

Abstract
Inosine 5-monophosphate dehydrogenase II, a key enzyme in the de novo synthesis of purine nucleotides, is expressed in prostate tumors and prostate cancer cells. AVN944 is a new, specific, noncompetitive IMPDH inhibitor. In this study, we investigated the effects of IMPDH inhibitor AVN944 on LNCaP, CWR22Rv1, DU145 and PC-3 human prostate cancer cells. AVN944 inhibited proliferation of these 4 prostate cancer cell lines and was associated with cell cycle G1 arrest of LNCaP cells and S-phase block of androgen-independent CWR22Rv1, DU145 and PC-3 cells. AVN944 induced caspase-dependent and caspase-independent cell death in LNCaP, CWR22Rv1, and DU145 cells. AVN944 induced expression of p53-target proteins Bok, Bax and Noxa in androgen-responsive cell lines and suppressed expression of survivin in prostate cancer cells regardless of their androgen sensitivity. AVN944 also induced differentiation of androgen-independent prostate cancer cells as indicated by morphological changes and increased expression of genes coding for prostasomal proteins, keratins and other proteins, including tumor suppressor genes MIG-6 and NDRG1. AVN944-differentiated androgen-independent DU145 and PC-3 cells are sensitized to TRAIL-induced apoptosis as demonstrated by induction of caspases and PARP cleavage. In summary, AVN944 inhibited the growth of human prostate cancer cells by inducing cell cycle arrest, cell death as well as differentiation. AVN944 is a novel, promising therapeutic agent that might be combined with other agents for treatment of human prostate cancer.
Keywords: AVN944, IMPDH, prostate cancer, cell death, differentiation, TRAIL
Prostate cancer is the most commonly diagnosed malignancy in men and is second only to lung cancer as the cause of cancer death in males. In 2008, it has been estimated that 186,320 men in the United States will be diagnosed with prostate cancer, and that 28,660 will die from the disease.1 Unfortunately, there is no cure for locally advanced or metastatic prostate cancer. Therefore, it is imperative that new strategies for the effective management and/or treatment of this disease be found.
Several lines of research have identified inosine 5-monophosphate dehydrogenase (IMPDH) as an attractive target for pharmacological intervention. IMPDH is a key enzyme in the de novo synthesis of purine nucleotides. Two isoforms of IMPDH were identified in humans, type I and type II.2 Each of the genes encodes a protein of 514 amino acids, with high homology between the isoforms (84% amino acid identity).
Jackson et al.3 and Weber4 linked IMPDH with proliferation and malignancy and observed that the enzyme is upregulated in rapidly proliferating tumor cells. Elevated activity of IMPDH is primarily caused by upregulation of IMPDH II. Allison et al.5 demonstrated that lymphocytes in particular are dependent on the de novo pathways of nucleotide biosynthesis, making IMPDH a target for immunosuppressive therapy. IMPDH I was also recently identified as an antiangiogenic drug target by Chong et al.6
IMPDH inhibition results in the depletion of guanine nucleotide pools, followed by decreased DNA and RNA synthesis. These events are associated with cell growth arrest, cell cycle block, differentiation and/or cell death. IMPDH inhibitor mycophenolate mofetil induces cell-cycle arrest and decreases T- and B-cell responses effectively, both in vitro and in vivo.7 IMPDH inhibitors tiazofurin, selenazofurin and benzamide riboside were previously tested for their antitumor properties8 and were found to induce differentiation and/or apoptosis in various cell systems, including leukemia HL-609,10 and K-562,11 melanoma12 and human lung cancer H520.13 Floryk et al. demonstrated that IMPDH inhibitors induced cell growth arrest, cell cycle block, differentiation and/or cell death in androgen-independent prostate cancer PC-314 and DU145.15
Selected IMPDH inhibitors with anticancer potential were previously tested in clinical trials. Tiazofurin demonstrated some objective responses, but further investigation was stopped due to its neurotoxicity.16 Hence, evaluation of more selective and well-tolerated IMPDH inhibitors is needed to determine the therapeutic potential of these compounds in the treatment of malignancies. A new, specific, noncompetitive IMPDH inhibitor AVN944 ((1-{3-[3-(Methoxy-4-oxazol-5-yl-phenyl)-ureido]-phenyl}-ethyl)-carbamic acid 2-cyano-1-ethyl-ethyl ester) was developed by Vertex (VX-944) and licensed by Avalon Pharmaceuticals (AVN944). VX-944 was observed to be 3- to 40-fold more potent than mycophenolic acid depending on the cell line.17 VX-944/AVN944 demonstrated cytotoxic effects against multiple myeloma in vitro18 and was shown to evade multidrug resistant pumps, and maintain potency in cancer cells bearing oncogenic mutations and in chemoresistant primary cancer cells.19 AVN944 was shown to be well-tolerated in humans and currently is being tested in clinical trials in patients with hematological malignances and in combination with gemcitabine in patients with pancreatic cancer (Avalon Pharmaceuticals).
To pursue the hypothesis that IMDHD II is a potential target in prostate cancer cells, IMPDH inhibitor AVN944 was tested for its antitumor properties. In this report, evidence is provided that AVN944 has antitumor properties in androgen-sensitive and androgen-independent prostate cancer cells. It is also shown that AVN944-differentiated androgen-independent prostate cancer cells respond to TRAIL treatment.
Material and methods
Reagents
Tetramethyl rhodamine methyl ester (TMRM) was obtained from Invitrogen (Carlsbad, CA). Recombinant TRAIL protein was purchased from Cell Sciences (Canton, MA). IMPDH inhibitor AVN944 was provided by Avalon Pharmaceuticals (Germantown, MA). Stock solution of 10 mM AVN944 was prepared in dimethyl sulfoxide (DMSO). Z-VAD-fmk was purchased from BD Biosciences (San Diego, CA). Other reagents were purchased from Sigma (St. Louis, MO).
Cell cultures and treatment
LNCaP, DU145 and PC-3 prostate cancer cells were obtained from American Type Culture Collection (Manassas, VA). CWR22Rv1 (22Rv1) prostate cancer cells were kindly provided by Dr. Francis Sirotnak (Sloan-Kettering Institute, New York, NY). Cells were cultured in 5% CO2 at 37°C in RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT), 2 mM l-glutamine, 100 U/ml penicillin and 100 U/ml streptomycin (Invitrogen). Normal prostate epithelial PrEC cells were purchased from Lonza (Walkersville, MD) and cultured in prostate epithelial growth medium (Lonza) supplemented with 2 mM l-glutamine, 100 U/ml penicillin and 100 U/ml streptomycin (Invitrogen). To determine cell numbers, cells were plated in 6-well plates at 2 × 105 cells per well 1 day prior to treatment. Attached cells were harvested with trypsin and counted using a Coulter counter (Beckman Coulter, Fullerton, CA).
Cell growth assay
Cells were plated in 12-well plate at 5 × 104 cells per well in triplicate and treated with the solvent or 5 μM AVN944 for the indicated period of time. At the end of treatment, cell viability was measured using an MTS proliferation assay (Promega, Madison, WI) according to the manufacturer's protocol.
