

Abstract
Human immunodeficiency virus type 1 (HIV-1) persists in a latent reservoir of infected resting memory CD4 cells in patients receiving antiretroviral therapy. We assessed whether multitarget therapy with enfuvirtide, 2 reverse-transcriptase inhibitors, and a ritonavir-boosted protease inhibitor leads to decay of this reservoir. Nineteen treatment-naive patients initiated this regimen; 9 experienced virologic suppression and continued enfuvirtide-containing therapy for at least 48 weeks. In enfuvirtide-treated patients with virological suppression, there was no decay of the latent reservoir (95% confidence interval for half-life, 11 months to infinity). The stability of the latent reservoir despite intensive therapy suggests that new strategies are needed to eradicate HIV-1 from this reservoir.
Human immunodeficiency virus type 1 (HIV-1) persists as integrated DNA in resting memory CD4 T cells in patients receiving antiretroviral therapy (ART) [1–3]; this latent reservoir has an estimated half-life of 44 months [4]. The lack of decay of this latent reservoir may reflect the intrinsic stability of the virus in resting memory CD4 cells, but it may also be due to ongoing low-level HIV-1 replication that replenishes the reservoir. Although most patients receiving standard antiretroviral regimens have low-level viremia detectable with special assays [5], there is controversy as to whether this residual viremia represents ongoing cycles of HIV-1 replication or episodic release of virus from stable reservoirs (eg, from cellular sources or anatomic compartments, such as the central nervous system). If current antiretroviral regimens completely block viral replication, then more intensive ART will not lead to decay of the latent reservoir. Alternatively, if ongoing viral replication contributes to maintenance of the latent reservoir, then increasing the potency of therapy by inhibiting additional viral targets may further diminish replication and lead to more rapid decay of the latent reservoir. Studies of the effect of more intensive ART on the size of the latent reservoir may help determine whether ongoing replication contributes to the stability of the latent pool.
Current first-line antiretroviral regimens inhibit HIV-1 enzymes that function inside the host cell, such as reverse transcriptase or protease. The antiretroviral activity of combination therapy with reverse-transcriptase and protease inhibitors may be increased by adding an additional agent—such as the fusion inhibitor enfuvirtide—that blocks an extracellular target [6]. Inhibition of HIV-1 via an extracellular site of action might also more effectively suppress replication if efflux pumps or other mechanisms result in cellular sanctuaries of viral replication. We assessed whether initiation of intensive ART with a multitarget regimen that includes fusion, protease, and reverse-transcriptase inhibitors leads to decay of the HIV-1 latent reservoir.
Methods
In a single-arm study (AIDS Clinical Trials Group A5173), treatment-naive HIV-1-infected patients with CD4 cell counts of ≥ 100 cells/μL, HIV-1 RNA loads of ≥ 1000 copies/mL, and no drug-resistance mutations (as determined by genotypic testing) initiated therapy with 90 mg of enfuvirtide administered by subcutaneous injection twice daily, 300 mg of tenofovir disoproxil fumarate administered daily, either 200 mg of emtricitabine or 300 mg of lamivudine administered daily, 1000 mg of saquinavir mesylate administered twice daily, and 100 mg of ritonavir administered twice daily. Patients who achieved a viral load of <50 copies/mL and continued enfuvirtide-containing ART were tested at week 24 and then every 24 weeks for the frequency of latently infected resting CD4 cells (measured in infectious units per million cells [IUPM]), using methods described elsewhere [7]; the planned study duration was 96 weeks. Viral load testing (Roche Amplicor HIV-1 Monitor assay, ultrasensitive version 1.5) was performed at a central laboratory for samples collected at study entry; on days 4 and 10; at weeks 4, 8, 12, and 16; and then every 8 weeks until week 96. CD4 cell counts were measured at entry; on day 10; at weeks 4, 8, 12, and 16; and then every 8 weeks until week 96. CD4 and CD8 cell activation was assessed at entry and every 24 weeks, by measuring the percentage of cells that expressed CD38 and by estimating CD38 cell surface density from the mean fluorescence intensity of this marker. CCR5 density on nonactivated (HLA-DR–negative) CD4 cells was measured at study entry.
