

Abstract
Caveolin-1 (cav-1) is a multifunctional protein and major component of caveolae membranes serving important functions related to signal transduction, endocytosis, transcytosis, and molecular transport. We previously showed that cav-1 is overexpressed and secreted by metastatic prostate cancer cells. We now report that cav-1 gene transduction (Adcav-1) or recombinant cav-1 (rcav-1) protein treatment of cav-1-negative prostate cancer cell line LP-LNCaP or cav-1-/- endothelial cells potentiated VEGF-stimulated angiogenic signaling.
Downregulation of cav-1 in prostate cancer cell line PC-3 or human umbilical vein endothelial cells (HUVECs) through cav-1 siRNA significantly reduced basal and VEGF-stimulated phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783) and/or Akt (S473 & T308) relative to those in control siRNA treated cells. Additionally rcav-1 stimulation of cav-1 siRNA treated HUVECs restored this signaling pathway. Confocal microscopy and immunoprecipitation analysis revealed association and colocalization of VEGFR2 and PLCγ1 with cav-1 following VEGF stimulation in HUVECs. Interestingly, treatment of HUVECs with cav-1 scaffolding domain (CSD) caused significant reduction in the VEGF-stimulated phosphorylation of VEGFR2, PLCγ1 and Akt suggesting that CSD inhibits cav-1-mediated angiogenic signaling. VEGF stimulation of HUVECs significantly increased tubule length and cell migration, but this stimulatory effect was significantly reduced by cav-1 siRNA and/or CSD treatment.
The present study demonstrates that cav-1 regulates VEGF-stimulated VEGFR2 autophosphorylation and activation of downstream angiogenic signaling, possibly through compartmentalization of specific signaling molecules. Our results provide mechanistic insight into the role of cav-1 in prostate cancer and suggest the use of CSD as a therapeutic tool to suppress angiogenic signaling in prostate cancer.
Keywords: caveolin-1, VEGFR2, PLCγ1, caveolin-1 scaffolding domain, angiogenesis
Introduction
Caveolin-1 (cav-1) is a multifunctional protein and major component of caveolae membranes, serving important regulatory functions for signal transduction, endocytosis, transcytosis and molecular transport.1,2 Specific proteins such as receptor tyrosine kinases, Ser/Thr kinases, phospholipases, G-protein-coupled receptors, and Src family kinases, are localized in lipid rafts and caveolar membranes, where they interact with cav-1 through the cav-1 scaffolding domain (CSD). CSD domain-mediated activities result in the generation of platforms for compartmentalization of discrete signaling events.3 We showed previously that cav-1 is overexpressed in metastatic prostate cancer, and demonstrated that virulent prostate cancer cells secrete biologically active cav-1 that is taken up by cav-1 negative tumor cells and/or endothelial cells (ECs), leading to stimulation of specific angiogenic activities through PI3K-Akt-eNOS signaling module.4-8 Thus, secreted cav-1 has both proangiogenic and anti-apoptotic roles in the metastatic progression of prostate cancer.
Angiogenesis is a vital function for the growth of normal tissues during embryogenesis, and for the malignant growth of solid tumors. This EC-focused process involves several distinct and sequential steps, including degradation of basement membrane by proteolytic enzymes, migration, proliferation, formation of vascular loops, maturation of neovessels and neo-synthesis of basement membrane constituents. However, abnormal angiogenesis often occurs in pathological conditions such as a malignant tumor, rheumatoid arthritis, diabetic retinopathy, and other chronic inflammatory diseases.9 A key angiogenic factor, vascular endothelial growth factor (VEGF), promotes the survival, permeability, migration and proliferation in ECs during neovascularization. At the surface of ECs, the VEGF receptor 2 (VEGFR2; also known as KDR or Flk1) receptor tyrosine kinase, has been identified as the major mediator of VEGF-dependent signaling and physiological and pathological angiogenic activities.10 Binding of the dimeric VEGF to the extracellular domains of two monomeric VEGFR2 receptors induces dimerization and activation of the tyrosine kinase and phosphorylation of multiple tyrosine residues (e.g., Y951, Y1175, Y1214, Y1054 and Y1059) which, in turn, stimulate binding, phosphorylation and activation of multiple downstream molecules involved in different signaling pathways such as PLCγ1, PKC and PI3K-Akt.11-14 The Y951 phosphorylation site binds T-cell-specific adapter and subsequently forms a complex with Src that leads to the regulation of cell migration.15 VEGFR2 (Y1175) autophosphorylation site in human is another site that serves as a docking site for PLCγ1, which indirectly mediates activation of the mitogen-activated protein (MAP) kinase pathway and thus regulates cell proliferation.13 VEGFR2 (Y1175) is also a binding site for Src homology 2 and thereby activates PI3K and promotes cell migration.14 Another VEGFR2 phosphorylation site is Y1214, which is involved in the activation of Cdc42 and p38 MAP kinase pathway that regulates cell motility.16 VEGFR2 is localized in endothelial caveolae through association with cav-1 which seems to play an important role in its activation and downstream signal transduction.
Dissociation of VEGFR2 from caveolae has been shown to be essential for its autophosphorylation and activation of downstream signaling events.17 Furthermore, reports have shown that upon VEGF stimulation, phosphorylation of both VEGFR2 and cav-1 (Y14) occur simultaneously, triggering their release from caveolae/lipid rafts and colocalization at focal complexes, at the edge of lamellipodia. Thus, phospho-cav-1 appears to function as a scaffolding protein for VEGF-mediated signaling by serving as a docking site for phospho-tyrosine-binding molecules at focal adhesion complexes.18,19 However, despite the importance of VEGFR2 in the orchestration of angiogenic response, the molecular mechanisms critical for the regulation of its signaling and biological activities are not well defined, and little is known about the role of cav-1 in VEGF-mediated angiogenesis.