Clonogenic assay
Cell survival after treatment with 5 μM AVN944 was measured by clonogenic assay. Single 22Rv1 and PC-3 cells were seeded into 10-cm culture dishes at 10,000 cells per plate in triplicate on day 0 and allowed to attach for another 24 hr. Cells were then treated with the solvent or 5 μM AVN944 for 3 days. Cells were washed 3 times with phosphate-buffered saline (PBS) and fresh culture medium was added. After 14 days, colonies were fixed in Crystal Violet Stain (Fisher Diagnostics, Middletown, VA) and visible colonies containing ∼50 or more cells were counted.
Cell cycle analysis
Cell cycle analysis was performed as described previously.14
Morphological changes
To obtain photographs of control and treated cells, cells were washed twice with PBS and fixed with 4% paraformaldehyde. Images of fixed cells were captured at ×20 magnification with a conventional Nikon inverted microscope using transmitted light and phase contrast imaging.
Cell death detection by ELISA
Cells were plated in 12-well plates at 5 × 104 cells per well in triplicate and treated with the solvent or 5 μM AVN944. When 50 μM z-VAD-fmk was used, cells were pretreated for 1 hr before AVN944 treatment. The cell death detection ELISA kit (Roche, Indianapolis, IN) was used to measure the level of histone-associated DNA fragments in the cytosol of dying cells according to the manufacturer's protocol and enrichment factor was calculated.
Protein extraction and western blot analysis
Western blot analysis was performed as described previously20 using following primary antibodies: IMDPH II (Atlas Antibodies AB, Sweden); β-actin (Sigma); cyclin A, B1 and D2 (Santa Cruz Biotechnology, Santa Cruz, CA); cyclin E and p53 (Neomarkers, Fremont, CA); p21Cip1 and GAPDH (Santa Cruz Biotechnology); caspase-2, caspase-3 p19/17, caspase-9 p37, PARP, Bcl-2 antiapoptotic proteins, Bcl-2 proapoptotic proteins, XIAP and survivin (Cell Signaling, Danvers, MA); and cytochrome c, AIF and Smac (BD Biosciences, San Jose, CA). Appropriate secondary antibodies (goat anti-mouse or anti-rabbit, Pierce, Rockford, IL) labeled with horseradish peroxidase were used. Chemiluminescence data were obtained by SuperSignal West Dura Extended Duration Substrate or West Femto kits (Pierce).
Release of mitochondrial proteins
Control and treated cells were harvested with trypsin, washed with PBS and counted. To obtain the cytosolic fraction, an aliquot of cells (2 × 105) was transferred to a 1.7-ml vial. Cells were centrifuged at 200g for 5 min, resuspended in 100 μl of mitochondrial buffer (80 mM KCl, 10 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 1 mM EDTA, 5 mM KH2PO4, 10 mM succinate, 1 μM rotenone) and treated with 10 μl of digitonin (2 mg/ml in mitochondrial buffer) for 5 min. The concentration of digitonin was optimized so as to not damage mitochondria. To analyze the content of cytochrome c, Smac and AIF, the western blotting was used as described earlier. Protein was loaded at 0.5 μg per lane. GAPDH was used to monitor equal loading.
Analysis of mitochondrial membrane potential
Mitochondrial membrane potential was analyzed by flow cytometry using TMRM (Invitrogen) as previously described21 with slight modifications. Mock- and AVN944-treated cells were permeabilized with digitonin as described earlier. At the end of digitonin treatment, 400 μl of mitochondrial buffer with TMRM (20 nM final concentration) was added to the cell suspension. The percentages of TMRM-negative and TMRM-positive cells were measured using Beckman Coulter Epics XL flow cytometer (Beckman Coulter).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed as previously described.20 To test the mRNA levels of differentiation markers, we used primers from the PrimerBank database22: CAV1 (15451856a2), MIG6 (10047086a1), NDRG1 (5174657a1), KLK5 (11244763a2); other primers were used as previously described.20 Primers were synthesized by MWG-Biotech (High Point, NC) and reactions were monitored by real-time PCR system ABI Prism 700 Sequence Detection System (Applied Biosystems, Foster City, CA), and relative changes in gene expression were calculated. Data were normalized to the expression levels of RPS25. Melting curve analysis was performed for each pair of primers.
Results
Expression of IMPDH II in prostate cancer cell lines
Western blotting was used to analyze IMPDH II levels in prostate cancer cells. Increased IMPDH II levels were detected in prostate cancer cells compared to normal prostate epithelial PrEC cells (Fig. 1).
Figure 1.
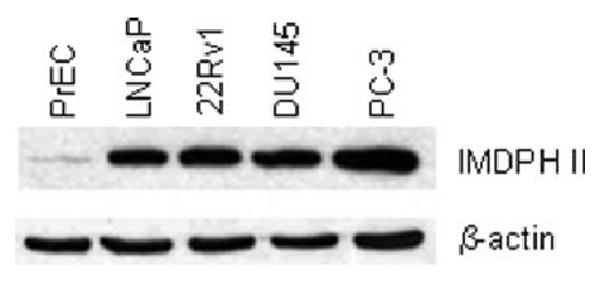
IMPDH II expression in normal prostate epithelial cells PrEC and prostate cancer LNCaP, 22Rv1, DU145 and PC-3 cells was determined by western blotting as described in “Material and methods.” β-actin was used to monitor equal loading.
Effects of AVN944 on prostate cancer cell growth and viability
To pursue the hypothesis that IMDHD II is a potential target in prostate cancer cells, IMPDH inhibitor AVN944 was tested for its antitumor properties in the following preclinical study.
Prostate cancer LNCaP (androgen-sensitive), 22Rv1 (androgen-responsive/-independent), DU145 and PC-3 (both androgen-independent) cells were subjected to increasing concentrations of AVN944 (0–20 μM) for 3 days. Treatment of prostate cancer cells with 1 μM AVN944 for 3 days already resulted in significant cell growth inhibition (Fig. 2a). To further study cytostatic/cytotoxic effects of AVN944 in prostate cancer cells, AVN944 at 5 μM was used to achieve maximum effects. Treatment of prostate cancer cell lines with 5 μM AVN944 completely inhibited cell growth of LNCaP, 22Rv1, DU145 and PC-3 cells after 2 days in culture (Fig. 2b). To test the cytotoxic potential of AVN944, prostate cancer cells were treated with 5 μM AVN944 for up to 3 days, and cell viability was measured using an MTS cell proliferation assay. Prostate cancer cell lines demonstrated different sensitivities to AVN944-induced cytotoxicity. The following percentages indicate viability of the cancer cell lines after 3 days of treatment, expressed as a percentage of control cell values: LNCaP, 15% ± 1%; 22Rv1, 40% ± 3%; DU145, 40% ± 1%; PC-3, 45% ± 3% (Fig. 2c). Cell viability was reduced to 64% ± 2%, 58% ± 3% and 48% ± 4% of control cells in normal prostate epithelial PrEC cells treated with 5, 10 and 20 μM AVN944 for 3 days as measured by the MTS assay.
Figure 2.
