano, CA).
Results were expressed as absolute values as well as the percent of patients outside the reference range for the referral laboratory. Reference ranges were not age-specific. Serum LDH and hs-CRP were measured as part of regular clinical care, and used as surrogate markers of hemolysis and inflammation, respectively. Liver iron was measured by MRI using established techniques [42]. These measurements were collected annually or biannually as part of routine chelation monitoring; only MRI liver iron estimates within 12 months of the laboratory draw were accepted.
All TM patients received simple transfusions of 10–15 cc/kg every 3 or 4 weeks to maintain a pretransfusion Hgb between 9–10 g/dl. Six patients with SCD received exchange transfusions and the rest received ~10 cc/kg of packed cells at the same intervals as the TM patients. Folate 1 mg/day and vitamin E 400U/day are routinely supplemented in our chronically transfused patients; none were receiving vitamin D or calcium replacement. All patients had normal BUN and creatinine levels. Spot urinary protein/creatinine ratios were rarely and intermittently elevated in patients on deferasirox but no patient had required dose modification or discontinuation because of proteinuria.
Parameter differences between genders and between diseases were evaluated using unpaired t-Test or Wilcoxin-Signed rank test when appropriate (JMP5.1, SAS Cary, NC). Predictors of abnormal vitamin levels were evaluated using linear regression with respect to age, HIC, LDH, and hs-CRP. Bonferroni correction was applied to correct for multiple comparisons.
Acknowledgments
The authors are grateful to Debbie Harris, Trish Peterson, Paola Pederzoli, Colleen McCarthy, and Janelle Miller for their support of the MRI program.
Contract grant sponsor: NHLBI; Contract grant number: 1 RO1 HL075592-01A1; Contract grant sponsor: General Clinical Research Center, Children’s Hospital Los Angeles; Contract grant number: RR000043-43; Contract grant sponsor: Center for Disease Control (Thalassemia Center); Contract grant number: U27/CCU922106; Contract grant sponsors: Novartis Pharma, Department of Pediatrics.
Footnotes
Conflict of interest: T.D.C. and J.C.W. have received honoria from Apotex. S.M.C. is a member of the Novartis speaker’s bureau for Deferasirox.
References
- 1.Repka T, Hebbel RP. Hydroxyl radical formation by sickle erythrocyte membranes: Role of pathologic iron deposits and cytoplasmic reducing agents. Blood. 1991;78:2753–2758. [PubMed] [Google Scholar]
- 2.Hebbel RP, Miller WJ. Phagocytosis of sickle erythrocytes: Immunologic and oxidative determinants of hemolytic anemia. Blood. 1984;64:733–741. [PubMed] [Google Scholar]
- 3.Scott MD, van den Berg JJ, Repka T, et al. Effect of excess alpha-hemoglobin chains on cellular and membrane oxidation in model beta-thalassemic erythrocytes. J Clin Invest. 1993;91:1706–1712. doi: 10.1172/JCI116380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter PB, Fung EB, Killilea DW, et al. Oxidative stress and inflammation in iron-overloaded patients with beta-thalassaemia or sickle cell disease. Br J Haematol. 2006;135:254–263. doi: 10.1111/j.1365-2141.2006.06277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amer J, Ghoti H, Rachmilewitz E, et al. Red blood cells, platelets and polymorphonuclear neutrophils of patients with sickle cell disease exhibit oxidative stress that can be ameliorated by antioxidants. Br J Haematol. 2006;132:108–113. doi: 10.1111/j.1365-2141.2005.05834.x. [DOI] [PubMed] [Google Scholar]
- 6.Brewer CJ, Coates TD, Wood JC. Spleen R2 and R2* in iron-overloaded patients with sickle cell disease and thalassemia major. J Magn Reson Imaging. 2009;29:357–364. doi: 10.1002/jmri.21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal JB, Miller ER, III, Brereton NH, Resar LM. Concentrations of B vitamins and homocysteine in children with sickle cell anemia. South Med J. 2004;97:149–155. doi: 10.1097/01.SMJ.0000051740.56511.93. [DOI] [PubMed] [Google Scholar]