We demonstrate here that induction of cav-1 expression or recombinant cav-1 (rcav-1) treatment of cav-1 negative LP-LNCaP prostate cancer cells or cav-1-/- ECs led to induction of VEGF/VEGFR2 mediated angiogenic signaling. In contrast, cav-1 knockdown in PC-3 prostate cancer cells or human umbilical vein endothelial cells (HUVECs) impaired VEGF/VEGFR2-induced signaling. In HUVECs cav-1 knockdown also reduced differentiation/tubule formation and cellular migration but these activities were restored in response to rcav-1 treatment. We further show increased physical association and colocalization of cav-1 with VEGFR2 or PLCγ1 in HUVECs following VEGF stimulation. Interestingly, CSD significantly reduced VEGF-stimulated phosphorylation of VEGFR2 and downstream signaling molecules and suppressed tubule formation and cell migration in HUVEC. Our results demonstrate that cav-1 plays a pivotal role in VEGF/VEGFR2-stimulated angiogenesis signaling and associated angiogenic biological activities, and suggest a potential therapeutic use of CSD to suppress angiogenic signaling in prostate cancer.
Results
Cav-1 regulates VEGF-stimulated angiogenesis signaling in prostate cancer cells and Cav-1-/- ECs
We previously demonstrated that cav-1 stimulates angiogenic responses in prostate cancer cells and ECs through a mechanism that involves the PI3K-Akt-eNOS pathway.7 To determine the role played by cav-1 in VEGF-stimulated VEGFR2 autophosphorylation and its downstream effects, we infected cav-1 negative LP-LNCaP cells with Adcav-1 or control AdRSV, and then treated the cells with VEGF. Cav-1 overexpression significantly increased the phosphorylation of VEGFR2 (Y951) (Fig. 1A). Increased phosphorylation of PLCγ1 (Y783), Akt (S473) and Akt (T308) was also demonstrated with no change in total protein in response to cav-1 overexpression and/or VEGF treatment. The observed significant increase in the phosphorylation of VEGFR2 and PLCγ1 in response to cav-1 could be due to the effect of cav-1-mediated increased expression and secretion of growth factors including VEGF in these cells.20 On the other hand treatment of the cells with VEGF had little or no effect on the phosphorylation in the control AdRSV infected cells at all time points (0, 5 and 15 min). These results indicate that in the absence of cav-1, VEGF stimulation of VEGFR2 autophosphorylation and its downstream effects is minimal, and that cav-1 is required for optimal VEGF-stimulated signaling (Fig. 1A).
Figure 1.
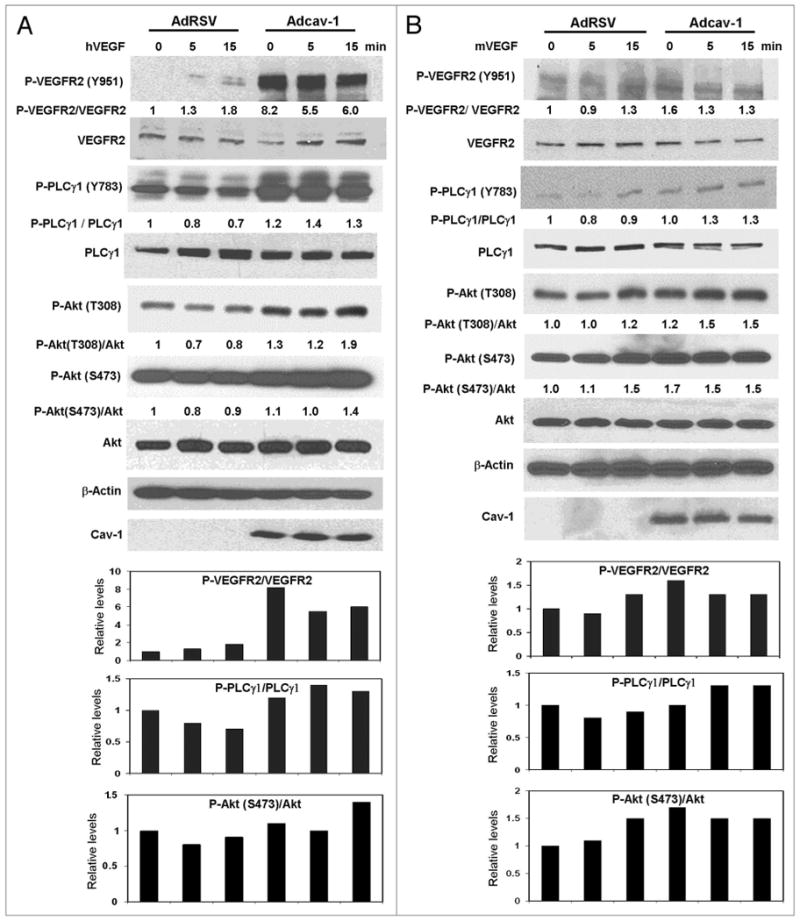

Figure 1A and B. Cav-1 stimulates the VEGF/VEGFR2 induced angiogenesis signaling pathway. (A) LP-LNCaP cells were infected with Adcav-1 or control AdRSV at an MOI of 10. Cells were incubated with SFM for 8 h, treated with hVEGF (25 ng/ml) for 0–15 min, lysed and analyzed by western blotting (B), cav-1-/- ECs were infected with Adcav-1 or control AdRSV at an MOI of 200. Cells were incubated in EBM-2 medium for 8 h, treated with mVEGF (50 ng/ml) for 0–15 min and lysed.
Figure 1C. Cav-1 stimulates the VEGF/VEGFR2 induced angiogenesis signaling pathway. (C) cav-1-/- ECs were plated overnight and incubated with 3.0 μg/ml of rcav-1 in EBM-2 for 8 h. The cells were then treated with mVEGF (50 ng/ml) for 0–15 min and lysed. In (A–C) introducing cav-1 to the cells significantly increased the phosphorylation of VEGFR2, PLCγ1 and Akt and it further stimulated their response to VEGF treatment. Blots shown (A-C) are representative of three independent experiments. Bar graphs represent densitometric data of ratio units of selected phosphorylated protein bands per total protein bands relative to that in the untreated and unstimulated controls.