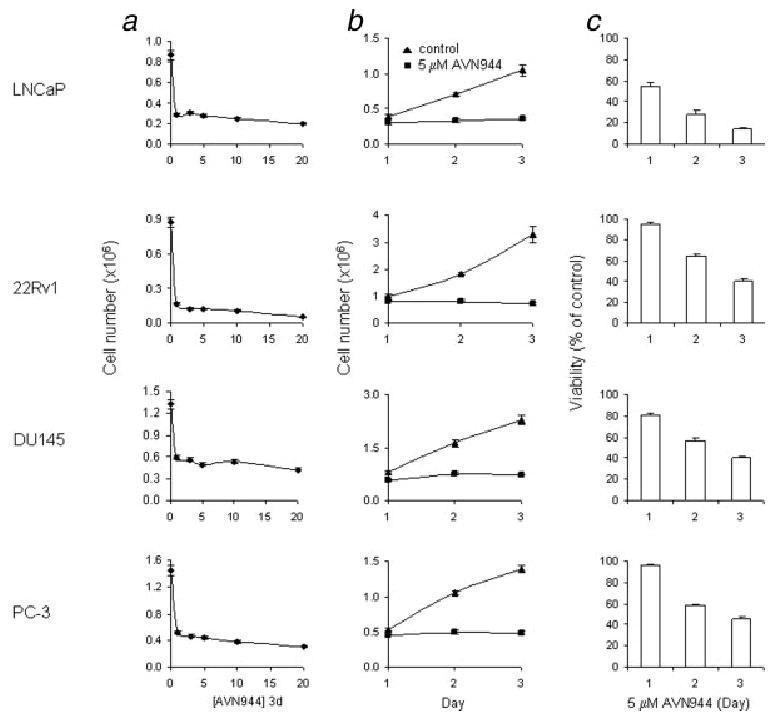
Dose response, cell growth in the presence of AVN944 and AVN944-induced cytotoxicity. (a) Prostate cancer LNCaP, 22Rv1, DU145 and PC-3 cells were treated with increasing doses (in μM) of AVN944 for 3 days in triplicate. Cells were counted at the end of each treatment. Bars indicate SD. (b) Cells were plated in triplicate and treated with 5 μM AVN944 for indicated periods of time. Cells were counted at the end of each treatment interval. Bars indicate SD. (c) Cells were plated and treated as in (b). Cell viability was determined by an MTS assay. Bars indicate SD.
Overall, these data suggest that AVN944 has strong cytostatic and cytotoxic effects in prostate cancer cells.
AVN944 treatment decreases clonogenic potential
To determine a long-term effect of AVN944 on cell growth, a clonogenic assay was used. Androgen receptor positive and androgen-responsive 22Rv1 cells and androgen receptor negative and androgen-independent PC-3 cells were selected for this assay. The clonogenic assay assessed the ability of prostate cancer cells to undergo several rounds of mitosis over a 14-day period after an initial 3-day treatment. AVN944 treatment resulted in the reduction in clonogenic potential of 22Rv1 and PC-3 cells by 90% ± 2% (p = .001) and by 92% ± 3% (p = 0.001), respectively.
AVN944-induced cell cycle arrest/block
AVN944-induced growth inhibition of androgen-independent prostate cancer cells was associated with cell cycle block in S phase (Fig. 3a). The following percentages of proliferating, mock-treated cells were in G0/G1 phase: 22Rv1, 43%; DU145, 61%; and PC-3, 58%. The following percentages were in S phase: 22Rv1, 29%; DU145, 21%; and PC-3, 20%. This composition shifted after treatment with AVN944 for 2 days; the percentage of 22Rv1, DU145 and PC-3 cells in S phase increased to 43, 50, and 42%, respectively. LNCaP cells treated with AVN944 for 2 days demonstrated only a slight change in distribution of cell cycle fractions; among the control LNCaP cells, 66% were in G0/G1 phase and 14% were in S phase. Treated cells yielded 70% G0/G1 phase and 9% in S phase, suggesting that AVN944 induced G1 arrest in LNCaP cells (Fig. 3a).
Figure 3.
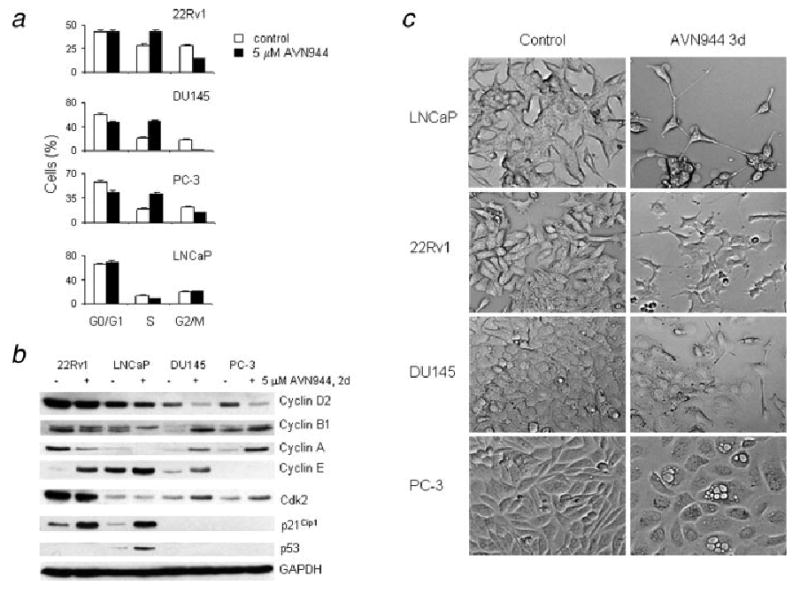
(a) AVN944-induced cell cycle block. Prostate cancer cells were treated with AVN944 for 2 days and cell cycle analysis was performed. (b) Changes in the expression of cell cycle regulators. Prostate cancer cells were treated with AVN944 for 2 days. The expression of cell cycle regulators was analyzed by western blot analysis. GAPDH was used to monitor equal loading. Experiments were repeated 3 times, and typical results are presented. (c) Morphological changes. Prostate cancer cells were treated with AVN944 for 3 days. At the end of treatment, attached cells were photographed using a conventional microscope at ×20 magnification.
Western blot analysis was used to study changes in the expression of cell cycle regulators in the AVN944-treated cell lines (Fig. 3b). AVN944 reduced levels of cyclin D2 in all studied cell lines. Changes in protein levels of cyclins A and B1 were cell-line specific; they were reduced in 22Rv1 and LNCaP cells and increased in DU145 and PC-3 cells. Cyclin E was induced in 22Rv1, LNCaP and DU145 cells but was not detected in PC-3 cells. AVN944 induced the expression of cyclin-dependent kinase inhibitor 1A (p21Cip1) in 22Rv1 and LNCaP cells. The induction of p53 protein was detected only in LNCaP cells.
Taken together, these data indicate that AVN944 induced G1 arrest in LNCaP cells and S-phase block in 22Rv1, DU145 and PC-3 cells. Cell cycle arrest/block was associated with specific expression of cell cycle modulators.
Morphological changes
Treatment with AVN944 for 3 days resulted in morphological changes (Fig. 3c). AVN944 induced the outgrowth of processes in LNCaP, 22Rv1 and DU145 cells. Cell enlargement and cytosolic vacuolization were detected in PC-3 cells. Although the driving force underlying these morphological changes remains unclear, these findings indicate a clear effect of AVN944 treatment on prostate cancer cells.
