

Abstract
Purpose
This study was undertaken to examine the role of the insulin-like growth factor (IGF) signaling pathway in the response of ovarian cancer cells to Taxol and to evaluate the significance of this pathway in human epithelial ovarian tumors.
Experimental design
The effect of Taxol treatment on AKT activation in A2780 ovarian carcinoma cells was evaluated using antibodies specific for phosphorylated AKT. To study the drug-resistant phenotype, we developed a Taxol-resistant cell line, HEY-T30, derived from HEY ovarian carcinoma cells. IGF2 expression was measured by real-time PCR. An IGF1R inhibitor, NVP-AEW541, and IGF2 siRNA were used to evaluate the effect of IGF pathway inhibition on proliferation and Taxol sensitivity. IGF2 protein expression was evaluated by immunohistochemistry in 115 epithelial ovarian tumors, and analyzed in relation to clinical/pathologic factors using the Chi-square or Fisher’s exact tests. The influence of IGF2 expression on survival was studied with Cox regression.
Results
Taxol-induced AKT phosphorylation required IGF1R tyrosine kinase activity and was associated with upregulation of IGF2. Resistant cells had higher IGF2 expression compared with sensitive cells, and IGF pathway inhibition restored sensitivity to Taxol. High IGF2 tumor expression correlated with advanced stage (p<0.001) and tumor grade (p<0.01), and reduced disease-free survival (p<0.05).
Conclusions
IGF2 modulates Taxol resistance, and tumor IGF2 expression is a candidate prognostic biomarker in epithelial ovarian tumors. IGF pathway inhibition sensitizes drug-resistant ovarian carcinoma cells to Taxol. Such novel findings suggest that IGF2 represents a therapeutic target in ovarian cancer, particularly in the setting of Taxol resistance.
Keywords: Taxol, IGF2, Ovarian cancer, Drug Resistance
Introduction
The high mortality rate of ovarian cancer is due to treatment failure in the setting of recurrent/progressive disease that is unresponsive to chemotherapy. Elucidation of the biological factors underlying drug resistance is critical for the development of more effective treatment. Exposure to diverse chemotherapeutic agents induces alterations in gene expression and in signaling cascades that can mediate resistance. Identifying and targeting these cell-specific adaptive responses represents a rational approach to the development of novel drug combination treatment strategies to circumvent resistance.
The widely used chemotherapeutic agent Taxol is indicated for first-line and subsequent treatment of ovarian carcinoma. Upon associating with its specific binding site on β-tubulin in the microtubule polymer, Taxol stabilizes microtubules and alters their dynamic properties, thereby perturbing their normal function in spindle assembly, cell division, motility, intracellular trafficking and signaling (1–5). Several mechanisms of Taxol resistance have been identified, including overexpression of the transporter p-glycoprotein, alterations in tubulin, and aberrant signal transduction pathways and/or cell death pathways (6). Despite these discoveries, there remains a critical need for the development of effective strategies to overcome clinical Taxol-resistance.
We and others have shown that Taxol exposure can activate proliferative and anti-apoptotic signaling pathways in cancer cells. For example, cell-specific activation of ERK activity has been observed after Taxol treatment (7). In cells that exhibit Taxol-induced ERK activation, MAPK pathway inhibitors potentiate Taxol response in vitro and in vivo (8). Activation of the serine-threonine kinase, AKT, which promotes cellular survival, has also been observed following Taxol treatment of ovarian cancer cells (9). However, the upstream signaling events that initiate Taxol-induced AKT activation have not been thoroughly investigated.
Ovarian carcinoma cells grown in tissue culture secrete insulin-like growth factor 2 (IGF2) and express its major receptor, the Type 1 IGF receptor (IGF1R), suggesting a role for autocrine/paracrine IGF2-IGF1R signaling in these cells (10). The IGF1R is a transmembrane tyrosine kinase receptor that undergoes autophosphorylation upon binding of either IGF1 or IGF2, leading to tyrosine kinase activation. Activated IGF1R initiates an anti-apoptotic signaling cascade mediated by increased phosphatidylinositol 3-kinase (PI3K) activity, resulting in activation of the downstream anti-apoptotic effector, AKT (11, 12). The IGF1R pathway is an attractive candidate for targeted therapy, and several small molecules and antibodies that specifically inhibit the IGF1R are undergoing clinical evaluation and may be approved for use in the clinic (13).
For these reasons, the present study was undertaken, to our knowledge the first to examine the role of the IGF signaling pathway in the cellular response of ovarian cancer cells to Taxol treatment, as well as the first to measure IGF2 protein expression in a sizeable cohort of patients with epithelial ovarian tumors. We report the novel finding that Taxol-induced AKT phosphorylation occurs in an IGF1R-dependent manner, and is associated with upregulation of IGF2 mRNA expression. Furthermore, in order to study the drug resistant phenotype, we developed a cell line model of acquired Taxol resistance and compared these cells with the parental, chemo-sensitive cell line. The Taxol-resistant cells exhibit significant upregulation of IGF2 gene expression. IGF pathway inhibition, by IGF1R blockade or IGF2 depletion, restores sensitivity to Taxol in these resistant cells. Furthermore, we assessed IGF2 protein expression levels by immunohistochemistry in 115 primary human epithelial ovarian tumors. High IGF2 expression was significantly associated with invasive carcinoma and disease progression, and correlated with shortened interval to disease recurrence. Thus, IGF2 is identified for the first time to be a crucial mediator of Taxol resistance in ovarian carcinoma cells, and its expression in primary epithelial ovarian tumors is associated with poor prognostic factors for recurrence; these findings offer significant potential for clinical application.
Materials and methods
A2780 and HEY ovarian carcinoma cells were maintained as subconfluent monolayer cultures in RPMI supplemented with 10% FBS (Atlanta, Lawrenceville, GA) and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). The Taxol resistant cell line, HEY-T30, was developed in our laboratory by exposure of HEY cells to stepwise escalating concentrations of Taxol over a 6-month period, and are maintained in media containing Taxol (30 nmol/L). The IGF1R inhibitor NVP-AEW541, a kind gift from Novartis Pharma AG (Basel, Switzerland), is a pyrrolo[2,3-d]pyrimidine derivative small molecular weight kinase inhibitor of the IGF1R (14).
Immunoblotting and Densitometry
Cells were treated as described in the figure legends. Cell lysates were prepared as previously described and protein concentration determined by the Lowry method (15). Cell lysates were separated by SDS-PAGE and transferred to nitrocellulose. Equal protein loading was confirmed by Ponceau staining. Blocking was done with 5% nonfat milk in tris-buffered saline containing 0.1% Tween-20 (TBST). Immunoblotting was performed with phospho-specific antibodies to pAKT-Ser473, pAKT-Thr308, pIGF1R-1135 (all from Cell Signaling Technology, Danvers, MA), followed by incubation in the appropriate HRP-conjugated secondary antibody (Pierce Biotechnology, Rockland, IL) and ECL™ detection (GE Healthcare, Piscataway, NJ). Membranes were placed in stripping buffer (62.5 mmol/L Tris-HCl pH 6.8, 2% SDS, 0.1 M beta-mercaptoethanol) at 50C for 15 minutes, followed by washing of the membrane with TBST. Absence of residual chemiluminescence on the membrane was confirmed by exposure of autoradiograph film. The stripped membranes were probed with antibodies to AKT and IGF1R (both from Santa Cruz Biotech, Santa Cruz, CA). For some experiments, both stripped and new membranes of the same lysates were used to confirm that immunoreactivity was similar when using stripped versus new membranes. All films were scanned and saved in unmodified TIFF format. Densitometry was performed using Image J software. Phospho-AKT expression was normalized to total AKT and shown as the fold-change relative to untreated cells. Anti-GAPDH antibody (Cell Signaling) was used to demonstrate equal protein loading.