- 8.Buison AM, Kawchak DA, Schall J, et al. Low vitamin D status in children with sickle cell disease. J Pediatr. 2004;145:622–627. doi: 10.1016/j.jpeds.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 9.Tangney CC, Phillips G, Bell RA, et al. Selected indices of micronutrient status in adult patients with sickle cell anemia (SCA) Am J Hematol. 1989;32:161–166. doi: 10.1002/ajh.2830320302. [DOI] [PubMed] [Google Scholar]
- 10.Schall JI, Zemel BS, Kawchak DA, et al. Vitamin A status, hospitalizations, and other outcomes in young children with sickle cell disease. J Pediatr. 2004;145:99–106. doi: 10.1016/j.jpeds.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 11.Gori AM, Sofi F, Marcucci R, et al. Association between homocysteine, vitamin B(6) concentrations and inflammation. Clin Chem Lab Med. 2007;45:1728–1736. doi: 10.1515/CCLM.2007.347. [DOI] [PubMed] [Google Scholar]
- 12.Hebbel RP. Special issue of microcirculation: Examination of the vascular pathobiology of sickle cell anemia. Foreword. Microcirculation. 2004;11:99–100. [PubMed] [Google Scholar]
- 13.Villamor E, Fawzi WW. Vitamin A supplementation: Implications for morbidity and mortality in children. J Infect Dis. 2000;182(Suppl 1):S122–S133. doi: 10.1086/315921. [DOI] [PubMed] [Google Scholar]
- 14.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 15.Wood JC, Claster S, Carson S, et al. Vitamin D deficiency, cardiac iron and cardiac function in thalassaemia major. Br J Haematol. 2008;141:891–894. doi: 10.1111/j.1365-2141.2008.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perelman N, Selvaraj SK, Batra S, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102:1506–1514. doi: 10.1182/blood-2002-11-3422. [DOI] [PubMed] [Google Scholar]
- 17.Selvaraj SK, Giri RK, Perelman N, et al. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102:1515–1524. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- 18.Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275:21048–21054. doi: 10.1074/jbc.M000636200. [DOI] [PubMed] [Google Scholar]
- 19.Fung EB, Harmatz PR, Lee PD, et al. Increased prevalence of iron-overload associated endocrinopathy in thalassaemia versus sickle-cell disease. Br J Haematol. 2006;135:574–582. doi: 10.1111/j.1365-2141.2006.06332.x. [DOI] [PubMed] [Google Scholar]
- 20.Chow LH, Frei JV, Hodsman AB, Valberg LS. Low serum 25-hydroxyvitamin D in hereditary hemochromatosis: Relation to iron status. Gastroenterology. 1985;88:865–869. doi: 10.1016/s0016-5085(85)80001-9. [DOI] [PubMed] [Google Scholar]
- 21.Wood JC, Tyszka JM, Ghugre N, et al. Myocardial iron loading in transfusion-dependent thalassemia and sickle-cell disease. Blood. 2004;103:1934–1936. doi: 10.1182/blood-2003-06-1919. [DOI] [PubMed] [Google Scholar]
- 22.Vaisman N, Akivis A, Sthoeger D, et al. Resting energy expenditure in patients with thalassemia major. Am J Clin Nutr. 1995;61:582–584. doi: 10.1093/ajcn/61.3.582. [DOI] [PubMed] [Google Scholar]
- 23.Hibbert JM, Creary MS, Gee BE, et al. Erythropoiesis and myocardial energy requirements contribute to the hypermetabolism of childhood sickle cell anemia. J Pediatr Gastroenterol Nutr. 2006;43:680–687. doi: 10.1097/01.mpg.0000228120.44606.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harmatz P, Heyman MB, Cunningham J, et al. Effects of red blood cell transfusion on resting energy expenditure in adolescents with sickle cell anemia. J Pediatr Gastroenterol Nutr. 1999;29:127–131. doi: 10.1097/00005176-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Barden EM, Zemel BS, Kawchak DA, et al. Total and resting energy expenditure in children with sickle cell disease. J Pediatr. 2000;136:73–79. doi: 10.1016/s0022-3476(00)90053-2. [DOI] [PubMed] [Google Scholar]