AVN944-induced apoptosis
To determine whether AVN944 induces apoptosis in prostate cancer cells, the level of histone-associated DNA fragments in the cytosol of dying cells was measured by the cell death detection ELISA kit. The enrichment of histone-associated DNA fragments was significantly increased in 22Rv1 cells (p = 0.001) and, to a lesser degree, in LNCaP (p < 0.005) and DU145 cells (p < 0.005) after 2 days of treatment with 5 μM AVN944. The lowest enrichment was detected in PC-3 cells (Fig. 4a). A pan-caspase inhibitor z-VAD-fmk was used to determine whether AVN944-induced apoptosis was caspase-dependent. Figure 3a shows that the pretreatment of 22Rv1 cells with z-VAD-fmk abolished the enrichment of histone-associated DNA fragments (p < 0.001) and significantly reduced such enrichment in LNCaP cells (p < 0.01). A slight reduction in the enrichment was detected in DU145 cells; however it was not statistically significant.
Figure 4.
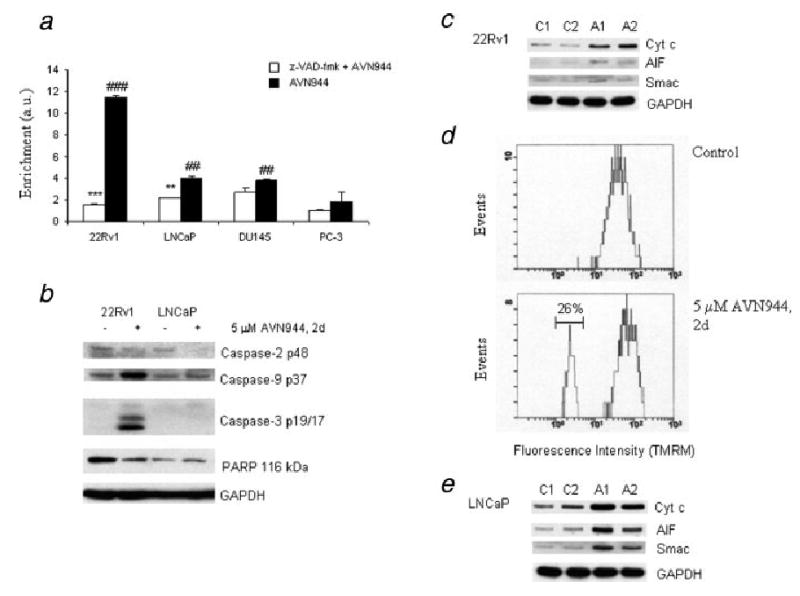
AVN944-induced apoptosis in prostate cancer cells. (a) AVN944-induced apoptosis in prostate cancer cells was measured by a cell death detection ELISA kit as described in “Material and methods.” Enrichment factor (Enrichment) indicating increased content of histone-associated DNA fragments in the cytosol was calculated as a fold increase compared to mock-treated cells in cells treated with z-VAD-fmk and/or AVN944. Experiments were done in triplicate. Bars indicate SD; ***p < 0.001; **p < 0.01 (significance of z-VAD-fmk + AVN944 vs. AVN944); ###p = 0.001; ##p < 0.005 (significance of AVN944 treatment vs. mock treatment). (b) Caspase activation and PARP cleavage. Lysates from 22Rv1 and LNCaP control (−) and AVN944 (+)-treated cells were prepared to analyze activation of caspases and PARP cleavage by western blotting; GAPDH was used as the loading control. Experiments were repeated 3 times, and a representative blot is presented. (c) Mitochondrial pathway. Mock-treated and AVN944-treated 22Rv1 cells for 1 or 2 days were permeabilized with digitonin. Western blot analysis was used to detect the presence of cytochrome c, AIF and Smac in the cytosol; GAPDH was used as a loading control. C, mock-treated cells for 1 (C1) and 2 (C2) days, A, AVN944-treated cells for 1 (A1) and 2 (A2) days. (d) Depletion of mitochondrial membrane potential in 22Rv1 cells. Flow cytometry was used to determine percentage of cells with depleted mitochondrial membrane potential (region, 26%). Experiments were repeated 3 times, and a representative result is presented. (e) Release of mitochondrial proteins in LNCaP cells. LNCaP cells were treated and analyzed as described in (c).
These data suggest that AVN944-induced cell death is caspase-dependent in 22Rv1, both caspase-dependent and independent in LNCaP, and caspase-independent in DU145 cells.
Western blotting was used to analyze the activation of caspases in attached cells after treatment of prostate cancer cells with 5 μM AVN944 for 2 days (Fig. 4b). Cleavage of caspase-2 was detected in 22Rv1 and LNCaP cells, while cleaved fragments of caspases-9 and -3 were detected only in 22Rv1 cells. A very weak activation of caspase-3 (p19) without further processing was detected in LNCaP cells (data not shown). PARP cleavage, a hallmark of apoptosis, was detected only in 22Rv1 cells (Fig. 4b). The activation of caspases-2, -9 and -3 and PARP cleavage were not detected in DU145 and PC-3 cells (data not shown).
AVN944 induces the mitochondrial apoptotic pathway in 22Rv1 cells
Detection of caspase-9 cleavage suggested that treatment of 22Rv1 cells with 5 μM AVN944 for 2 days resulted in the induction of the mitochondrial apoptotic pathway. To analyze the release of apoptotic mitochondrial proteins, the cytosolic fraction from control and AVN944-treated cells was prepared using digitonin as described in “Material and methods.” Western blot analysis revealed that AVN944 induced the release of cytochrome c and, to a lesser extent, AIF and Smac (Fig. 4c).
Changes in mitochondrial membrane potential after AVN944 treatment were measured by flow cytometry using TMRM as described in “Material and methods.” Analysis of obtained histograms (Fig. 4d) revealed that about 26% of 22Rv1 cells did not stain with TMRM after 2-day treatment with AVN944 as a result of depleted mitochondrial membrane potential.
AVN944 induces the AIF pathway in LNCaP cells
It was reported by Seth et al.23 that p53-dependent activation of caspase-2 plays a role in the release of AIF from mitochondria of cisplatin-treated renal tubular epithelial cells. An induction of p53 activity and caspase-2 cleavage in AVN944-treated LNCaP cells suggested that such a pathway may be activated in these cells as well. Western blot analysis confirmed that AVN944 induced the release of AIF, cytochrome c and Smac from mitochondria of LNCaP cells after 1 day of treatment (Fig. 4e). Interestingly, AVN944-induced mitochondrial injury did not result in significant caspase-9 activation (Fig. 4b) or in depletion of mitochondrial membrane potential (data not shown).
Taken together, these data indicate that AVN944 induces caspase-dependent apoptosis in 22Rv1 and LNCaP cells and also caspase-independent cell death in LNCaP and DU145 cells. AVN944 mediates induction of intrinsic apoptotic pathway in 22Rv1 cells and p53/caspase-2/AIF pathway in LNCaP cells.
Expression of antiapoptotic proteins
The pro- and anti-apoptotic Bcl-2 protein families may determine whether mitochondrial membrane permeabilization, which is the major event in the intrinsic cell death pathway, is activated upon cell death stimuli. In our study, the multidomain antiapoptotic Bcl-2 protein was only expressed in androgen-independent DU145 and PC-3 cells (Fig. 5a). AVN944 treatment of DU145 and PC-3 cells for 2 days resulted in reduced Bcl-2 levels. The prostate cancer cell lines constitutively expressed antiapoptotic proteins Bcl-XL and Mcl-1 (Fig. 5a). The expression of Bcl-XL was not altered by 2-day AVN944 treatment. The expression levels of Mcl-1 were reduced in 22Rv1 and PC-3 cells.