RNA isolation, reverse transcription, quantitative real-time PCR
Cell lysates were homogenized using Qiashredder columns (Qiagen Inc., Valencia, CA) and total RNA was isolated by RNeasy Mini Kit (Qiagen) with on-column DNase I (Qiagen) treatment. RNA quantity, purity and integrity were evaluated using the Nanodrop spectrophotometer (Fisher Thermo Scientific, Fremont, CA) and Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). Complementary DNA was synthesized from 1 µg of total RNA using Superscript III Reverse Transcriptase (Invitrogen).
To determine the expression level of IGF2, quantitative real-time PCR was performed using an Applied Biosystems 7900HT Fast Real-Time PCR System. Each reaction utilized 1/20th of the cDNA reaction, forward and reverse primers at a final concentration of 200 nmol/L, and PowerSYBR (Applied Biosystems, Foster City, CA) diluted in Ultrapure water (Invitrogen) to 1X final concentration. Forward and reverse primer sequences used for IGF1, IGF2, and cyclophilin B were previously validated (16–19). Melting curve analysis was performed to confirm a single amplicon corresponding to the PCR product size for each reaction. Real time PCR results were analyzed by the 2−ΔΔCt method to quantify the relative mRNA expression level, as previously described (20).
Cytotoxicity assay
Cells were seeded in 96-well plates at 2000 cells per well and after 24 hours, treated with serial dilutions of Taxol for an additional 72 hours. For drug combination experiments, cells were treated with serial dilutions of Taxol, with or without the addition NVP-AEW541 at 1 µmol/L. After 72 hours of treatment, the relative cell number in each well was determined using the sulforhodamine B (SRB) assay. Cell growth was expressed as %, defined as the ratio of the optical density in the treated well compared with the control well x 100, as previously described (21). Using Calcusyn software, dose-effect curves were generated, and the drug concentrations corresponding to a 50% decrease in cell number (IC50) were determined.
IGF2 depletion by small interfering RNA (siRNA)
Stealth® IGF2 siRNA oligonucleotides and nontargeting Negative Control High-GC Stealth siRNA were obtained from Invitrogen. The RNAiMax Lipofectamine (Invitrogen) reverse transfection was performed according to the manufacturer’s recommended protocol in 60 mm2 dishes, using the experimentally determined optimal siRNA oligonucleotide concentration of 20 nmol/L. After 24 hours of incubation in antibiotic-free OptiMEM (Invitrogen) with 10% FBS, cells were trypsinized and used for the experiments described below. For each experiment, IGF2 siRNA and negative control siRNA transfections were performed in parallel.
To determine the effect of IGF2 knockdown on cell proliferation and Taxol sensitivity, IGF2-siRNA or control-siRNA transfected cells were seeded 24 hours after transfection into 6-well plates at a density of 1.5 × 104 cells per mL, then treated with Taxol, or DMSO, for 72 hours. Adherent cells from each of three replicate wells per treatment were trypsinized and counted on a Coulter counter. The cell proliferation was determined and expressed relative to control-siRNA transfected cells.
To confirm the efficiency of RNA knockdown, 3 replicate wells of transfected cells from each 6-well plate were harvested at the termination of the experiment (96 hours after transfection) for RNA isolation and quantitative real-time PCR analysis of IGF2 expression.
Clinical Specimens
This study was reviewed and approved by the Institutional Review Boards of the Albert Einstein College of Medicine and the University of New Mexico Health Sciences Center. Formalin-fixed, paraffin embedded tumor specimens from 134 patients diagnosed with epithelial ovarian carcinoma or low malignant potential epithelial tumors, treated at the University of New Mexico Cancer Center between March 1996 and June 2006, were retrieved from the Human Tissue Repository. Specimens were obtained at the time of primary surgery. After secondary pathology review, a tissue microarray (TMA) was constructed, containing two cores from each specimen obtained at the time of primary surgery. Tissue cores were absent in 19 cases, leaving 115 cases available for evaluation. Pertinent clinical data were abstracted and a de-identified database was created. Investigators were blinded to clinical data until completion of staining and scoring.
Immunohistochemistry
Immunohistochemistry was performed using a rabbit polyclonal antibody directed at the IGF2 ligand (AB9574; Abcam Inc., Cambridge, MA). The optimal blocking and primary antibody conditions were determined using placental tissue. In brief, Target Retrieval Solution, Citrate pH 6 (Dako North America, Inc., Carpinteria, CA) was used for antigen retrieval, tris-buffered saline containing 5% goat serum and 2% bovine serum albumin was used for blocking, and the primary antibody was used at a 1:100 dilution with an incubation of 1 hour at room temperature. Secondary antibody and detection were performed using the DAKO Envision+ Polymer System (Dako North America), followed by counterstaining with hematoxylin. Staining of all tissue microarray slides was performed concurrently with staining of positive and negative control sections. Representative placenta sections are shown following staining with the IGF2 antibody, or with IGF2 antibody that was pre-absorbed with recombinant IGF2 (supplemental Figure S5). Representative stained ovarian tissue sections were photographed on a Zeiss Axioskop II, and images shown in Figure 5 depict the TIFF image files without modifications, other than cropping of the size. To evaluate assay reproducibility, IGF2 immunohistochemistry was repeated on an independently constructed tissue microarray that contained distinct tissue cores from 53 of the tumor specimens comprising the study population.
Figure 5. Representative images of IGF2 immunohistochemical staining.

A) Absence of staining in the surface epithelium of a normal ovary (40X)
B) Extremely low staining in a Stage 1 mucinous borderline tumor; H-score=0.5
C) Low staining in a Stage 3, grade 1 serous carcinoma; H-score=75
D) Medium staining in a Stage 3, grade 3 serous carcinoma; H-score=100
E) High staining in a Stage 3, grade 3 adenocarcinoma, not otherwise specified; H-score=250
F) High staining in a Stage 3, grade 3 serous carcinoma; H-score 300
A gynecologic pathologist (T.M.H.), who was blinded to all clinical data, graded the cytoplasmic staining intensity (0 negative, 1+ weak, 2+ moderate, 3+ strong) and the percentage of tumor cells with positive staining (1–100%). The H-score is defined as the product of the staining intensity and the percentage of positive staining; the mean H-score for each tumor was determined from the corresponding tissue cores.
Statistical analysis
For cell line experiments, numerical values (drug concentrations, combination index, mRNA levels) are expressed as the mean ± standard deviation, or mean ± standard error, as indicated. The differences in mean values between two groups were statistically analyzed using the two-tailed t-test, while the differences in mean values between multiple groups were analyzed using one-way or two-way ANOVA with the appropriate post-test, as indicated.