To further investigate the role of cav-1 in VEGF-stimulated angiogenic activities in ECs, we introduced cav-1 into cav-1-/- ECs either by Adcav-1 infection to the MOI 200, or by rcav-1 treatment, followed by analysis of the phosphorylation status of VEGF/VEGFR2 signaling pathway associated proteins. Increased phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783), Akt (S473) and Akt (T308) was observed after overexpression of cav-1 by Adcav-1, and a further increase in the phosphorylation status of these proteins as well as PLCγ1 (Y783) was observed after VEGF treatment (Fig. 1B). VEGF treatment of cells infected with control AdRSV showed a slight increase in the phosphorylation of VEGFR2 (Y951), Akt (S473) and Akt (T308) only at the 15-min time point with no increase in PLCγ1 (Y783), which indicates a low or limited response to VEGF stimulation in the absence of cav-1. These observations suggest that cav-1 plays an important role in both basal and VEGF-stimulated VEGFR2-mediated signaling.
We also tested the effect of rcav-1 on basal and VEGF-stimulated VEGFR2-mediated angiogenic signaling in cav-1-/- ECs. Rcav-1-treated ECs showed a significant increase in the basal phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783), Akt (S473) and Akt (T308) and VEGF stimulation led to significantly increased phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783), Akt (S473) and Akt (T308) in rcav-1-treated cells, compared to cells that were not treated with rcav-1 (Fig. 1C).
Cav-1 knockdown in PC-3 and HUVECs impairs VEGF-stimulated angiogenesis signaling
We investigated cav-1 regulation of basal and VEGF-stimulated VEGFR2-mediated angiogenic signaling in PC-3 cells and human ECs. We transfected PC-3 cells with cav-1-specific siRNA or siRNA of irrelevant target specificity as a non-specific control (NC), to downregulate cav-1 expression. In unstimulated cells the phosphorylation status of VEGFR2 (Y951), PLCγ1 (Y783) and Akt (S473 & T308) was not changed significantly after downregulation of cav-1. However, downregulation of cav-1 reduced the response to VEGF stimulation as shown in reduced phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783) and Akt (S473 & T308) at two time points (5 and 15 min) as compared with that of the NC. Interestingly, cav-1 phosphorylation (Y14) increased significantly in response to VEGF at both 5 and 15 min and in both NC and cav-1 siRNA (Fig. 2A).
Figure 2.


Figure 2A. Downregulation of cav-1 in PC-3 cells and HUVECs by cav-1 siRNA impaired the VEGF/VEGFR2 angiogenesis signaling pathway. (A) PC-3 cells were transfected with specific cav-1 siRNA or a control siRNA, followed by incubation with SFM for 8 h prior to hVEGF (25 ng/ml) treatment for 0–15 min.
Figure 2B. Downregulation of cav-1 in PC-3 cells and HUVECs by cav-1 siRNA impaired the VEGF/VEGFR2 angiogenesis signaling pathway. (B) HUVECs were transfected on two consecutive days and incubated with rcav-1 in EBM-2 for 8 prior to treatment with hVEGF (25 ng/ml) for 0–15 min. Cav-1 downregulation not only reduced the phosphorylation status of VEGFR2, PLCγ1 and Akt significantly but also reduced the VEGF stimulated specific phosphorylation sites of these proteins. Rcav-1 (3 μg/ml) treatment of the transfected HUVECs restored the phosphorylation and the VEGF stimulated phosphorylation levels of VEGFR2, PLCγ1 and Akt. Blots shown (A and B) are representative of three independent experiments. Bar graphs represent densitometric data of ratio units of selected phosphorylated protein bands per total protein bands relative to that in the untreated and unstimulated controls.
We also investigated the effect of cav-1 on VEGF/VEGFR2 signaling in HUVECs through downregulation of cav-1 using cav-1 siRNA transfection, and through rcav-1 treatment followed by stimulation with VEGF in HUVECs that had been previously treated with cav-1 siRNA. Using a double transfection protocol, the cav-1 levels were reduced significantly after specific cav-1 siRNA treatment (Fig. 2B). Downregulation of cav-1 in HUVECs significantly reduced basal and VEGF-stimulated phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783) and Akt (S473 & T308) compared to those treated with the NC. Interestingly, rcav-1 treatment of these cells restored the basal phosphorylation status of these signaling molecules and partially restored the response to VEGF stimulation, i.e., P-VEGFR2, PLCγ1 and P-Akt (Fig. 2B). These results demonstrate that cav-1 is an important regulator of basal and VEGF-stimulated angiogenesis signaling in prostate cancer cells and HUVECs. Importantly, rcav-1 restored VEGF-stimulated angiogenic signaling in cav-1 siRNA treated HUVECs, further demonstrating a potential role for secreted cav-1 in vivo.7
Cav-1 regulation of VEGF-stimulated angiogenesis signaling is associated with Cav-1 binding to VEGFR2 and PLCγ1
We investigated whether VEGF stimulation of angiogenesis signaling involves cav-1-VEGFR2 and/or cav-1-PLCγ1 interaction and direct binding that may sequester these molecules to specific cellular compartments. Amino acid sequence analysis revealed one potential cav-1-binding motif at the VEGFR2 C terminus, 1089WSFGVLLWEIF1099, and the PLCγ1 molecule revealed two potential cav-1-binding consensus sequence, one at the N terminus, 293FFLDEFVTF301, and the other at the C terminus, 1154FAFLRFVVY1162. We performed reversed coimmunoprecipitation using HUVEC lysates under high stringency in the presence of NP-40. VEGFR2 immunoprecipitation complexes contained cav-1, and cav-1 levels were increased (70%) following VEGF treatment (Fig. 3A). Similar results were obtained in the reversed coimmunoprecipitation complexes, in which cav-1 immunoprecipitation complexes contained VEGFR2, and VEGFR2 levels were increased (50%) following VEGF treatment (Fig. 3A). Interestingly, cav-1, and VEGFR2 colocalization was demonstrated using confocal microscopy. In unstimulated cells cav-1 and VEGFR2 were primarily colocalized in the plasma membrane, and after treatment with VEGF for 5 min both VEGFR2 and cav-1 were internalized to the cytoplasm as seen by intracellular punctuate staining (Fig. 3B). Cav-1 and PLCγ1 immunoprecipitation experiments also revealed PLCγ1 in the cav-1 coimmunoprecipitation complexes, and PLCγ1 levels were increased (60%) following VEGF treatment. Conversely, PLCγ1 immunoprecipitation complexes contained cav-1, and cav-1 levels were increased (60%) following VEGF treatment (Fig. 3C). Confocal microscopy also showed a similar pattern of PLCγ1 and cav-1 colocalization to those of VEGFR2 and cav-1 in unstimulated and VEGF-stimulated cells (Fig. 3D).