We analyzed the latent-reservoir decay rate in patients who attained a viral load of <50 copies/mL and received enfuvirtide-containing ART for at least 48 weeks (the analysis cohort). On the basis of previous estimates of assay and biologic variation and the correlation among repeated observations [8], in the initial study design we estimated that 10 evaluable patients, each with 4 latent-reservoir measurements, would provide 80% power to detect a change in the decay rate from the reported value of 44 months to <12 months. The original target enrollment was 40 patients, with the expectation that some would not attain a viral load of <50 copies/mL and that others would discontinue the study regimen prematurely. Because of both slow enrollment and data showing no evidence of decline in the latent reservoir, an independent review committee recommended an early stop to accrual.
The primary analysis of the latent reservoir was based on data from the analysis cohort; the estimated decay and 95% confidence intervals were obtained from a random-effects model of log-transformed latent-reservoir measurements [4]. Analyses of toxicities were based on data from all patients treated with enfuvirtide. Additional analyses were descriptive and used rank-based and partial correlations and repeated-measures models. Data from patients in the analysis cohort were compared with those from other patients by means of Wilcoxon rank sum and Fisher exact tests.
Results
Nineteen treatment-naive patients initiated intensive ART with enfuvirtide, tenofovir disoproxil fumarate, either emtricitabine or lamivudine, saquinavir mesylate, and ritonavir. The median age was 36 years; of the 19 patients, 17 (89%) were male, 11 (58%) were white, 5 (26%) were Hispanic, 2 (11%) were African American, and 1 (5%) was American Indian/Alaska Native. The median baseline CD4 cell count was 262 cells/μL (range, 146–597), and the median baseline viral load was 4.8 log10 (63,000) copies/mL.
Seven patients stopped taking enfuvirtide before week 48 (6 because of toxicities related to injection site reactions [at weeks 1–13] and 1 for personal reasons not related to toxicity); these 7 patients were excluded from the latent-reservoir analysis. Three patients did not experience virologic suppression or had virologic rebound before week 48 and were excluded from the reservoir analysis. Nine patients (the analysis cohort) both experienced virologic suppression and continued enfuvirtide-containing ART for at least 48 weeks. Patients in the analysis cohort were in general similar to those who discontinued enfuvirtide before week 48 (n = 7) and similar to those who did not have sustained virologic suppression (n = 3) in terms of age, sex, baseline CD4 cell count, and viral load, although the power to detect differences was small.
The patients in the analysis cohort had a median CD4 cell count increase at week 48 of 443 cells/μL, compared with 228 cells/μL in 6 patients who had a viral load of <50 copies/mL at week 48 but were no longer receiving enfuvirtide (P = .08). The median time to achieving a viral load of <50 copies/mL in the analysis cohort was 16 weeks; patients who did not remain on enfuvirtide had a similar median time to achieving an undetectable viral load (also 16 weeks).
Patients in the analysis cohort had a median of 4 latent-reservoir measurements each. Four patients had a slight decrease of the number of latently infected cells, and 5 patients had a slight increase (Figure 1). Overall, there was no evidence for decay of the latent reservoir (95% confidence interval for half-life, 11 months to infinity). The latent-reservoir decay rate observed in this trial was similar to that seen in previous studies in which patients were given less intensive antiretroviral regimens but on average were virologically suppressed for longer periods of time [4] (Figure 1).
Figure 1.
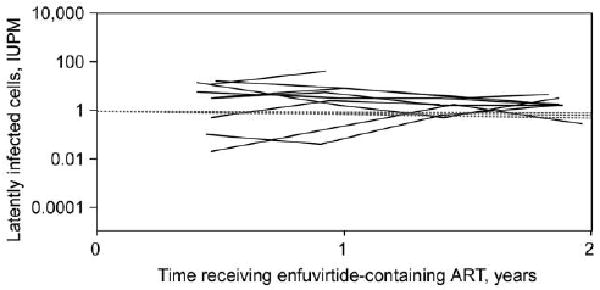
Latent reservoir in human immunodeficiency virus type 1 (HIV-1)–infected patients receiving intensive enfuvirtide-containing anti-retroviral therapy (ART) who had sustained virologic suppression and remained on enfuvirtide for at least 48 weeks (solid lines). The 95% confidence interval for the half-life of the latent reservoir is 11 months to infinity. Dashed lines represent the mean decay slope and 95% confidence intervals from measurements of the latent reservoir in 62 patients who were on ART regimens that did not contain enfuvirtide (historical controls) [4]. IUPM, infectious units per million cells.