For analysis of IGF2 expression in the ovarian tumor tissues, IGF2 expression was categorized as high or low using the median IGF2 H-score, and the Chi-square or Fisher’s exact tests were used to determine the association of IGF2 expression with clinical and pathological variables. Correlation of IGF2 H-scores obtained using two independently constructed and stained tissue microarrays was assessed by Spearman Rank correlation. Survival analysis was performed to study the effect of IGF2 expression on overall and disease-free death events using the Cox proportional hazards regression. Additional adjustment for age, grade, stage, extent of cytoreduction, performance status, and chemotherapy was achieved by including these variables in the Cox models. A predictor was considered as a potential candidate for the final model if the log-rank test of equality across strata had a p-value less than 0.25 in a univariate analysis. During final multivariate Cox model building, all the potential predictors were included in the model and the Wald test was used to measure the statistical significance of hazard ratios. All the possible interactions were tested and proportional hazards (PH) assumptions were checked. No significant interactions were found and the final model was stratified on the non-proportional predictors.
All p values are two-tailed, and p values less than 0.05 were considered statistically significant.
Results
Activation of AKT by Taxol
The effect of Taxol treatment on AKT activation in A2780 cells was evaluated by immunoblot analysis. Initially, a dose response experiment was performed to evaluate a range of Taxol concentrations (1, 5, and 50 nmol/L). The 5 nmol/L concentration produced the largest effect on AKT phosphorylation at 24 hours (data not shown); therefore, subsequent experiments utilized this drug concentration. As shown in Figure 1, Taxol (5 nmol/L) treatment for 24 hours resulted in increased AKT phosphorylation at the critical residues for activation, Threonine 308 and Serine 473. The mean fold-change in phosphorylation level after Taxol treatment, compared with basal phosphorylation level, was 8.5-fold and 2-fold, for Threonine 308 and Serine 473, respectively. ERK phosphorylation was not appreciably altered by Taxol treatment in these cells (data not shown).
Figure 1. Effect of Taxol treatment on AKT phosphorylation.

A2780 cells were maintained in complete media containing 10% FBS and treated for 24 hours with Taxol (5 nmol/L), NVP-AEW541 (1 µmol/L), or both drugs concurrently. Immunoblotting was performed to determine the effect of Taxol treatment on AKT phosphorylation.
A. Representative immunoblots are shown for phospho-AKT (Threonine 308) and phospho-AKT (Serine 473), along with the corresponding total AKT and GAPDH immunoblots.
B. The relative level of AKT phosphorylation at Threonine 308, as determined by densitometry, is shown in the graph (mean ±SE; 4 independent experiments). *p<0.05 (One-way ANOVA, Dunnett post-test)
C. The relative level of AKT phosphorylation at Serine 473, as determined by densitometry, is shown in the graph (mean ±SE; 7 independent experiments). **p<0.01 (One-way ANOVA, Dunnett post-test)
To determine whether Taxol-induced AKT phosphorylation is dependent on IGF1R activation, the small molecule IGF1R tyrosine kinase inhibitor NVP-AEW541 (Novartis Pharma AG, Basel, Switzerland) was used. With regard to selectivity, NVP-AEW541 is a 27-fold more potent inhibitor of the IGF1R kinase compared with inhibition of the insulin receptor kinase, over 50-fold more potent compared with inhibition of c-Kit, and over 100-fold more potent compared with inhibition of HER1, PDGFR, and Bcr-Abl (14). We evaluated a range of concentrations of NVP-AEW541 (0.1 to 10 µmol/L) for the ability to disrupt IGF1R signaling in ovarian carcinoma cells. NVP-AEW541 at 1 µmol/L effectively abrogates IGF2-induced IGF1R autophosphorylation and downstream AKT phosphorylation, and this concentration was used for subsequent experiments (Supplemental Figure S1).
Shown in Figure 1, Taxol-induced AKT phosphorylation was inhibited by concurrent treatment with the IGF1R inhibitor NVP-AEW541. Our results indicate that Taxol treatment results in increased AKT phosphorylation, and that phosphorylation of AKT due to Taxol treatment requires IGF1R tyrosine kinase activity.
IGF2 regulation by Taxol
To investigate whether the observed IGF1R activation is mediated by autocrine signaling, the effect of Taxol treatment on the expression of IGF1 mRNA and IGF2 mRNA was quantified in A2780 cells. A dose response experiment was performed to evaluate the effect of different Taxol concentrations (1, 5, 20, and 50 nmol/L) on IGF2 mRNA levels. Concordant with the immunoblot results, Taxol 5 nmol/L produced the greatest increase in IGF2 mRNA expression (data not shown). Following Taxol treatment (5 nmol/L), IGF2 mRNA levels progressively increased over 24 hours (Figure 2A). In contrast, cells treated with baccatin III, an inactive Taxol analog which does not bind to or stabilize microtubules (22), did not alter IGF2 mRNA levels compared with untreated cells. IGF1 mRNA expression was unchanged after Taxol treatment (data not shown).
Figure 2. Taxol-induced IGF2 mRNA expression.
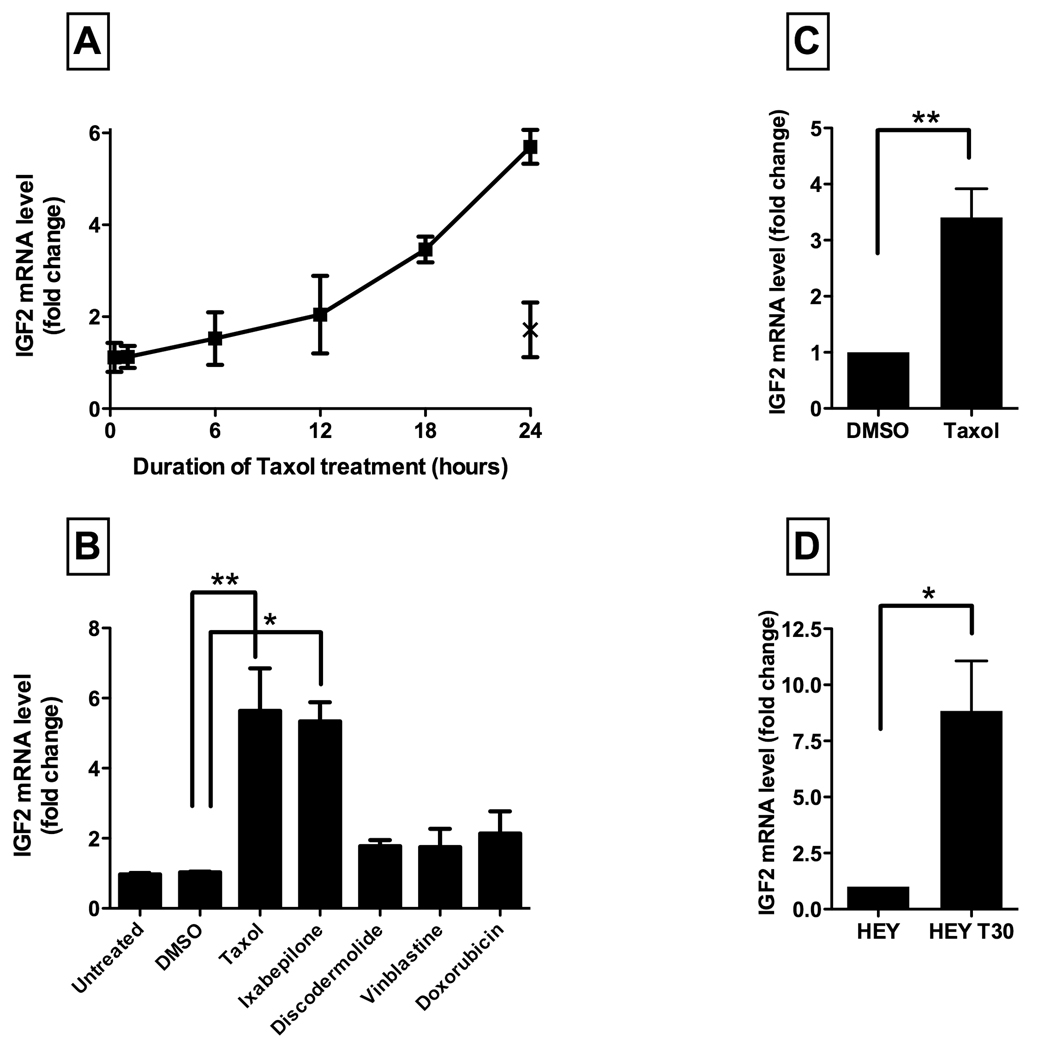
A. The time course of IGF2 expression following Taxol treatment (5 nmol/L) in A2780 cells is depicted with squares and a solid connecting line. The IGF2 mRNA level after Baccatin treatment (5 nmol/L) for 24 hours is also shown on the graph, marked by an X. Results shown are the mean ±SE of two independent experiments, each performed in triplicate.