Figure 3.

Cav-1 interacts with VEGFR2 and PLCγ1 in HUVEC. The cells were incubated with EBM-2 medium for 8 h and stimulated with 25 ng/ml of VEGF for 5 min. (A) Cell lysates were immunoprecipitated (IP) with either anti-cav-1 or anti-VEGFR2 rabbit pAb, and mock immunoprecipitates with IgG and Protein A/G Plus were also included. The coimmunoprecipitates were analyzed by western blotting (WB) with anti-cav-1 or anti-VEGFR2 mouse mAb. (B and D) HUVECs were incubated with EBM-2 for 8 h and treated with hVEGF (25 ng/ml) for 5 min. The cells were fixed, permeabilized and incubated with anti-cav-1 rabbit pAb, anti-VEGFR2 mouse mAb in (B), and anti-PLCγ1 mouse mAb in (D). The cells were dually stained with FITC anti-rabbit IgG, and TRITC anti-mouse IgG. Nuclei were visualized by Hoechst 33342 staining. (C) Cell lysates were immunoprecipitated with either anti-cav-1 or anti-PLCγ1 rabbit pAb, and analyzed by western blotting with anti-cav-1 or anti-PLCγ1 mouse mAb. Equal amounts of cell lysates were used to perform immunoprecipitation. Blots shown in (A and C) are representative of three independent experiments. Quatifitication by denisometry of the coimmunoprecipitated protein bands in (A and C) represent the ratio units of bound protein in VEGF treated preparations with respect to that in untreated controls.
These results demonstrate that VEGFR2 and PLCγ1 exist partially bound to cav-1 in HUVECs and that VEGF treatment initially increases the degree of association, colocalization and internalization, suggesting that cav-1 participates mechanistically in VEGF-stimulated angiogenic responses in this system.
CSD inhibits VEGF-stimulated angiogenesis signaling
We previously showed that rcav-1 lacking CSD failed to stimulate angiogenic activities in cav1-/- ECs, including tubule formation, migration and stimulation of eNOS phosphorylation (S1177).7 These results showed that endocytosis of exogenous rcav-1 and stimulation of angiogenic activities is mediated in part by CSD, which is critical for cellular internalization of the protein. CSD was also shown by others to inhibit VEGF-induced vascular leakage through the inhibition of eNOS.21 We first tested whether the CSD peptide can penetrate and become internalized in HUVECs without requiring a peptide carrier. Biotin-conjugated peptide and scrambled (s) control peptide were incubated with HUVECs in serum free culture medium. Both the CSD and the control sCSD became internalized in HUVECs and distributed throughout the cytoplasm (Fig. 4A), demonstrating that CSD readily penetrates and is internalized without requiring the antennapedia (AP) internalization sequence.21
Figure 4.
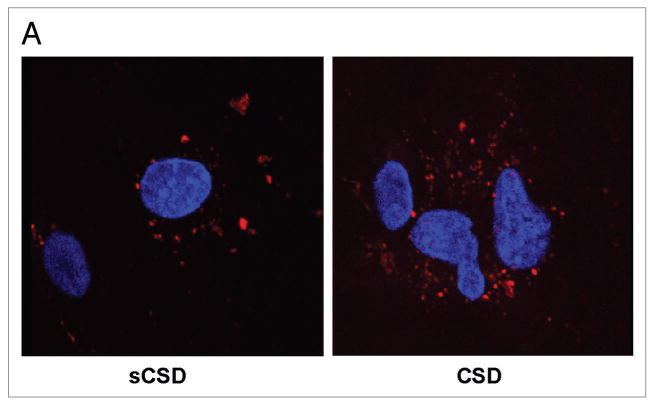
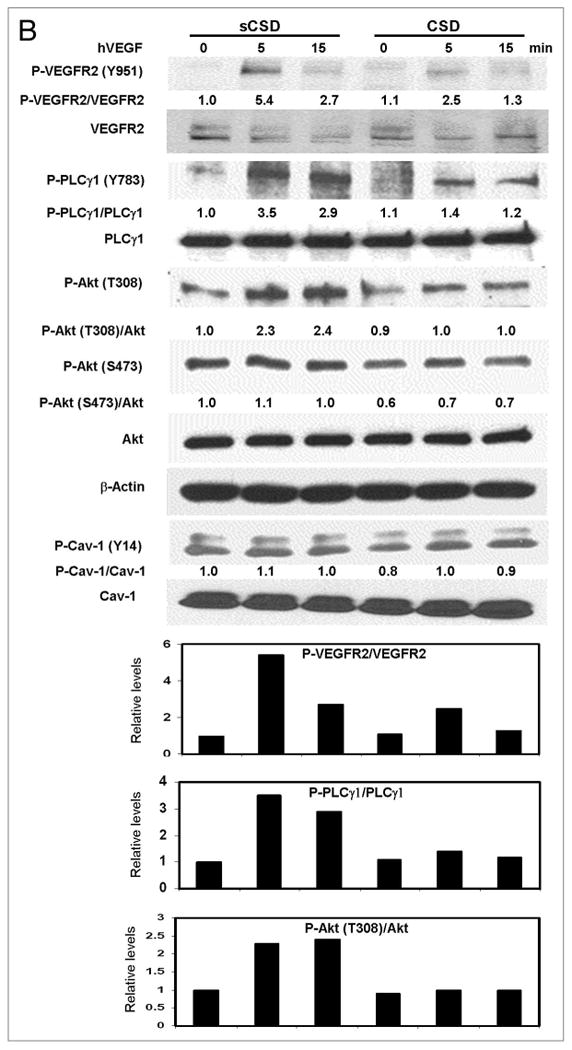
Figure 4A. CSD inhibits VEGF and cav-1 mediated angiogenesis signaling. (A) HUVECs were treated with biotin conjugated CSD or sCSD (5.0 μM) in EBM-2 for 8 h and were then washed, fixed, permeabilized and stained with TRITC-Streptavidin. The internalization of CSD and sCSD was detected on confocal microscopy and nuclei were visualized by using Hoechst 33342 staining.
