

Abstract
Background
P-glycoprotein (P-gp), the product of the multi-drug resistance gene (MDR), is an ATP-dependent transmembrane pump, which is expressed in multiple cell lineages including epithelial and hematopoetic cells. The human MDR gene is located on chromosome 7 (7q21.1), a susceptibility loci for inflammatory bowel disease (IBD). A significant number of IBD patients carry mutations in this gene and P-gp deficient FVB/N mice develop a severe spontaneous colitis, characterized by impaired intestinal barrier function and immune reactivity to intestinal bacterial antigens.
Methods
In this work we explored the role of mouse strain, as well as environmental insults on the development of colonic inflammation in the absence of P-gp. Among the induction methods utilized, dextran sodium sulfate (DSS) disrupts the intestinal epithelium, while Piroxicam is a non-steroidal anti-inflammatory (NSAID) drug which inhibits prostaglandin production, and initiates colitis in IL10 deficient animals. Helicobacter bilis (H.bilis) is a known mediator of bacterial induced colitis.
Results
We demonstrate that crossing this mutation onto the C57BL/6 strain confers protection from spontaneous colitis. C57BL/6.mdr1a deficient animals demonstrated increased histological inflammation, colonic shortening, fecal blood, and reduced body weight after 7 days of treatment with 2.25% DSS. C57BL/6.mdr1a deficient mice treated with piroxicam or infected with H. bilis showed no weight loss, or alterations in colonic histology.
Conclusions
These data indicate that the effects of P-gp deficiency are significantly modulated by background strain influences, but that the epithelium continues to have increased susceptibility to chemical injury in the C57BL/6 model.
Keywords: colitis, p-glycoprotein, mouse models, helicobacter
Inflammatory bowel diseases (IBD) are diseases of unknown etiology characterized by recurrent, spontaneous inflammation of the gastrointestinal tract (GI).(1) IBD consists of two disease phenotypes, ulcerative colitis (UC) and Crohn’s disease (CD). These diseases are proposed to be the result of genetic predispositions for either altered immune function leading to aberrant immune responses to natural stimuli, or overall autoimmune dysregulation.(2) Studies show that 20% of patients with IBD have a relative with either UC or CD, in fact a positive family history is thought to be the single greatest predictive factor associated with the disease.(1, 3)
Numerous genes are being examined as carriers of mutations that may result in a predisposition towards IBD, of particular interest is the human multi-drug resistance gene (MDR), an ATP-binding cassette subfamily B member 1A (Abcb1a). In humans the MDR gene is expressed on chromosome 7 (7q21.1) a susceptibility loci for IBD.(4) Studies to date have been inconsistent with regards to the significance of mutations in the MDR gene in relation to the development of IBD. Several recent meta-analysis studies have shown that a significant number of IBD patients carry mutations to this gene, and further studies have demonstrated that MDR mutations are associated with decreased P-gp expression and function.(5–8) Additionally, it was shown that mutations to this gene are strongly associated with early onset UC in German populations.(9) Conversely, it has been reported that subsets of IBD patients in Greek and European populations bear no correlation to MDR mutations.(10, 11)
The functional product of the MDR gene is P-glycoprotein (P-gp). P-gp is a glycosylated transmembrane protein, which functions as a drug efflux pump effectively removing hydrophobic and amphipathic substrates from the intracellular environment.(12) There are 2 MDR genes in humans (MDR1 and MDR3) and 3 mdr genes in mice (mdr1a (abcb1a), mdr1b (abcb1b,) and mdr2(abcb4)).(13, 14) The MDR3 (and mdr2) genes are expressed in the liver, and actively pump phosphatydlcholine into bile.(15) The MDR1 genes, those linked to colitis, are found in multiple tissue locations. They are present not only on the apical surface of epithelial cells in the colon and small intestine, but also on epithelial cells of the pancreas, kidney, and adrenal glands, on endothelial cells at the blood brain barrier, and on cells of the hematopoetic lineage.(16–18) Due to the expression of P-gp on the apical epithelium of organs with excretory function, and due to the known role of P-gp in drug extrusion; it has been postulated that P-gp’s physiologic function is to extrude xenobiotic substrates from the intracellular environment.(19)
A murine model of this genetic mutation has lent some insight into the potential physiological role of P-gp. FVB/N mice deficient in mdr1a gene expression develop a spontaneous colitis as early as 3–4 months of age.(20, 21) These FVB.mdr1a−/− mice have deficiencies in tight junction proteins in the intestinal epithelium, as well as an increased intestinal permeability prior to disease development.(21, 22) These mice have increased colonic IFNγ and IL8 and treatment of these animals with antibiotics was shown to be prophylactic and therapeutic in mitigating disease progression, confirming the role intestinal microbiota in inducing intestinal inflammation.(20, 23)
To expand our knowledge of the mechanisms of the spontaneous colitis in this disease model and to investigate the role of different genetic backgrounds in disease susceptibility, our lab endeavored to backcross the mdr1a genetic deletion on to the C57BL/6J (B6) mouse strain. B6.mdr1a−/− mice were established by 10-generations of backcross breeding to B6 control mice. We initially characterized disease development in these animals; however, the P-gp deficiency on the B6 genetic background did not result in any clinical or histological evidence of spontaneous colitis in animals up to one year of age.