B. A2780 cells were treated for 24 hours with vehicle alone (DMSO) or with cytotoxic drugs, at their equipotent molar concentrations. The IGF2 mRNA level is expressed as fold-change relative to untreated cells. Results shown are the mean ±SE of two independent experiments, each performed in triplicate.
*p<0.05; **p<0.01 (One-way ANOVA, Dunnett post-test)
C. HEY cells were treated with vehicle alone (DMSO) or Taxol 5 nmol/L for 24 hours. The IGF2 mRNA level, determined by reverse transcription quantitative real-time PCR, is expressed as fold-change relative to untreated cells in complete growth medium. Results shown are the mean ±SE of three independent experiments, each performed in triplicate. **p< 0.01 (t-test)
D. IGF2 mRNA expression in Taxol-resistant HEY-T30 cells, determined by reverse transcription quantitative real-time PCR, is expressed as fold-change relative to HEY parental cells. Results shown are the mean ±SE of three independent experiments, each performed in triplicate. *p<0.05 (t-test)
To evaluate whether treatment with other cytotoxic compounds induces IGF2 mRNA expression, A2780 cells were treated for 24 hours with each of several microtubule-interacting drugs and other compounds at their approximately equipotent drug concentrations (as determined by SRB proliferation assays), and IGF2 mRNA levels were measured (Figure 2B). Treatment with the microtubule-stabilizing agent ixabepilone (10 nmol/L), an epothilone compound with a chemical structure distinct from Taxol, resulted in an increase in IGF2 mRNA expression, to a similar degree as Taxol. However, treatment with discodermolide (20 nmol/L), also a microtubule-stabilizing agent but with a more complex mechanism of action (23, 24), did not significantly induce IGF2 mRNA at this time point. Treatment with the microtubule-destabilizing agent vinblastine (5 nmol/L) or the DNA-intercalating agent doxorubicin (50 nmol/L) did not result in significant IGF2 upregulation. These findings suggest that IGF2 upregulation by Taxol does not occur as a generalized response to cytotoxic drugs, but may require a specific interaction with microtubules that is shared between Taxol and ixabepilone.
Evaluation of paired sensitive and resistant HEY ovarian cancer cells
The Taxol resistant cell line, HEY-T30, was developed in our laboratory by repeated exposure of HEY ovarian carcinoma cells to Taxol. Like A2780 cells, HEY ovarian carcinoma cells exhibit upregulation of IGF2 mRNA levels following Taxol treatment for 24 hours (Figure 2C). The Taxol resistant HEY-T30 cells exhibit significantly elevated IGF2 mRNA levels compared with the parental HEY cells (Figure 2D). Under either normal growth or serum starvation conditions, constitutive IGF1R phosphorylation is observed in HEY-T30 cells whereas parental HEY cells do not exhibit constitutive IGF1R phosphorylation (Supplemental Figure S2). Cross-resistance to cisplatin, a platinum DNA-damaging chemotherapy drug, is not observed in HEY-T30 cells (Supplemental Figure S3).
The Taxol concentration that results in 50% growth inhibition (IC50) for parental and resistant cells is shown in Figure 3. We next assessed whether inhibition of IGF1R alters Taxol sensitivity in HEY and HEY-T30 cells. The growth inhibition resulting from combination drug treatment with Taxol and concurrent NVP-AEW541 was evaluated and compared with the effect of either drug alone (Figure 3). As in the previous experiment, NVP-AEW541 was used at a concentration of 1 µmol/L, a concentration which effectively blocks IGF1R phosphorylation. As a single agent, NVP-AEW541 (1 µmol/L) alone had a minimal effect on cell growth in either cell line. In the parental HEY cells, treatment with NVP-AEW514 (1 µmol/L) modestly potentiated the effect of Taxol. In these cells, the IC50 for Taxol is 2.6 nmol/L; however in the presence of NVP-AEW541, it is 1.7 nmol/L. The potentiation of Taxol efficacy by NVP-AEW541 in the resistant HEY-T30 cells was more dramatic. In the resistant cells, the IC50 for Taxol is 106 nmol/L, while in the presence of NVP-AEW541, it decreases to 17 nmol/L, representing a greater than 6-fold sensitization to Taxol. Thus, IGF1R inhibition was efficacious in sensitizing ovarian cancer cells to Taxol in this model of acquired drug resistance.
Figure 3. Potentiation of Taxol treatment by the IGF1R inhibitor NVP-AEW541.

Cytotoxicity assays for HEY and HEY-T30 ovarian carcinoma cells were performed using the sulforhodamine B method to determine the effect of Taxol treatment alone or with concurrent NVP-AEW541.
A. HEY parental cells: the cell number relative to untreated cells, expressed as cell growth (%), is plotted over a range of Taxol concentrations. Treatment with Taxol alone is depicted with squares and solid lines, compared with combination treatment with Taxol plus NVP-AEW541 1 µmol/L, depicted with triangles and dotted lines. The cell growth (%) resulting from NVP-AEW541 (1 µmol/L) alone is plotted on the Y-axis (triangle).
p<0.05 (Two-way ANOVA)
B. HEY-T30 resistant cells: the cell number relative to untreated cells, expressed as cell growth (%), is plotted over a range of Taxol concentrations, as in panel A. The cell growth (%) resulting from NVP-AEW541 (1 µmol/L) alone is plotted on the Y-axis (triangle).
p<0.001 (Two-way ANOVA)
C. For each cell line, the IC50 concentration of Taxol alone or with concurrent NVP-AEW541 1 µmol/L is plotted in the bar graph (mean ±SE; at least 3 independent experiments with 6 replicates each).
p<0.0001 (One-way ANOVA, Bonferroni post-test)
We have evaluated three additional ovarian carcinoma cell lines made resistant to Taxol and other microtubule-stabilizing agents (MSA) in our laboratory. As determined by SRB assay, the A2780-Tx15 cell line is 20-fold resistant to Taxol, the HEY-BMS20 cell line is 3-fold resistant to ixabepilone, and the OVCAR8-D30 cell line is 2-fold resistant to discodermolide. Each of these resistant cell lines is significantly sensitized to MSA treatment by concurrent IGF1R inhibition using 1 µmol/L NVP-AEW541.