We examined the relationship between baseline factors and the latent reservoir. Latent-reservoir size (measured by the mean log10 IUPM) was inversely correlated with baseline CD4 cell count (r = −0.77; P = .016) (Figure 2); that is, the lower the baseline CD4 cell count, the larger the mean log10 IUPM. This association persisted after adjustment for baseline viral load. After adjusting for baseline CD4 cell count, there was no relationship between baseline viral load and log10 IUPM (r = 0.36; P = .38). There was no evidence for an association between mean log10 IUPM and each of the following baseline factors: age, CCR5 density on CD4 cells, and CD4 or CD8 cell activation (measured by the percentage of T cells expressing CD38 and CD38 density) (r = −0.40 to 0.14; all P values are <.30).
Figure 2.
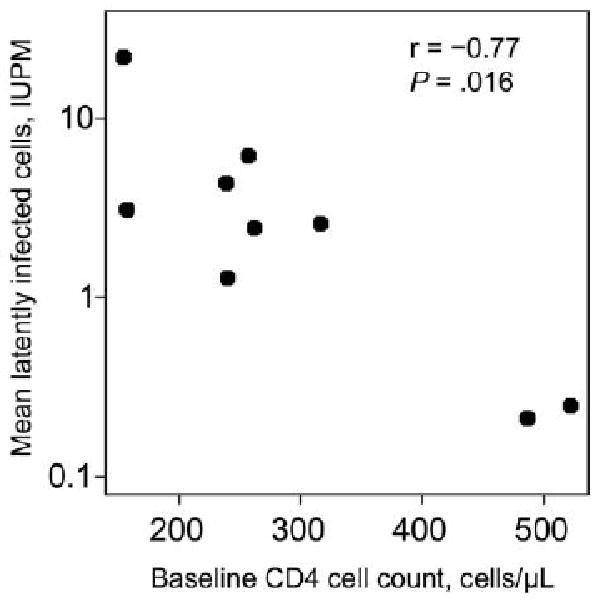
Inverse correlation between the mean latent-reservoir size and baseline CD4 cell count in human immunodeficiency virus type 1 (HIV-1)–infected patients in the analysis cohort. IUPM, infectious units per million cells.
We also evaluated the relationship between latent-reservoir size and immune activation during ART. Although in the analysis cohort log10 IUPM was inversely correlated with CD38 density on CD4 and CD8 cells at the time of latent-reservoir measurement (r = −0.4 and −0.24, respectively; P< .05), these associations were no longer statistically significant after adjustment for baseline CD4 cell count. There was also no statistically significant correlation between latent-reservoir size and the percentage of CD4 or CD8 cells expressing CD38.
To assess whether enfuvirtide use affected immune activation, we compared the percentage of T cells expressing CD38 and CD38 density of patients who continued to receive the fusion inhibitor with those of patients who stopped receiving the drug before the study was completed. Among patients with a viral load of <50 copies/mL, there was no evidence for a difference in immune activation at week 48 between those in the analysis cohort (who continued receiving enfuvirtide; n = 9) and those who had discontinued enfuvirtide (n = 5) before this time point. Similar results were seen at weeks 24, 72, and 96.
Finally, because CCR5 density on CD4 cells has been associated with in vitro activity of enfuvirtide [9] and response to ART [10], we hypothesized that lower CCR5 density might be associated with greater enfuvirtide activity. To test this hypothesis, we examined whether CCR5 cell surface levels affected how rapidly viral load declined after initiation of enfuvirtide-containing therapy. The estimated mean change in viral load from day 0 to day 7 was −1.26 log10 copies/mL (n = 18). There was no association (r = −0.13; P = .6) between baseline CCR5 density and change in viral load from day 0 to day 7 (range of days on which the viral load was measured, days 4–10).