Expression of genetic mutations may leave individuals predisposed towards colitis development in response to mitigating factors, such as a bacterial colonization or infection.(1) We therefore hypothesized that the B6.mdr1a−/− mice might be more susceptible to colitis induction after a secondary environmental insult. Environmental factors tested included Helicobacter bilis, and dextran sodium sulfate (DSS), which have both been previously reported to accelerate the development of colitis in the FVB.mdr1a−/− model.(24, 25) DSS has also been shown to alter mdr1a expression prior to clinical colitis symptoms.(26) Additionally, results from our B6.mdr1a−/− animals indicated that prostaglandin (PG) regulation may play a protective role in preventing colitis in this seemingly resistant strain. Therefore, we tested the affects of altering PG synthesis by blocking the key regulatory enzyme of the PG biosynthesis pathway, cyclooxygenase (COX) in our B6.mdr1a−/− animal model. We administered the non-steroidal anti-inflammatory drug (NSAID), Piroxicam, which inhibits both COX isoforms and rapidly induces colitis in IL10 deficient mice.(27)
MATERIALS AND METHODS
Animals
FVB.129P2-Abcba1tm1BorN7 (FVB.mdr1a−/−) as originally described by Schinkel et al. mice were purchased from Taconic Farms, Inc. (Hudson, NY).(28) Animals were bred and maintained under specific pathogen free (SPF) conditions in Thoren Isolator racks (Hazleton, PA) under positive pressure and were fed autoclaved NIH-31 rodent diet (Harlan Teklad, Madison, WI), and sterile drinking water ad libitum. Animals were acclimatized to our facility 2 weeks prior to mating. Mice were backcrossed onto the C57BL/6J background (Jackson Laboratories, Bar Harbor, ME), for >10 generations under a Research Crossbreeding Agreement with Taconic Farms, Inc., and genotype is confirmed by periodic genotyping by polymerase chain reaction (PCR). In brief, tail clippings are routinely collected, and DNA is extracted using Sigma’s RED Extract-N-Amp tissue PCR kit (St Louis, MO). Mdr1a phenotype is confirmed as indicated by Taconic using the primers MDR1A S2 CTCCTCCAAGGTGCATAGACC, MDR1A W2 CCCAGCTCTTCATCTAACTACCCTG, and MDR1A K02 CTTCCCAGCCTCTGAGCCCAG to amplify DNA using the PCR settings 1 cycle at 95° for 15 minutes, 35 consecutive cycles of 94° for 45 seconds, 60° for 1 minute, and 72° for 1 minute, followed by 1–5 minute cycle at 72°. PCR products are then run on a 1.5% agarose gel, and presence or absence of mdr1a gene expression is confirmed by analyzing banding patterns produced by amplification products (Invitrogen, Carlsbad, CA). Animals were routinely monitored for presentation of a colitic phenotype, including weight loss, diarrhea, and/or fecal blood. All experiments were approved by the Institutional Care and Use Committee of the University of Alabama at Birmingham. SPF conditions at UAB include absence of the following organisms, as determined by serological screening: mouse parvoviruses, including MPV-1, MPV-2, and minute virus of mice; mouse hepatitis virus, murine norovirus, Theiler's murine encephalomyelitis virus; mouse rotavirus (epizootic diarrhea of infant mice), Sendai virus; pneumonia virus of mice; reovirus; Mycoplasma pulmonis; lymphocytic choriomeningitis virus; mouse adenovirus; ectromelia (mousepox) virus; K polyoma virus; and mouse polyoma virus. Testing and other methods were as described at http://main.uab.edu/Sites/ComparativePathology/surveillance/.