- 26.Midiri M, Lo Casto A, Sparacia G, et al. MR imaging of pancreatic changes in patients with transfusion-dependent beta-thalassemia major. AJR Am J Roentgenol. 1999;173:187–192. doi: 10.2214/ajr.173.1.10397124. [DOI] [PubMed] [Google Scholar]
- 27.Montalto G, D’Angelo P, Lo Casto A, et al. Serum and fecal pancreatic enzymes in beta-thalassemia major. Int J Pancreatol. 1997;22:131–135. doi: 10.1007/BF02787471. [DOI] [PubMed] [Google Scholar]
- 28.Adelekan DA, Adekile AD, Thurnham DI. Dependence of pyridoxine metabolism on riboflavin status in sickle cell patients. Am J Clin Nutr. 1987;46:86–90. doi: 10.1093/ajcn/46.1.86. [DOI] [PubMed] [Google Scholar]
- 29.Nelson MC, Zemel BS, Kawchak DA, et al. Vitamin B6 status of children with sickle cell disease. J Pediatr Hematol Oncol. 2002;24:463–469. doi: 10.1097/00043426-200208000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Dhar M, Bellevue R, Brar S, Carmel R. Mild hyperhomocysteinemia in adult patients with sickle cell disease: A common finding unrelated to folate and cobalamin status. Am J Hematol. 2004;76:114–120. doi: 10.1002/ajh.20073. [DOI] [PubMed] [Google Scholar]
- 31.Ray D, Deshmukh P, Goswami K, Garg N. Antioxidant vitamin levels in sickle cell disorders. Natl Med J India. 2007;20:11–13. [PubMed] [Google Scholar]
- 32.Hasanato RM. Zinc and antioxidant vitamin deficiency in patients with severe sickle cell anemia. Ann Saudi Med. 2006;26:17–21. doi: 10.5144/0256-4947.2006.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilinc Y. Plasma, erythrocyte and urinary selenium levels in sickle cell homozygotes and traits. Turk J Pediatr. 1993;35:105–109. [PubMed] [Google Scholar]
- 34.Zemel BS, Kawchak DA, Fung EB, et al. Effect of zinc supplementation on growth and body composition in children with sickle cell disease. Am J Clin Nutr. 2002;75:300–307. doi: 10.1093/ajcn/75.2.300. [DOI] [PubMed] [Google Scholar]
- 35.Adewoye AH, Chen TC, Ma Q, et al. Sickle cell bone disease: Response to vitamin D and calcium. Am J Hematol. 2008;83:271–274. doi: 10.1002/ajh.21085. [DOI] [PubMed] [Google Scholar]
- 36.Kawchak DA, Schall JI, Zemel BS, et al. Adequacy of dietary intake declines with age in children with sickle cell disease. J Am Diet Assoc. 2007;107:843–848. doi: 10.1016/j.jada.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy TS, Fung EB, Kawchak DA, et al. Red blood cell folate and serum vitamin B12 status in children with sickle cell disease. J Pediatr Hematol Oncol. 2001;23:165–169. doi: 10.1097/00043426-200103000-00009. [DOI] [PubMed] [Google Scholar]
- 38.Marwah SS, Blann AD, Rea C, et al. Reduced vitamin E antioxidant capacity in sickle cell disease is related to transfusion status but not to sickle crisis. Am J Hematol. 2002;69:144–146. doi: 10.1002/ajh.10033. [DOI] [PubMed] [Google Scholar]
- 39.Chiou SS, Chang TT, Tsai SP, et al. Lipid peroxidation and antioxidative status in beta-thalassemia major patients with or without hepatitis C virus infection. Clin Chem Lab Med. 2006;44:1226–1233. doi: 10.1515/CCLM.2006.219. [DOI] [PubMed] [Google Scholar]
- 40.Livrea MA, Tesoriere L, Pintaudi AM, et al. Oxidative stress and antioxidant status in beta-thalassemia major: Iron overload and depletion of lipid-soluble antioxidants. Blood. 1996;88:3608–3614. [PubMed] [Google Scholar]
- 41.Napoli N, Carmina E, Bucchieri S, et al. Low serum levels of 25-hydroxy vitamin D in adults affected by thalassemia major or intermedia. Bone. 2006;38:888–892. doi: 10.1016/j.bone.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Wood JC, Enriquez C, Ghugre N, et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106:1460–1465. doi: 10.1182/blood-2004-10-3982. [DOI] [PMC free article] [PubMed] [Google Scholar]