Figure 4B. CSD inhibits VEGF and cav-1 mediated angiogenesis signaling. (B) HUVECs were plated overnight and treated with CSD or sCSD (5.0 μM) in EBM-2 for 8 h. The cells were then treated with hVEGF (25 ng/ml) for 0–15 min, lysed and western blotted. CSD treatment of HUVEC reduced the VEGF stimulated phosphorylation of VEGFR2, PLCγ1 and Akt compared with that obtained with sCSD treatment. Blots shown are representative of three independent experiments. Bar graphs represent densitometric data of ratio units of phophorylated protein bands per total protein bands relative to that in the untreated and unstimulated controls.
To test the effect of CSD on VEGF-stimulated angiogenesis signaling, we treated HUVECs with CSD peptide in serum free medium followed by treatment with VEGF. CSD treatment of HUVECs significantly reduced VEGF-stimulated phosphorylation of VEGFR2 (Y951), PLCγ1 (Y783) and Akt (S473 &T308) as compared to those treated with control sCSD (Fig. 4B). Modest reductions in basal phosphorylation levels of Akt were observed in CSD-treated compared to sCSD-treated HUVECs (Fig. 4B). These data demonstrate that CSD inhibits VEGF-stimulated angiogenesis signaling through inhibition of endogenous cav-1 function.
VEGF-stimulated tubule formation and cell migration is mediated by Cav-1 and inhibited by CSD
EC migration and differentiation are important events for angiogenesis which involve new capillary formation from preexisting vessels. To further demonstrate the role of cav-1 in VEGF-stimulated angiogenesis in vitro, we used two biological assays, tubule formation and wound healing migration, in which HUVECs were transfected with cav-1 siRNA or NC and then treated with VEGF or left untreated. VEGF treatment of HUVECs increased the tubule length and cell migration in NC transfected cells compared to their untreated counterparts, but this stimulatory effect of VEGF was significantly impaired when cav-1 was downregulated (Fig. 5A–D). Downregulation of cav-1 by transfection with specific siRNA in unstimulated and VEGF-stimulated HUVECs caused a significant reduction in tubule length (p < 0.05, and p < 0.01, respectively; Fig. 5B) and the number of migrated cells compared with that of NC (p < 0.01, and p < 0.01, respectively; Fig. 5D). An interesting finding was that CSD treatment of unstimulated and VEGF-stimulated HUVECs reduced the tubule length in NC-transfected cells (p < 0.05, and p < 0.01, respectively; Fig. 5B), but not in cav-1 siRNA transfected cells (Fig. 5B) compared to treatment with sCSD. Additionally, CSD treatment of unstimulated and VEGF-stimulated HUVECs significantly reduced cell migration in NC-transfected cells (p < 0.05, and p < 0.01, respectively; Fig. 5D), and in cav-1 siRNA transfected cells (p < 0.05, and p < 0.05, respectively; Fig. 5D) compared to treatment with sCSD. These results show that downregulation of cav-1 through cav-1 siRNA or CSD treatment alone led to significant reductions of angiogenic activities in HUVECs, and that combining the two treatments yields additive or synergistic effects.
Figure 5.


Figure 5A and B. VEGF-stimulated tubule formation and cell migration is mediated by cav-1 and inhibited by CSD. (A) Representative micrograph shows newly formed tubules of HUVECs transfected with cav-1 siRNA or NC and cultured on Matrigel matrix for 18 h in the presences and absence of hVEGF (25 ng/ml), and treated with sCSD or CSD (5 μM). (B) Bar graph represents the relative tubule lengths of HUVECs incubated as in (A).
Figure 5C and D. VEGF-stimulated tubule formation and cell migration is mediated by cav-1 and inhibited by CSD. (C) Representative micrograph shows wound healing migration of HUVECs transfected with siRNA or NC and treated with or without VEGF (25 ng/ml) and with sCSD or CSD (5μM). (D) Bar graph represents the relative numbers of HUVECs transfected with cav-1 siRNA or NC that migrated into the cleared area after wounding of the culture. Cells were treated with sCSD or CSD and hVEGF for 24 h and the number of migrated cells were counted. The ratios in (B and D) represent tubule lengths and numbers of migrated cells relative to those in cells transfected with NC and treated with sCSD and hVEGF condition ± SD of three independent experiments. p values in (B and D) were determined by two-sided t test. *statistically significant difference (*p < 0.05, **p < 0.01), NS Columns, mean data; error bars, SD.
Discussion
The molecular mechanisms involved in VEGFR2-mediated signaling of VEGF stimulated angiogenic responses in cancer cells and ECs are still poorly understood. The present study demonstrated that cav-1 plays a critical role in VEGF-stimulated VEGFR2 autophosphorylation and activation of downstream signaling in prostate cancer cells and ECs, possibly through compartmentalization of the signaling molecules. Despite a number of reports on the involvement of cav-1 in VEGF-stimulated angiogenesis, whether cav-1 is a stimulator or an inhibitor of angiogenesis is still controversial. Cav-1 was shown to play an important role in VEGF-stimulated angiogenic activities by acting both as a negative regulator of VEGFR-2 activity under resting conditions and as a substrate that is tyrosine-phosphorylated under activating conditions.17 In addition, cav-1 overexpression was reported to enhance endothelial capillary tubule formation.22 We and others found that production of nitric oxide and tubule formation in cav-1-/- ECs were significantly reduced compared with those in cav-1+/+ with or without VEGF treatment.7,23 Localization of VEGFR2 within caveolae was found to be essential in coupling VEGF-stimulated VEGFR2 phosphorylation and downstream angiogenic signaling, and it was also demonstrated that phosphorylated VEGFR2 dissociates rapidly from caveolae following stimulation.17 Recently, Ikeda et al. reported that VEGF stimulation of ECs results in the phosphorylation of both VEGFR2 and cav-1 (Y14) and that the two molecules remain associated following their release from caveolae/lipid raft.19 These reports emphasize the multiple and complex functions of cav-1 in VEGF-stimulated angiogenic responses. They further reveal that the mechanisms through which these responses are mediated are still incompletely understood.