Helicobacter Infection
H. bilis (ATCC# 51630) was obtained from the American Type Tissue Collection (Manassas, VA). Organisms were streaked onto Brucella blood agar plates and grown at 37° under microaerophilic conditions (90% N2, 5% H2 and 5%CO2) for 24–28 hours. Bacteria were then inoculated into Brain Heart Infusion broth (supplemented with 5% fetal bovine serum (Hyclone, Thermo Fisher Scientific), 2.5 µg/ml amphopeteracin B, 3µg/ml vancomycin, and 10 µg/ml Trimethoprim) and grown under micoraerophilic conditions at 37° for 24 hours in a continuous shaker. Viability was confirmed via phase microscopy. Organisms were pelleted and quantified at an optical density of 450nm (one OD450=109 bacteria.). Mdr1a−/− and control strain mice of 6–10 weeks of age were orally inoculated with 2×107 cfu bacteria three times over the course of one week. Mice were monitored weekly for presentation of a colitic phenotype, including weight loss, diarrhea, and/or fecal blood. Animals were sacrificed at 4 or 12 weeks post infection.
DSS Treatment
B6 or B6.mdr1a−/− animals from 6–10 weeks of age were given 2.25% w/v DSS (m.w. 36,000–50,000, MP Biomedicals, Solon Ohio) in drinking water ad libitum for 1 week prior to sacrifice (acute). Alternatively animals were treated with 2% w/v DSS in drinking water for 4 alternating cycles of: 1 week of treatment, followed by 2 weeks of recovery during which DSS is replaced with sterile drinking water (chronic). Mice were monitored for presentation of a colitic phenotype, including weight loss, diarrhea, and/or fecal blood.
Piroxicam Diet
B6 or B6.mdr1a−/− animals from 6–10 weeks of age were given 280 ppm piroxicam, (Sigma, Aldrich, St Louis, MO) in pelleted rodent diet NIH-31 from Harlan Tecklad (Madison, WI) as described by Lynch et al.(27) Animals were given access to chow ad libitum, and were either treated for 1 week prior to sacrifice (acute), or treated for 2 weeks and sacrificed 4 weeks post treatment (chronic). Mice were monitored for presentation of a colitic phenotype including weight loss, diarrhea, and/or fecal blood.
Histological Analysis
At time of sacrifice mice were anesthetized with isoflourane and then euthanized by cervical dislocation. Distal small intestine, colon, and cecal segments were flushed with PBS and the colon was assessed for changes in weigh and length. The tissue was then opened longitudinally, and oriented as strips, mucosa up in tissue cassettes. Tissue was fixed in 10% buffered formalin for 24 hours and embedded in paraffin. Tissue was cut into 5µm sections and stained with standard H&E for histologic examination. Experimental conditions were concealed until after the slides were examined. Cecum, proximal colon, middle colon, and distal colon were evaluated separately. For each segment, crypt epithelial hyperplasia, goblet cell loss, superficial and crypt epithelial degeneration and loss, crypt exudate, inflammatory cell accumulation in lamina propria and submucosa, submucosal edema, mucosal ulceration, transmural inflammation, fibrosis, and dysplasia were evaluated. Severity of each change was scored 0, 1, 2, or 3 for absent (normal), mild, moderate, and severe, respectively. The distribution of each change present also was scored 1, 2, 3, or 4 for ≤25%, 25%–50%, 50%–75%, or 75%–100%, respectively, of the segment affected. Lesion scores for each segment were calculated as the sum of severity scores multiplied by distribution scores, with changes indicating severe inflammation or injury, including crypt epithelial degeneration, ulceration, transmural inflammation, and dysplasia, weighted by a factor of 2. An overall lesion score was calculated as the average of the scores for the four segments.