Discussion
In treatment-naive HIV-1-infected patients who received enfuvirtide-containing multitarget ART and who achieved and sustained a plasma viral load of <50 copies/mL, we did not detect a decline in latent-reservoir size over 96 weeks. Although the small sample size limits our power to detect associations—particularly in the secondary analyses—we can, with 95% certainty, exclude the possibility that the half-life of the latent reservoir is <11 months in patients receiving this intensive regimen. Our trial examined the latent-reservoir decay rate in treatment-naive patients who initiated their first antiretroviral regimen, whereas most previous studies measured the reservoir in patients who were already receiving therapy. Despite this difference, the latent-reservoir decay rate observed in our trial was similar to that seen in a study of patients receiving less intensive ART regimens (Figure 1), which were generally combinations of nucleoside reverse-transcriptase inhibitors with nonnucleoside reverse-transcriptase inhibitors or protease inhibitors [4]. Our results suggest that addition of a fusion inhibitor to a regimen that includes reverse-transcriptase and protease inhibitors does not dramatically affect the rate of latent-reservoir decay.
Are there factors that influence the size of the latent reservoir that might impact the ability to deplete this pool? We found that baseline CD4 cell count was inversely associated with the size of the latent reservoir, even after adjustment for baseline viral load. This observation is consistent with the hypothesis that a longer duration of uncontrolled viremia—which leads to more extensive CD4 cell count decline—might result in a larger latent reservoir. However, the latent reservoir is established during acute HIV-1 infection and persists even in patients who initiate therapy during this period [11, 12]. Alternatively, the latent reservoir may be smaller in patients with more robust immune systems. Whether earlier initiation of ART in patients with chronic HIV-1 infection might result in a smaller latent reservoir is not known. The finding of an inverse relationship between baseline CD4 cell count and latent-reservoir size must be confirmed in larger study populations.
Is the size of the latent reservoir related to the amount of persistent immune activation? After adjusting for baseline CD4 cell count, we did not observe any association between T cell activation and latent-reservoir size. We also did not find evidence that use of enfuvirtide had an effect on immune activation in those patients whose viral load was <50 copies/mL, although the power to detect differences was small.
There are several implications of our study. The stability of the latent reservoir in patients initiating therapy with a multitarget regimen that includes fusion, protease, and reverse-transcriptase inhibitors suggests that this strategy will not lead to depletion of this reservoir, an important step toward eradication of HIV-1 infection. It remains to be seen whether intensification of therapy with agents that have other mechanisms of action—such as integrase inhibitors or CCR5 antagonists—will impact residual viremia or the latent reservoir; studies are ongoing. If persistent viral replication does not contribute to maintenance of the latent reservoir [13], then novel strategies— such as reactivation of latent HIV-1 provirus or acceleration of the death rate of latently infected cells [14, 15]—may be needed to successfully eradicate HIV-1 infection. Studies of new approaches must be urgently pursued.
Acknowledgments
We acknowledge John Schmitz, Christopher Pilcher, Lynne Peeples, Camlin Tierney, Anne Kmack, Lynette Purdue, Tianxi Cai, David Wininger, Elizabeth Adams, Carla Pettinelli, Cheryl Marcus, Melissa Kerkau, James Rooney, James Thommes, and all the members of the A5173 team. We also thank the patients and the staff who participated in this study. In particular, we acknowledge the following individuals: Dr David A. Wininger and Mark D. Hite (Ohio State University), Dr Jorge L. Santana Bagur and Dr Santiago Marrero (Puerto Rico AIDS Clinical Trials Unit), David Currin and Susan Pedersen (University of North Carolina), Cathi Basler and Graham Ray (University of Colorado Hospital), Amy Sbrolla and Teri Flynn (Massachusetts General Hospital AIDS Clinical Trials Group), Dr Judith Aberg and Karen Cavanagh (New York University and New York City Health and Hospitals Corporation at Bellevue Hospital Center), and Michael C. Royal and Dr Michael K. Klebert (Washington University). We thank the following companies for providing the study drug: Gilead, Hoffmann–La Roche, Abbott, and Trimeris.