IGF2 knockdown in Taxol-resistant cells
IGF1R inhibition would be expected to block signaling transduced not only by IGF2 but also by IGF1 ligand-binding. Therefore, to determine whether inhibition of IGF2-transduced signaling alone is sufficient to sensitize ovarian cancer cells to Taxol, IGF2 depletion by siRNA was examined. The parental HEY cells and the Taxol-resistant HEY-T30 cells were transfected with IGF2-siRNA or control-siRNA. As determined by real-time PCR, the efficacy of IGF2 mRNA knockdown by IGF2 siRNA was decreased by approximately 90% at 48 and 96 hours; immunoblotting confirmed that IGF2 protein expression was decreased by approximately 70% by IGF2 siRNA treatment (Supplemental Figure S4).
For each cell line, the proliferation and the sensitivity of the IGF2-depleted cells to Taxol was compared with control-siRNA transfected cells. In parental HEY cells (Figure 4A), IGF2 depletion does not significantly affect the cell growth nor does it significantly potentiate the effect of Taxol in these cells, compared with control-siRNA transfection. In contrast, the effect of IGF2 depletion in the resistant HEY-T30 cells is substantial (Figure 4B). Unlike IGF1R inhibition, which alone minimally affects cellular proliferation of HEY-T30 cells, IGF2 depletion by siRNA robustly suppressed cellular proliferation. The relative cell number following IGF2-siRNA transfection was 27% compared with control-siRNA transfected cells, corresponding to an approximately 4-fold reduction in cell number (P <0.01). Treatment of IGF2-depleted HEY-T30 cells using approximately the IC50 concentration of Taxol (100 nmol/L) was highly efficacious. As shown in Figure 4B, IGF2-siRNA plus Taxol treatment resulted in 10% cell growth after a 72 hour incubation, compared with 45% cell growth following control-siRNA plus Taxol treatment (p<0.05).
Figure 4. Effect of IGF2 depletion by siRNA transfection in HEY and HEY-T30 cells.

IGF2 siRNA transfection was used to evaluate the effect of IGF2 depletion on cell proliferation and Taxol sensitivity. Cells were transfected with IGF2 siRNA (IGF2-SI) or control siRNA (c-SI). Twenty-four hours after transfection, cells were counted, reseeded, and incubated for an additional 72 hours, with or without the addition of Taxol at the IC50 concentration of Taxol for each cell line, followed by cell counting. Cell numbers are expressed as % cell growth relative to DMSO-treated, c-SI-transfected cells. Results shown are the mean ±SEM of two independent experiments, each performed in triplicate for HEY parental cells (Panel A) and HEY-T30 cells (Panel B).
*p< 0.05, **p<0.01 (one-way ANOVA, Bonferroni post-test)
IGF2 expression in epithelial ovarian tumors
To assess the potential clinical application of these findings, IGF2 protein expression was analyzed in primary ovarian tumors. The clinical and pathologic characteristics of the study population are shown in Table 1. Thirty-six (31.3%) patients had epithelial ovarian tumors of low malignant potential (LMP) while 79 (68.7%) had invasive epithelial ovarian carcinoma (EOC). Patients with EOC were older (p=0.001) and were more likely to have advanced stage disease (p<0.001). The distribution of histologic types differed between the LMP and EOC groups (p<0.001). In the EOC group, serous histology (55.7%) was the most frequent histology. Mucinous histology was more frequent in the LMP group than in the EOC group (47.2% versus 13.9%).
Table 1.
Characteristics of the study population
| Variable | LMP N | (%) | EOC N | (%) | Total N | (%) | p |
|---|---|---|---|---|---|---|---|
| Age | 0.001 | ||||||
| ≤ 50 years | 22 | 61.1 | 23 | 29.1 | 45 | 39.1 | |
| > 50 years | 14 | 38.9 | 56 | 70.9 | 70 | 60.9 | |
| Race/Ethnicity | 0.142 | ||||||
| Non-Hispanic White | 21 | 58.3 | 36 | 45.6 | 57 | 49.6 | |
| Hispanic | 11 | 30.6 | 28 | 35.4 | 39 | 33.9 | |
| American Indian | 2 | 5.6 | 14 | 17.7 | 16 | 13.9 | |
| Black | 1 | 2.8 | 1 | 1.3 | 2 | 1.7 | |
| Asian | 1 | 2.8 | 0 | 0.0 | 1 | 0.9 | |
| FIGO Stage | <0.001 | ||||||
| I | 32 | 88.9 | 24 | 30.4 | 56 | 48.7 | |
| II | 2 | 5.6 | 7 | 8.9 | 9 | 7.8 | |
| III | 2 | 5.6 | 38 | 48.1 | 40 | 34.8 | |
| IV | 0 | 0.0 | 10 | 12.7 | 10 | 8.7 | |
| Histology | <0.001 | ||||||
| Serous | 17 | 47.2 | 44 | 55.7 | 61 | 53.0 | |
| Mucinous | 17 | 47.2 | 11 | 13.9 | 28 | 24.3 | |
| Endometrioid | 0 | 0.0 | 6 | 7.6 | 6 | 5.2 | |
| Other | 2 | 5.6 | 18 | 22.8 | 20 | 17.4 | |
| Grade | N/A | ||||||
| 0 | 36 | 100.0 | 0 | 0.0 | 36 | 31.3 | |
| 1 | 0 | 12 | 15.2 | 12 | 10.4 | ||
| 2 | 0 | 19 | 24.1 | 19 | 16.5 | ||
| 3 | 0 | 48 | 60.8 | 48 | 41.7 | ||
| Total | 36 | 79 | 11 5 |
LMP, epithelial ovarian tumors of low malignant potential; EOC, invasive epithelial ovarian carcinoma; Race/ethnicity as defined by SEER coding; N/A, not applicable.
Representative tissue sections of benign and malignant clinical specimens demonstrating a range of IGF2 protein expression, with corresponding H-scores noted in the figure legends, are shown in Figure 5. Positive and negative control sections used for quality assurance are shown in supplemental Figure S5. To assess potential batch effects and biological variation due to tissue sampling, a second tissue microarray was used that included distinct tissue cores from 53 tumors from the original study population. The correlation of IGF2 H-scores from the two independently constructed and stained tissue microarrays was high (Spearman Rank correlation, R=0.664).
Table 2 depicts the association of IGF2 expression with selected clinical and pathologic variables. IGF2 tumor expression was not correlated with age or ethnicity. Shown in Figure 6A, IGF2 expression was significantly associated with FIGO stage, with higher expression in advanced stage tumors compared with early stage tumors (p<0.001). The histological subtypes demonstrated distinct patterns of expression (Figure 6B): mucinous tumors had lower IGF2 expression compared with serous or endometrioid tumors (p=0.011). Shown in Figure 6C, IGF2 expression varied significantly with tumor grade, with higher expression observed in invasive epithelial ovarian cancers compared with borderline ovarian tumors (p=0.003).