Barrier Permeability
Barrier permeability was assessed by analyzing serum concentrations of FITC-dextran following oral gavage as described.(29) Briefly, B6 and B6.mdr1a−/− mice were orally gavaged 4 hours prior to sacrifice using an 80mg/ml stock of FITC-dextran, for a total dose of 60mg/100g of body weight (MW 4,000; Sigma-Aldrich, St. Louis, MO). Total blood volume was collected at sacrifice via cardiac puncture. Blood samples were coagulated at 4° in the dark for one hour, prior to centrifugation and serum harvest. Samples were analyzed using the Synergy™ HT Multi-Mode Microplate Reader (Bio-Tek Technologies, Winooski, Vermont) and standardized to FITC loaded control serum, FITC was detectable between 25−0.195ng/ml.
RNA Isolation
Colon segments were snap frozen in liquid nitrogen and RNA was isolated from tissue using Trizol® as previously described (Invitrogen®, Carlsbad, CA).(30) Genomic DNA contamination was cleared from samples using the Turbo DNA-free kit from Applied Biosystems®. cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit® (Roche, Pensburg, Germany). Quantitative real-time reverse-transcriptase polymerase chain reaction (RT-PCR) was performed using TaqMan Universal PCR Mix® (Invitrogen®, Carlsbad, CA) in combination with Applied Biosystems® gene specific primer probe sets. RNA expression level was calculated using the crossing threshold of detectable fluorescence level as determined by the RT cycler MX3000P® (Stratagene®, La Jolla, CA). Crossing thresholds were then averaged to get a gene specific value which was then normalized to the average expression of the 18S housekeeping gene for each strain and experimental condition studied. The 18S housekeeping gene was utilized as the housekeeping gene in this study as published results indicate it is expressed with relatively stability under inflammatory conditions.(31–33) Changes in gene expression in the experimental groups were then calculated as an average fold change when compared to control strain values, and expressed on a log 2 scale as fold changes from the control baseline (=1). This data analysis is as described in the Applied Biosystems manufacturer’s instructions (4371095 Rev A, PE Applied Biosystems, Foster City, CA). Data is considered physiologically relevant if changes in expression exceed a 2 fold change from control strain values.
Elisa for H. bilus-Specific Serum IgG and Fecal IgA
ELISAs were performed to determine the specific IgG response to H. bilus in serum samples. In brief, a 96-well Immunlon Assay Plate (Fisher Scientific, Pittsburgh, PA) was coated with H. bilis sonicate (10 µg/mL in PBS) overnight at 4°C. Plates were washed 5 times with PBS with 0.05% Tween and nonspecific binding sites were blocked with 5% bovine serum albumin (BSA, Fisher Scientific, Pittsburgh, PA) in PBS for one hour at room temperature (RT). The plate was washed as before and then samples diluted in 1% BSA in PBS were incubated for two hours at room temperature. After a wash of five cycles, alkaline phosphatase-linked goat anti-mouse IgG (diluted 1:2000 in 1% BSA in PBS) was added to the wells and incubated for 2 hours at RT (IgG, Cat. #1030-04, 1ml stock solution, Southern Biotech; Birmingham, AL). The plate was washed another five times, and then the bound secondary antibody was detected using pNPP substrate solution (N-2770, Sigma, St. Louis, MO). The plates were read on a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA) at 405 nm. To determine the concentration of IgG anti-H. bilis, a standard curve of the corresponding IgG (5300-01; Southern Biotech) was run on each plate as described above with the exception of coating the plate with 10µg/mL goat anti-mouse Ig (#1010-01, Southern Biotech). To determine H. bilis specific fecal IgA, a similar protocol was followed with the exception that alkaline phosphatase-linked goat anti-mouse IgA (diluted 1:2000 in 1% BSA in PBS) was added to the wells and incubated for 2 hours at RT (IgA, Cat. #1040-04, 1ml stock solution, Southern Biotech), and the standard curve utilized IgA (5300-01; Southern Biotech).