Figure 5.
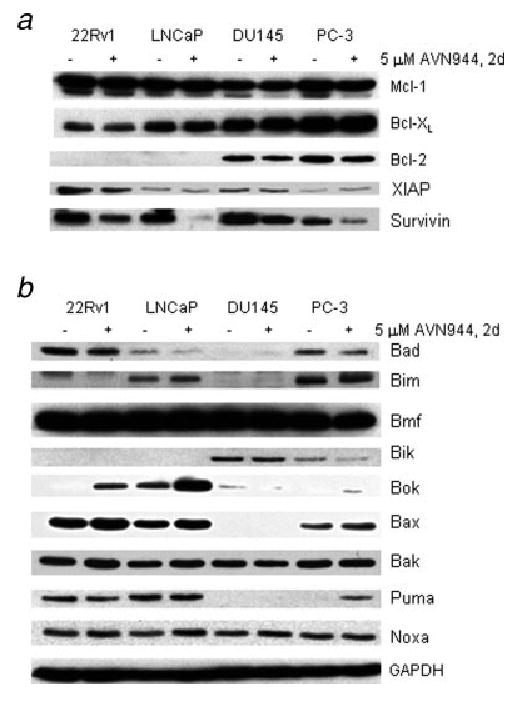
Expression of antiapoptotic (a) and proapoptotic (b) proteins. Cells were cultured in the absence (−) or the presence (+) of AVN944 for 2 days. (a) The expression of the indicated Bcl-2 family of antiapoptotic proteins, XIAP (inhibitor of apoptotic proteins) and survivin was analyzed by the western blot method as described. (b) The expression of proapoptotic proteins was analyzed as described in (a). GAPDH is indicated as the loading control. Experiments were repeated 3 times, and representative blots are presented.
Members of the inhibitor apoptosis proteins (IAP) family play a fundamental role in apoptosis control, development and homeostasis, and they have been implicated in cancer progression and increased resistance to chemotherapeutic and radiation treatments.24 We determined the effect of AVN944 on the suppression of IAP members, including XIAP and survivin. XIAP and survivin were expressed in all 4 cell lines (Fig. 5a). Constitutive expression levels of survivin were greater than those of XIAP. AVN944 caused a marked decrease of survivin in all 4 cell lines; the biggest reduction was detected in LNCaP and PC-3 cells.
Expression of proapoptotic proteins
Proapoptotic Bcl-2 members include the BH3 domain-only proteins, such as Bad, Bid, Bim, Bik, Bmf, Noxa and Puma, and the multidomain Bax-like proteins, such as Bak, Bax and Bok. The BH3-only proteins seem to act as damage sensors and direct antagonists of Bcl-2 and the other prosurvival proteins, whereas the Bax-like proteins, once activated, play an active role in the permeabilization of the mitochondrial outer membrane.
In 22Rv1 cells, the proapoptotic proteins Bmf, Bax, Bad, Noxa and Puma were constitutively expressed (Fig. 5b). AVN944 induced Bok protein levels and increased the expression of the Bax and Noxa proteins after 2-day treatment. LNCaP cells expressed Bmf, Bax, Puma, Bim, Box, Noxa and Bad. AVN944 strongly induced the expression of the Bok protein and increased the expression of Bax, Noxa and Bim. DU145 cells constitutively expressed Bmf, Bik and Noxa proteins, and AVN944 did not alter their levels or induce expression of other proapoptotic proteins. PC-3 cells constitutively expressed Bmf, Bim, Noxa, Bax, Bad and Bik. No expression of Bok or Puma was detected. PC-3 cells treated with AVN944 expressed increased levels of Bax and Puma and reduced levels of Bad.
AVN944 induces differentiation of androgen-independent prostate cancer cells
Floryk et al. reported that IMPDH inhibitors induce differentiation of androgen-independent PC-314 and DU14515 cells. The differentiation is characterized by cell growth inhibition, cell cycle block, morphological changes and the expression of differentiation markers, including prostasomal proteins and keratins.
AVN944-induced cell growth inhibition, cell cycle block and morphological changes suggest that AVN944 induces differentiation in DU145 and PC-3 cells. The induction of previously characterized differentiation markers was tested by qRT-PCR. Treatment of DU145 and PC-3 cells induced the expression of CC3, CD10, CD55, CEACAM5, GDF15, GLA, KLK5, KRT2, KRT4, LGALS3, MIG6, NDRG1 and TNFSF9 genes in both cell lines at various levels. Expression of CAV1, CPA4, CTSB, GAL, GRP78, KRT6B and KRT4 genes was induced only in DU145 cells (Table I).
TABLE I. AVN944-Induced Expression of Differentiation Markers in DU145 and PC-3 Cells.
| Gene | DU145 | PC-3 |
|---|---|---|
| CAV1 | 6.0 | 1 |
| CC3 | 33.2 | 20.0 |
| CD10 | 6.3 | 5.0 |
| CD55 | 15.0 | 2.4 |
| CEACAM5 | 11.6 | 25.8 |
| CPA4 | 17.7 | 1 |
| CTSB | 5.6 | 1 |
| Cyr61 | 1 | 4.9 |
| GAL | 2.5 | 1 |
| GDF15 | 12.8 | 32.1 |
| GLA | 2364.72 | 30.4 |
| GRP78 | 3.6 | 1 |
| KLK5 | 46.1 | 16.2 |
| KRT2 | 18.4 | 4.3 |
| KRT4 | 2315.42 | 1033.52 |
| KRT6B | 5.4 | 1 |
| KRT19 | 5.9 | 1 |
| LGALS3 | 12.3 | 2.2 |
| MIG6 | 3.2 | 4.9 |
| NDRG1 | 5.9 | 19.4 |
| TNFSF9 | 5.2 | 3.9 |
Expression levels of studied genes were normalized to those of RPS25.
The change in the mRNA level was in the interval 1.0–1.7.
Ct for the mock-treated cells were set arbitrarily to 50, which represents a number of cycles ran during the qRT-PCR analysis.
AVN944 sensitizes androgen-independent prostate cancer cells to TRAIL
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), similar to TNF-α and FasL, triggers apoptosis through binding with death receptors (DR4 and DR5).25,26 Among these 3 death ligands, TRAIL has the most potential for cancer therapy because it can selectively induce apoptosis in cancer cells but not in most normal cells.27–30 The results of some studies demonstrated that prostate cancer cells are resistant to the apoptotic effects of TRAIL alone.31 However, other studies have shown that TRAIL is synergistic with specific chemotherapeutic drugs.29,30,32–39 Thus, we hypothesized that androgen-independent, AVN944-apoptosis resistant and AVN944-differentiated prostate cancer cells are sensitized to TRAIL-induced apoptosis. To test this hypothesis, DU145 and PC-3 cells treated with AVN944 for 3 days were subjected to 20 ng/ml TRAIL for 1 d. Viability of AVN944-differentiated DU145 and PC-3 cells after TRAIL treatment was reduced to 18% ± 2% and 17% ± 3%, respectively, as measured by the MTS assay. Treatment of DU145 and PC-3 cells with 20 ng/ml TRAIL alone had a minimal impact on their viability (Fig. 6a). Analysis of cell lysates from mock-treated, AVN944, TRAIL and AVN944-differentiated-TRAIL-treated cells revealed that 6-hour TRAIL treatment of AVN944-differentiated cells induced apoptosis as demonstrated by the expression of cleaved caspase-9, caspase-3, and PARP (Fig. 6b). It is noteworthy that TRAIL alone triggered caspase-9 and caspase-3 cleavage; however, these cells were not committed to cell death, as further caspase-3 processing and PARP cleavage were not detectable.