In this study we demonstrated that introduction of cav-1 through adenoviral vector-mediated gene transduction or rcav-1 treatment caused a significant increase in VEGFR2 phosphorylation and its downstream signaling effectors in the cav-1-deficient prostate cancer cell line LP-LNCaP and in cav-1-/- ECs, in both the presence and absence of exogenously added VEGF. We previously found that treating cav-1-/- ECs with rcav-1 led to cellular internalization of rcav-1 followed by stimulation of angiogenic activities through the PI3K-Akt-eNOS pathway.7 Importantly, we further showed that rcav-1 internalization and stimulation of angiogenic responses were mediated by CSD. We demonstrated here that cav-1 is critical for ECs to maintain maximal angiogenic signaling, and that VEGF stimulation of angiogenesis is impaired without moderate levels of intracellular cav-1. These data are supported by the demonstration of angiogenic responses in cav-1-/- ECs when relatively low levels of cav-1 were introduced by transfection of cav-1 cDNA into these cells.23 By using immunoprecipitation and immunostaining we showed that cav-1 interacts directly with VEGFR2 or PLCγ1 and that this interaction increases in cells treated with VEGF (25 ng/ml) for 5 min. Cav-1 was reported to interact with VEGFR2 in the resting state but to dissociate from VEGFR2 when cells are treated with VEGF, which also leads to the phosphorylation of both molecules.17 Another group reported that cav-1 is associated with VEGFR2 within caveolae and that VEGF treatment leads to phosphorylation of both molecules and their release from the caveolae as a complex.19 The differences between the results of these studies may be explained by variations in VEGF concentration and treatment times or by the methods used in the analysis.
In the past decade, the list of proteins localized to the caveolae or bound to cav-1 has grown and includes G-protein-coupled receptors, growth factors receptors, tyrosine kinases, Ser/Thr kinases, enzymes, cellular proteins/adaptors, nuclear proteins, and structural proteins.24 Most of the reports that point to VEGFR2 or PLCγ1 colocalization within caveolae or binding to cav-1 have been based on immunofluorescence, or density-gradient centrifugation, often in combination with extraction of membrane fractions with cold Triton X-100 to isolate detergent-resistant, cav-1-rich, low buoyancy membranes. Co-fractionation of a protein with these detergent-resistant membranes might indicate caveolar localization. However, such fractionation leads to the isolation of low-buoyant-density fractions and has the disadvantage that the isolated fractions contain both caveolae and non-caveolar lipid rafts. Thus additional studies that utilize coimmunoprecipitation combined with immunofluorescence or electron microscopy are necessary to confirm the caveolar localization or direct binding of a given protein to cav-1.
In primary ECs VEGFR2 is localized to both the plasma membrane and endosomes. VEGF binding stimulates VEGFR2 autophosphorylation, internalization and the subsequent ubiquitination necessary for endosomal sorting events that lead to lysosomal degradation.25 Our results showing direct cav-1 binding to VEGFR2 before and after VEGF stimulation suggest an important role for cav-1 in stabilizing VEGFR2 after ligand binding. These data are supported by cav-1-VEGFR2 colocalization and internalization 5 min after VEGFR2 stimulation (Fig. 3). Additional studies that address the kinetics and compartmentalization of cav-1 following VEGF stimulation will further clarify this role. It is also of interest that cav-1 binds to PLCγ1 and that cav-1-PLCγ1 colocalization and internalization follow a pattern that is similar to cav-1 and VEGFR2 following VEGF stimulation in HUVECs (Fig. 3). These data imply that cav-1 is involved in the organization and compartmentalization of multiple signaling molecules during VEGF-stimulated angiogenic signaling.
It is noteworthy that exogenously added rcav-1 functioned similarly to endogenously expressed cav-1 in cav-1-/- ECs and HUVECs (Figs. 1 and 2). Although, in general, the signaling responses to rcav-1 were less than those elicited by cav-1 gene transduction, rcav-1 stimulation of VEGFR2 signaling was clear. It is particularly interesting that rcav-1 treatment significantly increased VEGFR2 and Akt phosphorylation in cav-1-/- ECs and HUVECs (Figs. 1C and 2B). This result extends our previous analysis of the effects of rcav-1 on angiogenic signaling that showed virulent prostate cancer cells secrete biologically active cav-1.6 Rcav-1 is taken up by cav-1 negative tumor cells and/or ECs leading to stimulation of specific angiogenic activities through the PI3K-Akt-eNOS signaling module.7 Activation of VEGFR2 signaling in prostate cancer cells and prostate cancer associated ECs by prostate cancer-derived, secreted cav-1 presents an interesting paradigm for understanding the engagement of the tumor microenvironment by cav-1. Additional studies in this area are warranted.
In this paper we also report that CSD treatment can inhibit VEGF-stimulated angiogenic signaling, tubule formation and cell migration. To date, the available data on the effect of CSD in angiogenesis are controversial, with no consensus yet reached on a clearly defined mechanism of action. One group used CSD conjugated to the C terminus of the AP and found that treatment of ECs with CSD led to enhanced capillary tubule formation.22 On the other hand, the same peptide, cavtratin, was reported to inhibit eNOS-dependent vascular leakage in established tumors by enhancing apoptosis, and decreasing tumor angiogenesis.21 Both of those reports suggested that the mechanism of cavtratin's action is similar to that of molecular cav-1, in that the peptide functions as an enhancer of capillary tubule formation22 and as a negative regulator of eNOS;21 in other words, the CSD acts as a surrogate of cav-1.