Statistical Analysis
Statistics on continuous data was performed using the unpaired t-test in GraphPad InStat 3® (San Diego, CA). Statistics on continuous data was performed using Student’s T-test, and non-continuous data was analyzed using the Mann Whitney U test. Comparisons having a p≤0.05 were considered significant and are indicated by asterisks.
RESULTS
Absence of Spontaneous Colitis in B6.mdr1a−/− mice
B6.mdr1a−/− mice were sacrificed at 2, 5, 8, and 12 month of age, and examined for histological and phenotypic evidence of colitis. These time points were selected based on previous data from our lab indicating the significance of these times in the development of colitis in FVB.mdr1a−/− animals. FVB.mdr1a−/− in our facility demonstrate no clinical symptoms and are histologically free of colitis at 2 months (Fig. 1 B/C). By 5 months of age FVB.mdr1a−/− animals begin to demonstrate weight loss and diarrhea and upon sacrifice 4 out of 10 animals showed histological evidence of colitis (Fig. 1 E/F). At 8 months of age, FVB.mdr1a−/− demonstrate a 66% mortality rate (Fig. 2C) and 6 out of the 7 animals analyzed demonstrated histologically significant colitis (Fig 1 H/I)). Significant numbers of FVB.mdr1a−/− mice at 12 month of age proved difficult to attain, as few of the animals survived to this time point; however, the three mice that were analyzed all had significant colitis (Fig. 1 K/L).