Figure 6.
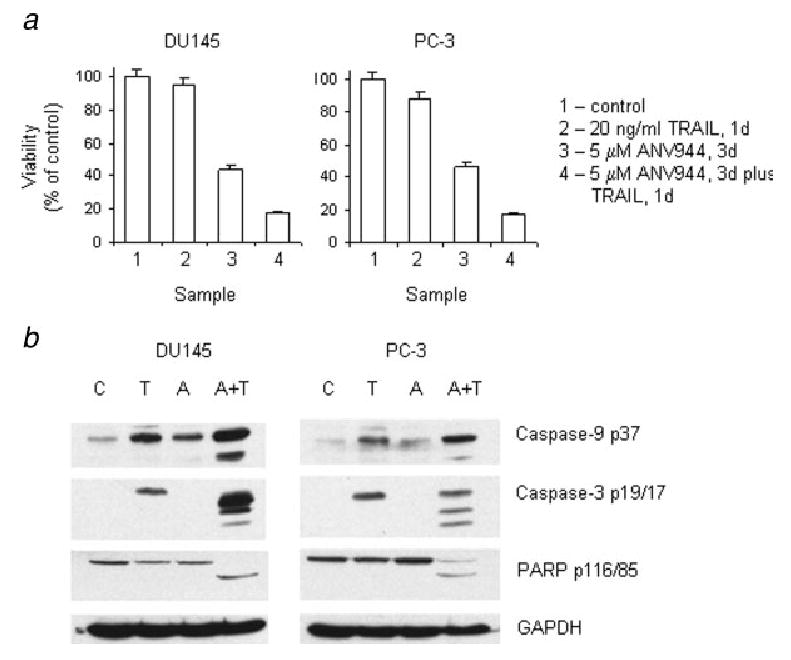
Cell viability of AVN944-differentiated DU145 and PC-3 cells treated with TRAIL (a) and TRAIL-induced apoptosis in AVN944-differentiated PC-3 and DU145 cells (b). (a) DU145 and PC-3 cells were treated with AVN944 for 3 days. At the end of treatment, cells were subjected to TRAIL for 1 d. Cell viability was determined by the MTS assay. Bars indicate SD. (b) DU145 and PC-3 cells were treated with AVN944 for 3 days. At the end of treatment, cells were subjected to TRAIL for 6 hr. Cell lysates were analyzed for the expression of apoptotic markers, including cleaved caspase-9, caspase-3 and PARP. Experiments were repeated 3 times, and typical results are presented. C, control; T, TRAIL; A, AVN944; A+T, AVN944+TRAIL.
In summary, AVN944-differentiated androgen-independent DU145 and PC-3 cells are sensitized to TRAIL-induced apoptosis.
Discussion
Based on our observations of IMPDH II increased expression in prostate cancer cells, we tested a new IMPDH inhibitor, AVN944, for its antitumor properties in prostate cancer using androgen-sensitive LNCaP, androgen-responsive/-independent 22Rv1 and androgen-independent DU145 and PC-3 prostate cancer cells.
AVN944 treatment resulted in dose-dependent cell growth inhibition in all studied prostate cancer cells, independently of their androgen sensitivity. The results of the MTS proliferation assay demonstrated that LNCaP cells were the most sensitive to AVN944-induced cell growth arrest. Cell growth inhibition was associated with cell cycle arrest or cell cycle block. AVN944 induced G1 arrest in LNCaP cells and S-phase block in androgen-independent 22Rv1, DU145 and PC-3 cells. As previously published, mycophenolic acid induced S-phase block in PC-314 and DU14515 cells.
The clonogenic assay allows for assessment of the ability of prostate cancer cells not only to survive but also to retain reproductive potential after treatment. Androgen receptor positive, androgen-responsive 22Rv1 cells representing a model of prostate cancer progression to the androgen-independent stage and androgen negative, androgen-independent PC-3 cells were used for this assay. A reduced clonogenic potential of these cells after AVN944 treatment may indicate that AVN944 could be a valid treatment option for either localized or metastatic disease.
Pankiewicz40 suggested that IMPDH inhibitors mimic many of the effects of p53 expression, including induction of cell growth inhibition and p53-mediated G1 arrest after induced DNA damage. On the other hand, cells with mutated p53 protein are able to escape G1 arrest. The p53 status of cell lines used in the present study may explain differences in the AVN944-induced cell cycle arrest/block. Van Bokhoven et al.41 reported that LNCaP cells express wild-type p53 protein, 22Rv1 and DU145 express mutated p53 proteins and PC-3 cells do not express p53 protein. The same study also reported that about 10% of 22Rv1 cells stained positive for p53 nuclear protein, and the authors suggested that Q331R mutation does not lead to a stabilized protein. In our study, western blot analysis showed that AVN944 induced detectable p53 levels in LNCaP cells. Although an induced p53 protein was not detected in 22Rv1 cells under selected experimental conditions, our data suggest that AVN944 stimulates p53 activity in 22Rv1 cells. Both LNCaP and 22Rv1 cells induced p21Cip1 protein, a known p53 downstream effector, and p53 proapoptotic targets as discussed below. In future studies it would be useful to analyze whether reduced stability of p53 protein enables AVN944-treated 22Rv1 cells to escape G1 arrest to be blocked in the S-phase.
AVN944 also induced morphological changes in prostate cancer cells. The outgrowth of processes was detected in LNCaP, 22Rv1 and DU145 cells. IMPDH inhibition by AVN944 triggered cytosolic vacuolization in PC-3 cells. Induction of similar cytosolic vacuolization in PC-3 cells by other IMPDH inhibitors, such as mycophenolic acid and tiazofurin, has been previously described.14 Future studies should characterize signaling pathways leading to described morphological changes in prostate cancer cells treated with IMPDH inhibitors.
To test whether AVN944 induces apoptosis in prostate cancer cells, the enrichment of histone-associated DNA fragments in the cytosol was measured in attached cells 2 days after AVN944 treatment. Results of this assay revealed that AVN944 induced apoptosis in 22Rv1 cells and, to a lesser degree, in LNCaP and DU145 cells. The use of pan-caspase inhibitor z-VAD-fmk suggested that AVN944-induced death in 22Rv1 and LNCaP cells was caspase-dependent, while cell death in DU145 cells was caspase-independent. Published data15 and findings presented here suggest that DU145 cells may serve as a model system to study IMPDH-inhibitor-induced, caspase-independent cell death. Our analysis of caspase activation in AVN944-treated cells revealed that caspase-2 cleavage was activated in LNCaP and 22Rv1 cells. Caspase-9 and -3 were strongly activated only in 22Rv1 cells. The activation of caspase-9 and -3 was not detected in DU145 or PC-3 cells.