We show in this paper that CSD treatment led to a reduction in the VEGF-stimulated phosphorylation of VEGFR2, PLCγ1 and Akt (Fig. 4). We further show that CSD inhibition of angiogenesis signaling is associated with inhibition of two critical biological activities in angiogenesis, i.e., EC tubule formation and migration (Fig. 5). Our results are not in conflict with those of the previous study that showed CSD-mediated inhibition of angiogenesis.21 However, our results, in aggregate, are consistent with a mechanism of CSD inhibition of cav-1-mediated, VEGF-stimulated angiogenesis, rather than a mechanism through which CSD acts as a “cav-1 surrogate” for angiogenesis stimulation22 or inhibition.21
Overall, our results show that endogenously expressed or exogenously added cav-1 plays an important role in VEGFR2 autophosphorylation, VEGF mediated signaling, and EC tubule formation and migration in prostate cancer cells and ECs. These activities are associated with cav-1 binding to and compartmentalization with VEGFR2 and PLCγ1. Finally, our data present a novel mechanism for potential therapeutic use of CSD to suppress angiogenic signaling in prostate cancer.
Materials and Methods
Cells, antibodies and reagents
ECs from cav-1-/- mouse aortas were isolated and prepared and grown in endothelium growth medium (EGM) as described previously.7 HUVECs (Lonza, Inc., Walkersville, MD) were cultured at 37°C in 5% CO2 in EGM-2 Bullet kit medium (Lonza). Cells from passages 4 to 7 were used for these studies. LNCaP and PC-3 cells were obtained from ATCC and maintained at 37°C in 5% CO2 in complete RPMI1640 medium supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA). Anti-cav-1 and anti-VEGFR2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-PLCγ1, anti-phospho PLCγ1 (Y783), anti-phospho-Akt (T308), and anti-phospho Akt (S473) were purchased from Cell Signaling Technology (Danvers, MA) and anti-Phospho-cav-1, anti-PLCγ1 (mAb), and Akt were purchased from BD Biosciences (San Jose, CA). Anti-phospho-VEGFR2 (Y 951) and recombinant human and mouse VEGF were purchased from Invitrogen (Carlsbad, CA). Rcav-1 was purified by the modified procedure described previously.7 The CSD (DGI WKA SFT TFT VTK YWF YR) peptide and its control scrambled CSD (WGI DKA FFT TST VTY KWF RY) conjugated to biotin through its N terminal were purchased from Biosynthesis, Inc., (Lewisville, TX).
Small interfering RNA
PC-3 cells or HUVECs were transfected with cav-1 specific small interfering RNA (siRNA) and negative control siRNA constructs (Ambion, Inc., Austin, TX) using siPORT™ Amine transfection reagent according to the manufacturer's instruction (Ambion). Briefly, cells were transfected with 20 nM cav-1 siRNA (on two consecutive days for HUVECs), and after 48 h, cells were incubated with endothelium basal medium-2 (EBM-2) (Lonza, Inc.,) or serum free medium (SFM, for PC-3 cells) for 8 h followed by recombinant human (rh) VEGF (Invitrogen) treatment for 0–15 min. The cells were lysed in RIPA buffer containing protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN), and phosphatase inhibitor cocktails I and II (Sigma, Inc., St. Louis, MO).
Overexpression of Cav-1 in low passage LNCaP and Cav-1-/- EC
Recombinant adenoviral vectors containing human cav-1 cDNA (Adcav-1) or control AdRSV without a cDNA were used to infect low passage LNCaP (LP-LNCaP) cells at a multiplicity of infection (MOI) of 10 or cav-1-/- ECs at MOI of 200. Cells were infected in SFM for 3 h, after which the medium was replaced with complete medium and incubated for 48 h. The medium was then removed and cells were incubated in SFM (LP-LNCaP cells) or EBM-2 (cav-1-/- ECs) for 8 h. Recombinant human or mouse VEGF were added to the cells, incubated for 0 to 15 min, and then cells were lysed with RIPA lysis buffer containing the protease and phosphatase inhibitor cocktails.
Immunoprecipitation and western blotting
For immunoprecipitation, cells were solubilized with lysis buffer containing 1% NP-40, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 1 mM EGTA, protease inhibitor cocktail, and phosphatase inhibitor cocktails I and II. Soluble proteins (0.5 mg) were precleared and incubated with primary antibody (2 μg) overnight at 4°C with shaking. Protein A/G Plus Agarose (Santa Cruz Biotechnology) was added and incubated for 1 h, after which the bound proteins were washed three times with lysis buffer, boiled in SDS sample buffer and analyzed by western blotting.
The proteins were transferred onto nitrocellulose membrane, and blotted with specific antibody and antibody detection was performed by using a chemiluminescence-based detection system (Pierce Biotechnology, Rockford, IL). Quantification was carried out using Nikon's NIS-Elements AR 3.0 imaging and quantification software; data expressed as the ratio units of phosphorylated protein per total protein relative to that in the untreated and unstimulated controls.
Immunofluoresence microscopy
Fixed cells were permeabilized and immunostained using the following primary antibodies: Rabbit polyclonal anti-cav-1 antibody (pAb), mouse monoclonal (mAb) anti-VEGFR2 (Santa Cruz Biotechnology) and anti-PLCγ1 mAb (BD Biosciences) as described previously.7 Cav-1 was detected with anti-rabbit fluorescein isothiocyanate conjugated (FITC) (Jackson Immuno Research, West Grove, PA), and VEGFR2 and PLCγ1 were detected with anti-mouse tetramethyl rhodamine isothiocyanate conjugated (TRITC) (Invitrogen). Nuclei were visualized using Hoecgst 33342 staining. Immunostaining was analyzed using an Olympus IX71 FV500 laser-scanning confocal microscope with photomultiplier tubes (PMTs). Images were visualized using an Olympus 60× PlanApo oil immersion objective and captured using FluoView v5.0 software and the PMTs. All images were captured using the same PMT voltages, which were determined by the brightest experimental conditions.