Table 2.
IGF 2 expression in LMP tumors and epithelial ovarian carcinoma (EOC)
| Variable | LMP tumors (36) |
EOC (79) |
Totals (115) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IGF2-Low N | (%) | IGF2-High N | (%) | p | IGF2-Low N | (%) | IGF2-High N | (%) | p | IGF2-Low N | (%) | IGF2-High N | (%) | p | |
| Age | 1.000 | 0.873 | 0.335 | ||||||||||||
| ≤ 50 years | 17 | 60.7 | 5 | 62.5 | 9 | 28.1 | 14 | 29.8 | 26 | 43.3 | 19 | 34.5 | |||
| > 50 years | 11 | 39.3 | 3 | 37.5 | 23 | 71.9 | 33 | 70.2 | 34 | 56.7 | 36 | 65.5 | |||
| Race/Ethnicity | 0.544 | 0.339 | 0.166 | ||||||||||||
| Non-Hispanic White | 15 | 53.6 | 6 | 75.0 | 12 | 37.5 | 24 | 51.1 | 27 | 45.0 | 30 | 54.6 | |||
| Hispanic | 10 | 35.7 | 1 | 12.5 | 14 | 43.8 | 14 | 29.8 | 24 | 40.0 | 15 | 27.3 | |||
| American Indian | 1 | 3.6 | 0 | 0.0 | 1 | 3.1 | 0 | 0.0 | 6 | 10.0 | 10 | 18.2 | |||
| Black | 1 | 3.6 | 1 | 12.5 | 5 | 15.6 | 9 | 19.1 | 2 | 3.3 | 0 | 0.0 | |||
| Asian | 1 | 3.6 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 1 | 1.7 | 0 | 0.0 | |||
| FIGO Stage | 0.028 | 0.124 | <0.001 | ||||||||||||
| I | 27 | 96.4 | 5 | 62.5 | 12 | 37.5 | 12 | 25.5 | 39 | 65.0 | 17 | 30.9 | |||
| II | 0 | 0.0 | 2 | 25.0 | 4 | 12.5 | 3 | 6.4 | 4 | 6.7 | 5 | 9.1 | |||
| III | 1 | 3.6 | 1 | 12.5 | 15 | 46.9 | 23 | 48.9 | 16 | 26.7 | 24 | 43.6 | |||
| IV | 0 | 0.0 | 0 | 0.0 | 1 | 3.1 | 9 | 19.1 | 1 | 1.7 | 9 | 16.4 | |||
| Histology | 0.055 | 0.664 | 0.011 | ||||||||||||
| Serous | 10 | 35.7 | 7 | 87.5 | 17 | 53.1 | 27 | 57.4 | 27 | 45.0 | 34 | 61.8 | |||
| Mucinous | 16 | 57.1 | 1 | 12.5 | 6 | 18.8 | 5 | 10.6 | 22 | 36.7 | 6 | 10.9 | |||
| Endometrioid | 0 | 0.0 | 0 | 0.0 | 3 | 9.4 | 3 | 6.4 | 3 | 5.0 | 3 | 5.5 | |||
| Other | 2 | 7.1 | 0 | 0.0 | 6 | 18.8 | 12 | 25.5 | 8 | 13.3 | 12 | 21.8 | |||
| Tumor Grade | N/A | 0.933 | 0.003 | ||||||||||||
| 0 | 28 | 100.0 | 8 | 100.0 | 0 | 0.0 | 0 | 0.0 | 28 | 46.7 | 8 | 14.6 | |||
| 1 | 0 | 0.0 | 0 | 0.0 | 5 | 15.6 | 7 | 14.9 | 5 | 8.3 | 7 | 12.7 | |||
| 2 | 0 | 0.0 | 0 | 0.0 | 7 | 21.9 | 12 | 25.5 | 7 | 11 . 7 | 12 | 21.8 | |||
| 3 | 0 | 0.0 | 0 | 0.0 | 20 | 62.5 | 28 | 59.6 | 20 | 33.3 | 28 | 50.9 | |||
| Total | 28 | 8 | 32 | 47 | 60 | 55 | |||||||||
IGF2-Low, H-score≤105; IGF2-High, H-score>105; N/A, not applicable
Figure 6. Correlation between IGF2 expression and clinicopathologic factors in ovarian tumors.

A) IGF2 staining is correlated with FIGO stage, with higher frequency of IGF2 overexpression in advanced stage tumors; p<0.001 (Fisher’s exact test)
B) Serous (Ser) and endometrioid (Endo) histologic types are correlated with high IGF2 staining, compared with mucinous (Muc) tumors; p=0.011 (Chi-square test)
C) IGF2 staining is correlated with tumor grade, with low malignant potential (LMP; borderline) tumors demonstrating the lowest frequency of IGF2 overexpression; p=0.003 (Fisher’s exact test)
D) Patients with high IGF2 expression (red line) had a significant reduction in progression-free survival (PFS) compared with patients with low IGF2 expression (black line); p=0.03 (log-rank test).
E) When LMP tumors are excluded from the analysis, the association of IGF2 expression and progression-free survival (PFS) is not statistically significant.
On univariate survival analysis (Figure 6D), high IGF2 expression was significantly associated with reduced disease-free survival (p=0.03). Age (p=0.02), grade (p<0.0001), stage (p<0.0001), extent of cytoreduction (p<0.0001), and performance status (p=0.0002), and chemotherapy (p=0.0002), but not race or histology, were risk factors associated with disease-free survival on univariate analysis. When LMP tumors were excluded from the analysis, the association of IGF2 expression and disease-free survival was no longer statistically significant (Figure 6E). On multivariate analysis, stage (p=0.001), extent of cytoreduction (p=0.002), and grade (p=0.04) were independently associated with disease-free survival.
With regard to overall survival, there have been 29 deaths. There have been 12 deaths (among 60 patients; 20%) in the low IGF2 group, compared with 17 deaths (among 55 patients; 31%) in the high IGF2 group; the effect of IGF2 expression on overall survival was not significant on univariate analysis (p=0.19). Age (p=0.005), grade (p<0.0001), stage (p<0.0001), extent of cytoreduction (p<0.0001), performance status (p<0.0001), and chemotherapy (p=0.009) were risk factors associated with overall survival on univariate analysis. For multivariate analysis, stratification by grade (LMP vs EOC) was performed, as no deaths occurred in the LMP group. On multivariate survival analysis, stage (p=0.007), extent of cytoreduction (p=0.001), and chemotherapy (p=0.0003) were independently associated with overall survival.
Discussion
This study is the first, to our knowledge, to evaluate the role of the IGF signaling pathway in the response of ovarian carcinoma cells to Taxol. Our findings demonstrate that Taxol treatment results in upregulation of IGF2 expression associated with activation of AKT. As shown in a cell line model of acquired drug resistance in ovarian carcinoma, Taxol-resistance is associated with elevated IGF2 expression, while IGF1R inhibition or IGF2 depletion restores drug sensitivity. In clinical tumor specimens, high IGF2 protein expression is significantly associated with poor prognostic factors for recurrence and death.