The detection of caspase-9, caspase-3 and PARP cleavage in AVN944-treated 22Rv1 cells suggested an induction of the mitochondrial cell death pathway. Further analysis confirmed this finding, as cytochrome c was detected in the cytosol of AVN944-treated cells. These events were accompanied by the reduction of mitochondrial membrane potential, as a population of 22Rv1 cells without TMRM signal was detected.
Seth et al.23 reported that p53-mediated activation of caspase-2 leads to the release of AIF from mitochondria of cisplatin-treated renal tubular epithelial cells. Similar results were obtained in AVN944-treated LNCaP cells. AVN944 treatment mediated activation of p53 and subsequent caspase-2 cleavage followed by the release of AIF, cytochrome c and Smac from mitochondria. Interestingly, the release of cytochrome c led to minimal caspase-9 activation. In addition, a cell population without TMRM signal was not detected (data not shown). Further studies are necessary to understand the role of p53 in AVN944-treated LNCaP cells.
To understand differences in the sensitivity of prostate cancer cells to the AVN944-induced mitochondrial apoptotic pathway, analysis of antiapoptotic and proapoptotic proteins was performed in control and AVN944-treated cells. Androgen-responsive LNCaP and 22Rv1 cells demonstrated similar changes in the expression of these proteins. It is noteworthy that AVN944 mediated induction of p53 mediators Bax,42 Bok and Noxa43 in LNCaP and 22Rv1 cells. In DU145 and PC-3 cells, a reduction of antiapoptotic proteins Mcl-1 and Bcl-2 was detected. However, minimal apoptotic responses were detected in these cells within 2 to 3 days of AVN944 treatment.
Most importantly, survivin, a member of a family of apoptosis-inhibitor proteins,44 was downregulated by AVN944 in the prostate cancer cells. Recent studies have targeted survivin expression as a therapeutic approach,24 as the expression of survivin is a predictor of both poor prognosis and decreased survival time and is implicated in conferring chemotherapeutic and radiotherapeutic resistance phenotypes to tumor cells.
In accordance with previous studies,14,15 IMPDH inhibition resulted in the induction of differentiation of PC-3 and DU145 cells. AVN944 induced the expression of numerous differentiation markers, including prostasomal proteins and keratins. The majority of tested differentiation markers were induced in both DU145 and PC-3 cells. Interestingly, AVN944 treatment resulted in the induction of the CEACAM5 gene, which codes for a secretory protein. AVN944 also increased expression of gene coding for caveolin-1 in DU145 cells. The exact role of caveolin-1 in this system is not known; however we suggest that caveolin-1 may be a survival factor45 during differentiation of DU145 cells. We do not rule out a possibility that AVN944 induces differentiation in androgen-sensitive prostate cancer cells; however, markers have not been characterized yet for this cell type. It is of interest to identify a group of secretory proteins that can be used as predictive biomarkers of AVN944 activity in prostate cancer during clinical trials.
We also hypothesized that AVN944 would sensitize prostate cancer cells to TRAIL-induced apoptosis. Our data demonstrated that AVN944-differentiated androgen-independent prostate cancer cells were sensitized to TRAIL-induced apoptosis as demonstrated by the presence of cleaved caspase-9, caspase-3 and PARP. It is noteworthy that TRAIL was the most effective to reduced viability of cells pretreated with AVN944 for 3 days (data not shown) suggesting that this time period is necessary for particular changes in the genome/proteome of prostate cancer cells. AVN944-pretreated androgen-sensitive LNCaP cells were not sensitized to TRAIL treatment as they were unable to activate caspase-9 and caspase-3 (data not shown). It is important to conduct further studies to identify changes in the genome/proteome of AVN944-differentiated prostate cancer cells that lead to TRAIL sensitivity.
The preclinical data presented here suggest that prostate cancer cells are responsive to IMPDH inhibition. IMPDH inhibition mediates induction of p53 activity, cell growth inhibition, cell cycle arrest/block, differentiation and/or cell death. AVN944 is a novel, promising therapeutic agent that might be combined with other agents for treatment of human prostate cancer.
Acknowledgments
Grant sponsor: Specialized Program of Research Excellence; Grant number: P50-58204; Grant sponsor: National Institutes of Health; Grant number: RO1 CA68814.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Natsumeda Y, Ohno S, Kawasaki H, Konno Y, Weber G, Suzuki K. Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem. 1990;265:5292–5. [PubMed] [Google Scholar]
- 3.Jackson RC, Weber G, Morris HP. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature. 1975;256:331–3. doi: 10.1038/256331a0. [DOI] [PubMed] [Google Scholar]
- 4.Weber G. Biochemical strategy of cancer cells and the design of chemotherapy: G. H. A. Clowes Memorial Lecture. Cancer Res. 1983;43:3466–92. [PubMed] [Google Scholar]
- 5.Allison AC, Hovi T, Watts RW, Webster AD. The role of de novo purine synthesis in lymphocyte transformation. Ciba Found Symp. 1977;48:207–24. doi: 10.1002/9780470720301.ch13. [DOI] [PubMed] [Google Scholar]
- 6.Chong CR, Qian DZ, Pan F, Wei Y, Pili R, Sullivan DJ, Jr, Liu JO. Identification of type 1 inosine monophosphate dehydrogenase as an antiangiogenic drug target. J Med Chem. 2006;49:2677–80. doi: 10.1021/jm051225t. [DOI] [PubMed] [Google Scholar]
- 7.Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. 2005;80:S181–S190. doi: 10.1097/01.tp.0000186390.10150.66. [DOI] [PubMed] [Google Scholar]
- 8.Yalowitz JA, Jayaram HN. Molecular targets of guanine nucleotides in differentiation, proliferation and apoptosis. Anticancer Res. 2000;20:2329–38. [PubMed] [Google Scholar]
- 9.Sokoloski JA, Blair OC, Sartorelli AC. Alterations in glycoprotein synthesis and guanosine triphosphate levels associated with the differentiation of HL-60 leukemia cells produced by inhibitors of inosine 5′-phosphate dehydrogenase. Cancer Res. 1986;46:2314–19. [PubMed] [Google Scholar]
- 10.Goldstein BM, Leary JF, Farley BA, Marquez VE, Levy PC, Rowley PT. Induction of HL60 cell differentiation by tiazofurin and its analogues: characterization and efficacy. Blood. 1991;78:593–8. [PubMed] [Google Scholar]
- 11.Olah E, Natsumeda Y, Ikegami T, Kote Z, Horanyi M, Szelenyi J, Paulik E, Kremmer T, Hollan SR, Sugar J. Induction of erythroid differentiation and modulation of gene expression by tiazofurin in K-562 leukemia cells. Proc Natl Acad Sci USA. 1988;85:6533–7. doi: 10.1073/pnas.85.17.6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiguchi K, Collart FR, Henning-Chubb C, Huberman E. Induction of cell differentiation in melanoma cells by inhibitors of IMP dehydrogenase: altered patterns of IMP dehydrogenase expression and activity. Cell Growth Differ. 1990;1:259–70. [PubMed] [Google Scholar]