Tubule formation assays
The tubule formation assay was performed as described previously.7 Briefly, Cells were trypsinized counted and plated on Matrigel™ Matrix (growth factor-reduced matrigel; BD Biosciences) coated 24 well-plates in EGM-2 (Lonza, Inc.,) medium containing 1% fetal calf serum. After 16–20 h of incubation at 37°C in 5% CO2, images of the tubules formed were captured by phase contrast microscopy. The tubule lengths in each well were measured in 5 low-power fields using Nikon's NIS-Elements AR 3.0 imaging and quantification software.
Wound-healing migration assay
The wound-healing migration assay was also performed as described previously.7 Briefly, HUVECs in EGM-2 medium were incubated for 16 h in 24-well plates to 70–80% confluence. A straight longitudinal incision was made on the monolayer using a pipette tip, and the cells were washed with EGM-2. The cells were then reincubated for 24 h in EGM-2 and stained using the HEMA3 stain set (Biochemical Sciences Inc., Swedesboro, NJ) according to the manufacturer's instructions. The cells that had migrated into the cleared area were counted using Nikon's NIS-Elements AR 3.0 colony count software.
Statistical analysis
Differences in tubule length and the number of migrated cell were determined by unpaired two-sided t test, using StatView software (Version 5.0; SAS Institute).
Acknowledgments
This work was supported in part by NIH RO1 CA68814 and Department of Defense grant DAMD PC051247.
Abbreviations
- Cav-1
caveolin-1
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
- LP-LNCaP
low passage-LNCaP
- HUVEC
human umbilical vein endothelial cell
- CSD
caveolin-1 scaffolding domain
- FITC
fluorescein isothiocyanate conjugated
- TRITC
tetramethyl rhodamine isothiocyanate conjugated
- EGM-2
endothelial growth medium-2
- EBM-2
endothelial basal medium-2
References
- 1.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 2.Shaul PW, Anderson RG. Role of plasmalemmal caveolae in signal transduction. Am J Physiol. 1998;275:L843–51. doi: 10.1152/ajplung.1998.275.5.L843. [DOI] [PubMed] [Google Scholar]
- 3.Shatz M, Liscovitch M. Caveolin-1: a tumor-promoting role in human cancer. Int J Radiat Biol. 2008;84:177–89. doi: 10.1080/09553000701745293. [DOI] [PubMed] [Google Scholar]
- 4.Nasu Y, Timme TL, Yang G, Bangma CH, Li L, Ren C, et al. Suppression of caveolin expression induces androgen sensitivity in metastatic androgen-insensitive mouse prostate cancer cells. Nat Med. 1998;4:1062–4. doi: 10.1038/2048. [DOI] [PubMed] [Google Scholar]
- 5.Yang G, Truong LD, Timme TL, Ren C, Wheeler TM, Park SH, et al. Elevated expression of caveolin is associated with prostate and breast cancer. Clin Cancer Res. 1998;4:1873–80. [PubMed] [Google Scholar]
- 6.Tahir SA, Yang G, Ebara S, Timme TL, Satoh T, Li L, et al. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res. 2001;61:3882–5. [PubMed] [Google Scholar]
- 7.Tahir SA, Yang G, Goltsov AA, Watanabe M, Tabata K, Addai J, et al. Tumor cell-secreted caveolin-1 has proangiogenic activities in prostate cancer. Cancer Res. 2008;68:731–9. doi: 10.1158/0008-5472.CAN-07-2668. [DOI] [PubMed] [Google Scholar]
- 8.Yang G, Addai J, Wheeler TM, Frolov A, Miles BJ, Kadmon D, et al. Correlative evidence that prostate cancer cell-derived caveolin-1 mediates angiogenesis. Hum Pathol. 2007;38:1688–95. doi: 10.1016/j.humpath.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 11.Dougher M, Terman BI. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene. 1999;18:1619–27. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 12.Kendall RL, Rutledge RZ, Mao X, Tebben AJ, Hungate RW, Thomas KA. Vascular endothelial growth factor receptor KDR tyrosine kinase activity is increased by autophosphorylation of two activation loop tyrosine residues. J Biol Chem. 1999;274:6453–60. doi: 10.1074/jbc.274.10.6453. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20:2768–78. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem. 2004;279:22267–75. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005;24:2342–53. doi: 10.1038/sj.emboj.7600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamalice L, Houle F, Jourdan G, Huot J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene. 2004;23:434–45. doi: 10.1038/sj.onc.1207034. [DOI] [PubMed] [Google Scholar]
- 17.Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14:334–47. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaverina I, Krylyshkina O, Small JV. Regulation of substrate adhesion dynamics during cell motility. Int J Biochem Cell Biol. 2002;34:746–61. doi: 10.1016/s1357-2725(01)00171-6. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda S, Ushio-Fukai M, Zuo L, Tojo T, Dikalov S, Patrushev NA, et al. Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2005;96:467–75. doi: 10.1161/01.RES.0000158286.51045.16. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Ren C, Yang G, Goltsov AA, Tabata K, Thompson TC. Caveolin-1 promotes autoregulatory, Akt-mediated induction of cancer-promoting growth factors in prostate cancer cells. Mol Cancer Res. 2009;7:1781–91. doi: 10.1158/1541-7786.MCR-09-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gratton JP, Lin MI, Yu J, Weiss ED, Jiang ZL, Fairchild TA, et al. Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell. 2003;4:31–9. doi: 10.1016/s1535-6108(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Wang XB, Park DS, Lisanti MP. Caveolin-1 expression enhances endothelial capillary tubule formation. J Biol Chem. 2002;277:10661–8. doi: 10.1074/jbc.M110354200. [DOI] [PubMed] [Google Scholar]
- 23.Sonveaux P, Martinive P, DeWever J, Batova Z, Daneau G, Pelat M, et al. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–61. doi: 10.1161/01.RES.0000136344.27825.72. [DOI] [PubMed] [Google Scholar]
- 24.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 25.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, et al. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–82. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]