The pyrrolo[2,3-d]pyrimidine compound NVP-AEW541 used in this study, and the closely related compound NVP-ADW742, were the first IGF1R-specific small molecule tyrosine kinase inhibitors reported in the literature (14, 25). Using doses corresponding to specific inhibition of the IGF1R tyrosine kinase, we demonstrated that NVP-AEW541 effectively blocks Taxol-induced AKT phosphorylation. Although NVP-AEW541 alone did not suppress proliferation in ovarian carcinoma cells, treatment with NVP-AEW541 significantly potentiated the effect of Taxol in both sensitive and resistant cells. While the compound NVP-AEW541 is not undergoing clinical development at this time, a variety of small molecule inhibitors and monoclonal antibodies that target the IGF1R are presently undergoing evaluation in clinical trials, and it is anticipated that one or more of these compounds may be approved for use in the clinic (13).
An alternate therapeutic strategy is to target the aberrantly expressed ligand, rather than its receptor. As IGF2 has been shown to bind not only the IGF1R, but also the insulin receptor isoform A and hybrid IGF1R-insulin receptors, signaling by IGF2 could potentially circumvent the IGF1R in the presence of IGF1R-specific inhibitors (26, 27). In the present study, the combination of IGF2 depletion and Taxol was found to be highly effective at inhibiting cell growth in the setting of acquired drug resistance. Interestingly, IGF2 depletion alone resulted in potent suppression of proliferation distinct from the minimal antiproliferative effect of IGF1R inhibition. The potent effect of IGF2 depletion suggests possible dependency on IGF2 for proliferation in these Taxol-resistant cells.
Previously, it was reported that PI3K and AKT inhibition could enhance sensitivity to Taxol (9, 28, 29). As PI3K/AKT are major downstream effectors of IGF1R activity, the concordant effects resulting from blockade at downstream targets in the IGF signaling cascade validates the findings of the present study. From a clinical perspective, significant metabolic toxicity has been encountered with direct AKT inhibition (29), highlighting a potential disadvantage to blockade at the downstream intracellular targets where multiple signaling pathways converge. Targeting a specific ligand-cell surface receptor may reduce these undesired effects on the physiology of normal cells. However, targeting at the receptor level, such as IGF1R or EGFR inhibition, has been shown to promote activation of the reciprocal receptor in cancer cell lines via crosstalk mechanisms (30). Thus, it is likely that optimal combination treatment strategies will require consideration of compensatory feedback signals that occur in a cell-specific manner.
In the clinic, multi-drug resistance is a hallmark of progressive ovarian cancer. In addition to Taxol, platinum compounds are widely used in the treatment of primary and recurrent ovarian cancer. Interestingly, it was recently reported that IGF1R inhibition may restore drug sensitivity in platinum-resistant ovarian cancer cells (31). Another study demonstrated activity of an IGF1R-targeting antibody in a fluorouracil-resistant colorectal carcinoma cell line (32). With regard to tubulin-targeting agents, resistance to the microtubule stabilizing agent docetaxel was associated with predicted PI3K pathway activation, by gene expression profiling, in a variety of carcinoma cell lines (33). These studies, in conjunction with the present study, strongly support the relevance of the IGF pathway to chemotherapy resistance.
IGF2 upregulation has been implicated in carcinogenesis and disease progression in ovarian cancer. Previous studies utilized a gene expression microarray approach to identify IGF2 mRNA to be significantly over-expressed in ovarian cancer compared with benign ovary (34, 35). In addition, high IGF2 mRNA expression was associated with advanced stage, high grade, and poor survival in a cohort of patients with serous epithelial ovarian cancer (36). Interestingly, IGF2 protein levels in the serum of patients with ovarian cancer were decreased compared with unaffected women (37, 38). The present study is, to our knowledge, the first to evaluate the significance of IGF2 expression at the protein level in ovarian tumors from patients. Consistent with the prior mRNA expression studies, high IGF2 protein expression was observed more frequently in patients with advanced stage and high-grade tumors, and was associated with clinical/pathologic predictors of worse prognosis. In the study sample, approximately half of the tumors were serous histology, which is the most common type of epithelial ovarian cancer; however, early stage cancer, less common histological types, and LMP tumors were also present at relatively high proportions in this cohort. Based on our findings, subsequent studies should prospectively evaluate IGF2 protein expression in primary and recurrent epithelial ovarian tumors for evaluation of its utility as a potential biomarker, and to confirm its frequent overexpression in ovarian cancer. If IGF2 is found to be a useful biomarker, the standard methods used for IGF2 immunohistochemistry in this study are within the capability of many surgical pathology laboratories and therefore readily translatable to clinical application.
In summary, activation of autocrine IGF signaling occurs following Taxol treatment of ovarian carcinoma cells, and is observed in association with the acquisition of Taxol resistance. Inhibition of the IGF signaling pathway significantly potentiates Taxol efficacy and sensitizes drug-resistant cells. In primary ovarian tumors, high IGF2 expression is strongly associated with known clinicopathologic risk factors, including stage and grade, and its overexpression is associated with reduced disease-free survival. These findings strongly support the clinical relevance of the IGF pathway in ovarian cancer, and suggest a potential therapeutic role for inhibitors of this pathway in the management of this disease.
Statement of Translational Relevance.
Elucidation of biological factors underlying drug resistance is critical for developing more effective treatments for ovarian cancer. Activation of the serine-threonine kinase, AKT, which promotes cellular survival, occurs following Taxol treatment; however, the upstream signaling events have not been thoroughly investigated. In this study, we examined the role of the insulin-like growth factor (IGF) signaling pathway in the response of ovarian cancer cells to Taxol. Taxol-induced AKT phosphorylation required IGF1R activity, and was associated with upregulation of IGF2. Resistant cells exhibited increased IGF2 mRNA compared with sensitive cells. IGF pathway inhibition, by IGF1R blockade or IGF2 depletion, restored Taxol sensitivity. High IGF2 expression in ovarian tumors was associated with advanced stage and high grade; patients with high IGF2 tumor expression had reduced disease-free survival. Thus, IGF2 modulates Taxol-resistance, and its expression correlates with poor prognosis in patients. These novel findings suggest that IGF2 is a therapeutic target for ovarian cancer.
Supplementary Material
Acknowledgements
We acknowledge the use of the Genomics Shared Resource and the Analytical Imaging Facility of the Albert Einstein Cancer Center, supported by the National Cancer Institute Cancer Center Support Grant (2P30CA013330). The authors would like to thank Drs. Charles R. Key and Mary Lipscomb, Co-Directors of the Human Tissue Repository at the University of New Mexico Hospital/Cancer Center, supported by the National Cancer Institute Cancer Center Support Grant (P30 CA118110), for providing tissue samples and clinical data. The authors appreciate helpful discussions with Drs. Chia-Ping Huang Yang and Hayley McDaid.
Financial support: This work was supported by the National Cancer Institute-National Institute of Child Health and Human Development Reproductive Scientist Development Program (RSDP) Grant 5K12HD000849, the Gynecologic Cancer Foundation/Kea Simon Ovarian Cancer Research Grant, and the Dew Point Scholars Program, Department of Obstetrics & Gynecology and Women’s Health at Albert Einstein College of Medicine (to G.S.H.); National Cancer Institute Grant CA124898 and the National Foundation for Cancer Research (to S.B.H.); and National Cancer Institute Grant CA118743 (to E.R.P.).