Financial support: National Institute of Allergy and Infectious Diseases (grants AI-38858 and AI-68636 to the AIDS Clinical Trials Group; grants AI-38855 and AI-68634 to the Statistical and Data Analysis Center; Clinical Trials Unit [CTU] grant AI-069474 to Ohio State University [site 2301]; CTU grants 5-U01-AI-069415-03 and P20-RR-11126 to Puerto Rico AIDS Clinical Trials Unit [site 5401]; CTU grant 5-U01-AI-069423-03 and General Clinical Research Center [GCRC] grant M01-RR-000046-48 to University of North Carolina [site 3201]; CTU grant AI-69450 and GCRC grants RR-025780 and AI-54907 to University of Colorado Hospital [site 6101]; CTU grant U01-A1-069472 to Massachusetts General Hospital AIDS Clinical Trials Group Clinical Research Site [CRS; site 101]; CTU grants AI-27665 and AI-069532 to New York University and New York City Health and Hospitals Corporation at Bellevue Hospital Center [site 401]; CTU grant U01-AI-069495 to Washington University CRS [site 2101]; and grants U01-AI-069511, P30-AI-078498, and N01-AI-38858 [subcontract 200VC007] to University of Rochester School of Medicine and Dentistry); Hoffmann–La Roche.
Footnotes
Presented in part: 16th Conference on Retroviruses and Opportunistic Infections, Montreal, 8–11 February 2009 (poster 424).
Potential conflicts of interest: M.A. has served as a consultant to Pfizer. L.M.D. has a family member who is receiving royalties from Merck and GlaxoSmithKline for products not related to this study. J.J.E. has been an investigator for Merck, Panacos, GlaxoSmithKline, and Abbott on research grants to the University of North Carolina; has served as a paid consultant or speaker for Merck, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Tibotec, Roche, and Pfizer; and has been a consultant to Trimeris in the past. R.T.G. has received research grants or unrestricted educational grants from Tibotec, Abbott, and Gilead and has served as a paid speaker at an advisory board meeting for GlaxoSmithKline. All other authors report no potential conflicts.
Disclaimer: The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
References
- 1.Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 2.Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siliciano JD, Kajdas J, Finzi D, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 5.Palmer S, Maldarelli F, Wiegand A, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A. 2008;105:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molto J, Ruiz L, Valle M, et al. Increased antiretroviral potency by the addition of enfuvirtide to a four-drug regimen in antiretroviral-naive, HIV-infected patients. Antivir Ther. 2006;11:47–51. [PubMed] [Google Scholar]
- 7.Siliciano JD, Siliciano RF. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol Biol. 2005;304:3–15. doi: 10.1385/1-59259-907-9:003. [DOI] [PubMed] [Google Scholar]
- 8.Finzi D, Blankson J, Siliciano JD, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 9.Heredia A, Gilliam B, DeVico A, et al. CCR5 density levels on primary CD4 T cells impact the replication and enfuvirtide susceptibility of R5 HIV-1. AIDS. 2007;21:1317–1322. doi: 10.1097/QAD.0b013e32815278ea. [DOI] [PubMed] [Google Scholar]
- 10.Gervaix A, Nicolas J, Portales P, et al. Response to treatment and disease progression linked to CD4+ T cell surface CC chemokine receptor 5 density in human immunodeficiency virus type 1 vertical infection. J Infect Dis. 2002;185:1055–1061. doi: 10.1086/339802. [DOI] [PubMed] [Google Scholar]
- 11.Blankson JN, Finzi D, Pierson TC, et al. Biphasic decay of latently infected CD4+ T cells in acute human immunodeficiency virus type 1 infection. J Infect Dis. 2000;182:1636–1642. doi: 10.1086/317615. [DOI] [PubMed] [Google Scholar]
- 12.Chun TW, Justement JS, Moir S, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis. 2007;195:1762–1764. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- 13.Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog. 2007;3:e122. doi: 10.1371/journal.ppat.0030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 15.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]