- 13.Khanna N, Jayaram HN, Singh N. Benzamide riboside induced mitochondrial mediated apoptosis in human lung cancer H520 cells. Life Sci. 2004;75:179–90. doi: 10.1016/j.lfs.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Floryk D, Tollaksen SL, Giometti CS, Huberman E. Differentiation of human prostate cancer PC-3 cells induced by inhibitors of inosine 5′-monophosphate dehydrogenase. Cancer Res. 2004;64:9049–56. doi: 10.1158/0008-5472.CAN-04-1553. [DOI] [PubMed] [Google Scholar]
- 15.Floryk D, Huberman E. Mycophenolic acid-induced replication arrest, differentiation markers and cell death of androgen-independent prostate cancer cells DU145. Cancer Lett. 2006;231:20–9. doi: 10.1016/j.canlet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Tricot GJ, Jayaram HN, Lapis E, Natsumeda Y, Nichols CR, Kneebone P, Heerema N, Weber G, Hoffman R. Biochemically directed therapy of leukemia with tiazofurin, a selective blocker of inosine 5′-phosphate dehydrogenase activity. Cancer Res. 1989;49:3696–701. [PubMed] [Google Scholar]
- 17.Jain J, Ma J, Furey B, Recher C, Demur C, Poondru S, Zhang J, Li S, Firestone B, Olson K, Murphy M, Taylor J, et al. The IMPDH inhibitor VX-944 demonstrates in vivo efficacy in an aggressive leukemia model. ASH Annual Meeting Abstracts. 2004;104:2530. [Google Scholar]
- 18.Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Podar K, Le Gouill S, Shiraishi N, Yasui H, Roccaro AM, Tai YZ, Chauhan D, et al. Novel inosine monophosphate dehydrogenase inhibitor VX-944 induces apoptosis in multiple myeloma cells primarily via caspase-independent AIF/Endo G pathway. Oncogene. 2005;24:5888–96. doi: 10.1038/sj.onc.1208739. [DOI] [PubMed] [Google Scholar]
- 19.Jain J, Almquist S, Hoover R, Shlyakhter D, Ford P, Markland W, van Drie J, Rasmussen R, Weber P, Dauffenbach L, Kerfoot C, Mosher R. VX-944: an inosine monophosphate dehydrogenase inhibitor with unique anti-cancer activity. 18th EQRTC-NCI-AACR Symposium on “Molecular Targets and Cancer Therapeutics”; Geneva, Switzerland. 2004. [Google Scholar]
- 20.Floryk D, Huberman E. Differentiation of androgen-independent prostate cancer PC-3 cells is associated with increased nuclear factor-kappaB activity. Cancer Res. 2005;65:11588–96. doi: 10.1158/0008-5472.CAN-05-1831. [DOI] [PubMed] [Google Scholar]
- 21.Floryk D, Houstek J. Tetramethyl rhodamine methyl ester (TMRM) is suitable for cytofluorometric measurements of mitochondrial membrane potential in cells treated with digitonin. Biosci Rep. 1999;19:27–34. doi: 10.1023/a:1020193906974. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem. 2005;280:31230–9. doi: 10.1074/jbc.M503305200. [DOI] [PubMed] [Google Scholar]
- 24.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 25.Sheikh MS, Huang Y. Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets. 2004;4:97–104. doi: 10.2174/1568009043481597. [DOI] [PubMed] [Google Scholar]
- 26.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–44. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Van Ophoven A, Ng CP, Patel B, Bonavida B, Belldegrun A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) for treatment of prostate cancer: first results and review of the literature. Prostate Cancer Prostatic Dis. 1999;2:227–33. doi: 10.1038/sj.pcan.4500373. [DOI] [PubMed] [Google Scholar]
- 28.Nagane M, Huang HJ, Cavenee WK. The potential of TRAIL for cancer chemotherapy. Apoptosis. 2001;6:191–7. doi: 10.1023/a:1011336726649. [DOI] [PubMed] [Google Scholar]
- 29.Ray S, Almasan A. Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res. 2003;63:4713–23. [PubMed] [Google Scholar]
- 30.Shankar S, Chen X, Srivastava RK. Effects of sequential treatments with chemotherapeutic drugs followed by TRAIL on prostate cancer in vitro and in vivo. Prostate. 2005;62:165–86. doi: 10.1002/pros.20126. [DOI] [PubMed] [Google Scholar]
- 31.Guseva NV, Taghiyev AF, Rokhlin OW, Cohen MB. Death receptor-induced cell death in prostate cancer. J Cell Biochem. 2004;91:70–99. doi: 10.1002/jcb.10707. [DOI] [PubMed] [Google Scholar]
- 32.Nimmanapalli R, Perkins CL, Orlando M, O'Bryan E, Nguyen D, Bhalla KN. Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels. Cancer Res. 2001;61:759–63. [PubMed] [Google Scholar]
- 33.Ng CP, Zisman A, Bonavida B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: role of XIAP in resistance. Prostate. 2002;53:286–99. doi: 10.1002/pros.10155. [DOI] [PubMed] [Google Scholar]
- 34.Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol Ther. 2002;1:520–7. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- 35.Jeon KI, Rih JK, Kim HJ, Lee YJ, Cho CH, Goldberg ID, Rosen EM, Bae I. Pretreatment of indole-3-carbinol augments TRAIL-induced apoptosis in a prostate cancer cell line, LNCaP. FEBS Lett. 2003;544:246–51. doi: 10.1016/s0014-5793(03)00473-3. [DOI] [PubMed] [Google Scholar]
- 36.Wu XX, Ogawa O, Kakehi Y, Mizutani Y, Nishiyama H, Kamoto T, Megumi Y, Ito N, Kinoshita H, Isogawa Y, Terachi T. TRAIL and chemotherapeutic drugs in cancer therapy. Vitam Horm. 2004;67:365–83. doi: 10.1016/S0083-6729(04)67019-1. [DOI] [PubMed] [Google Scholar]
- 37.Deeb D, Xu YX, Jiang H, Gao X, Janakiraman N, Chapman RA, Gautam SC. Curcumin (diferuloyl-methane) enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in LNCaP prostate cancer cells. Mol Cancer Ther. 2003;2:95–103. [PubMed] [Google Scholar]
- 38.Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J, Kraft AS. The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim. Mol Cancer Ther. 2005;4:443–9. doi: 10.1158/1535-7163.MCT-04-0260. [DOI] [PubMed] [Google Scholar]
- 39.Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005;65:6364–70. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- 40.Pankiewicz KW. Novel nicotinamide adenine dinucleotide analogues as potential anticancer agents: quest for specific inhibition of inosine monophosphate dehydrogenase. Pharmacol Ther. 1997;76:89–100. doi: 10.1016/s0163-7258(97)00092-2. [DOI] [PubMed] [Google Scholar]
- 41.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–25. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 42.Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, Hoffman B, Liebermann D. Immediate early up-regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9:1791–8. [PubMed] [Google Scholar]
- 43.Yakovlev AG, Di Giovanni S, Wang G, Liu W, Stoica B, Faden AI. BOK and NOXA are essential mediators of p53-dependent apoptosis. J Biol Chem. 2004;279:28367–74. doi: 10.1074/jbc.M313526200. [DOI] [PubMed] [Google Scholar]
- 44.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 45.Li L, Ren CH, Tahir SA, Ren C, Thompson TC. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol. 2003;23:9389–404. doi: 10.1128/MCB.23.24.9389-9404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]