Footnotes
A portion of this data was presented at the Annual Meeting of the American Association for Cancer Research (AACR), Los Angeles, CA, April 2007 and the Annual Meeting of the Society of Gynecologic Oncologists (SGO), Tampa, FL, March 2008.
Conflicts of interest: None
References
- 1.Giannakakou P, Nakano M, Nicolaou KC, et al. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc Natl Acad Sci U S A. 2002;99:10855–10860. doi: 10.1073/pnas.132275599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994;5 Suppl 6:S3–S6. [PubMed] [Google Scholar]
- 3.Rao S, Orr GA, Chaudhary AG, Kingston DG, Horwitz SB. Characterization of the taxol binding site on the microtubule. 2-(m-Azidobenzoyl)taxol photolabels a peptide (amino acids 217–231) of beta-tubulin. J Biol Chem. 1995;270:20235–20238. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- 4.Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 5.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 6.Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDaid HM, Horwitz SB. Selective potentiation of paclitaxel (taxol)-induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol. 2001;60:290–301. doi: 10.1124/mol.60.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDaid HM, Lopez-Barcons L, Grossman A, et al. Enhancement of the therapeutic efficacy of taxol by the mitogen-activated protein kinase kinase inhibitor CI-1040 in nude mice bearing human heterotransplants. Cancer Res. 2005;65:2854–2860. doi: 10.1158/0008-5472.CAN-04-4391. [DOI] [PubMed] [Google Scholar]
- 9.Mabuchi S, Ohmichi M, Kimura A, et al. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem. 2002;277:33490–33500. doi: 10.1074/jbc.M204042200. [DOI] [PubMed] [Google Scholar]
- 10.Gotlieb WH, Bruchim I, Gu J, et al. Insulin-like growth factor receptor I targeting in epithelial ovarian cancer. Gynecol Oncol. 2006;100:389–396. doi: 10.1016/j.ygyno.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 11.Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 12.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodon J, DeSantos V, Ferry RJ, Jr, Kurzrock R. Early drug development of inhibitors of the insulin-like growth factor-I receptor pathway: lessons from the first clinical trials. Mol Cancer Ther. 2008;7:2575–2588. doi: 10.1158/1535-7163.MCT-08-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Echeverria C, Pearson MA, Marti A, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–239. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 15.Yang CP, Horwitz SB. Taxol mediates serine phosphorylation of the 66-kDa Shc isoform. Cancer Res. 2000;60:5171–5178. [PubMed] [Google Scholar]
- 16.Lu L, Katsaros D, Wiley A, et al. Promoter-specific transcription of insulin-like growth factor-II in epithelial ovarian cancer. Gynecol Oncol. 2006;103:990–995. doi: 10.1016/j.ygyno.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Lu L, Katsaros D, Wiley A, et al. The relationship of insulin-like growth factor-II, insulin-like growth factor binding protein-3, and estrogen receptor-alpha expression to disease progression in epithelial ovarian cancer. Clin Cancer Res. 2006;12:1208–1214. doi: 10.1158/1078-0432.CCR-05-1801. [DOI] [PubMed] [Google Scholar]
- 18.Matsuyama R, Togo S, Shimizu D, et al. Predicting 5-fluorouracil chemosensitivity of liver metastases from colorectal cancer using primary tumor specimens: three-gene expression model predicts clinical response. Int J Cancer. 2006;119:406–413. doi: 10.1002/ijc.21843. [DOI] [PubMed] [Google Scholar]
- 19.Alexiadis M, Mamers P, Chu S, Fuller PJ. Insulin-like growth factor, insulin-like growth factor-binding protein-4, and pregnancy-associated plasma protein-A gene expression in human granulosa cell tumors. Int J Gynecol Cancer. 2006;16:1973–1979. doi: 10.1111/j.1525-1438.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 20.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR Chapter 15. Curr Protoc Mol Biol. 2006 doi: 10.1002/0471142727.mb1508s73. Unit 15 8. [DOI] [PubMed] [Google Scholar]
- 21.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 22.He L, Jagtap PG, Kingston DG, Shen HJ, Orr GA, Horwitz SB. A common pharmacophore for Taxol and the epothilones based on the biological activity of a taxane molecule lacking a C-13 side chain. Biochemistry. 2000;39:3972–3978. doi: 10.1021/bi992518p. [DOI] [PubMed] [Google Scholar]
- 23.Klein LE, Freeze BS, Smith AB, 3rd, Horwitz SB. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle. 2005;4:501–507. doi: 10.4161/cc.4.3.1550. [DOI] [PubMed] [Google Scholar]
- 24.Huang GS, Lopez-Barcons L, Freeze BS, et al. Potentiation of taxol efficacy by discodermolide in ovarian carcinoma xenograft-bearing mice. Clin Cancer Res. 2006;12:298–304. doi: 10.1158/1078-0432.CCR-05-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004;5:221–230. doi: 10.1016/s1535-6108(04)00050-9. [DOI] [PubMed] [Google Scholar]
- 26.Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–39695. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 27.Sciacca L, Mineo R, Pandini G, Murabito A, Vigneri R, Belfiore A. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene. 2002;21:8240–8250. doi: 10.1038/sj.onc.1206058. [DOI] [PubMed] [Google Scholar]
- 28.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3'-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–1092. [PubMed] [Google Scholar]
- 29.Luo Y, Shoemaker AR, Liu X, et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 30.Haluska P, Carboni JM, TenEyck C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008;7:2589–2598. doi: 10.1158/1535-7163.MCT-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckstein N, Serva K, Hildebrandt B, Politz A, von Jonquieres G, Wolf-Kummeth S. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69:2996–3003. doi: 10.1158/0008-5472.CAN-08-3153. [DOI] [PubMed] [Google Scholar]
- 32.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potti A, Dressman HK, Bild A, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12:1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- 34.Sawiris GP, Sherman-Baust CA, Becker KG, Cheadle C, Teichberg D, Morin PJ. Development of a highly specialized cDNA array for the study and diagnosis of epithelial ovarian cancer. Cancer Res. 2002;62:2923–2928. [PubMed] [Google Scholar]
- 35.Lancaster JM, Dressman HK, Whitaker RS, et al. Gene expression patterns that characterize advanced stage serous ovarian cancers. J Soc Gynecol Investig. 2004;11:51–59. doi: 10.1016/j.jsgi.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Sayer RA, Lancaster JM, Pittman J, et al. High insulin-like growth factor-2 (IGF-2) gene expression is an independent predictor of poor survival for patients with advanced stage serous epithelial ovarian cancer. Gynecol Oncol. 2005;96:355–361. doi: 10.1016/j.ygyno.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Visintin I, Feng Z, Longton G, et al. Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res. 2008;14:1065–1072. doi: 10.1158/1078-0432.CCR-07-1569. [DOI] [PubMed] [Google Scholar]
- 38.Mor G, Visintin I, Lai Y, et al. Serum protein markers for early detection of ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:7677–7682. doi: 10.1073/pnas.0502178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.