FIGURE 1.
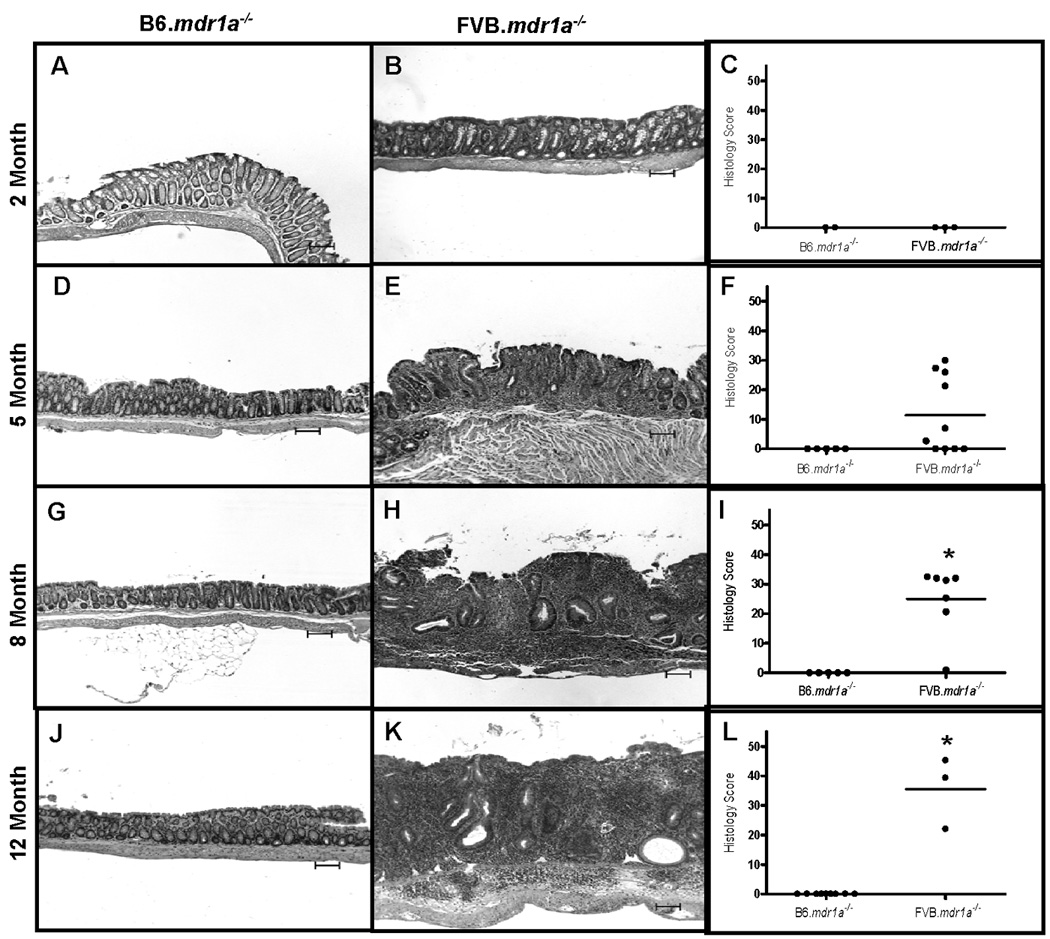
B6.mdr1a−/− and FVB.mdr1a−/− colonic histology. Representative examples of histology and histological scores are given for B6 and FVB P-gp deficient mice at 2 (A–C), 5 (D–F), 8 (G–I), and 12 (K–L) months of age. Images captured at 100X magnification. Bar=100µm.* Indicates statistical significance of P<0.05 as determined by Mann Whitney analysis. Values were significantly different when compared both between P-gp deficient strains, and when compared to control strains.
FIGURE 2.
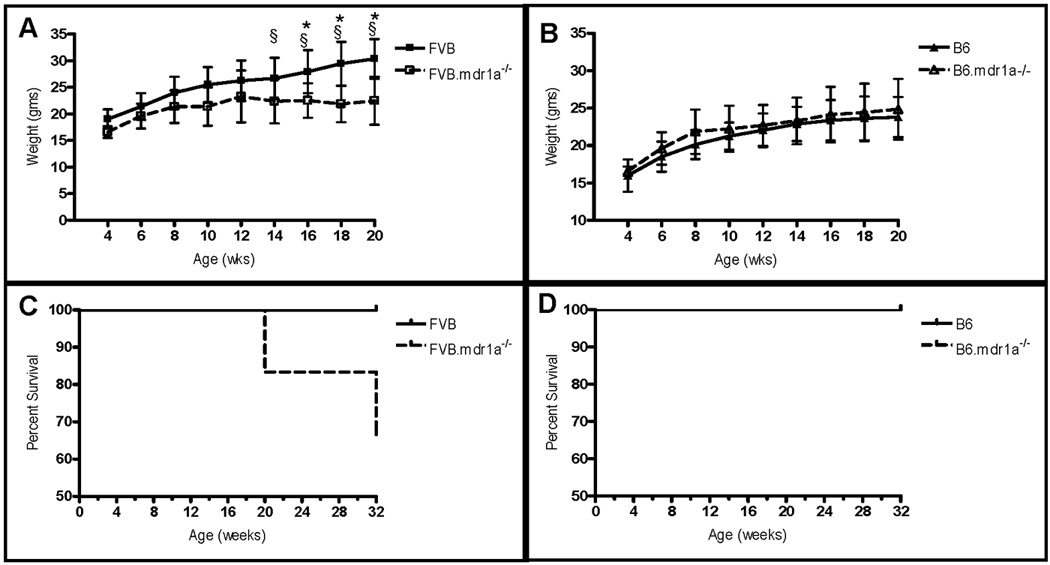
Weight changes and survival curves of B6 and FVB animals deficient in P-gp expression. Changes in body weight (A, B) are expressed from weaning at 4 weeks of age. Survival (C, D) was determined up to 8 months of age. Weights and survival were derived from an N of 8 for the B6, B6.mdr1a−/− and FVB animals, and 7 for the FVB.mdr1a−/−. § Indicates the incidence of diarrhea in FVB.mdr1a−/− animals. * Indicates statistically significant weight loss of P<0.05 in FVB.mdr1a−/− animals when compared to wild type controls, as determined by a Student’s T-test.
Over the lifespan of B6.mdr1a−/− mice, clinical and gross observation and histologic examination showed that they were free of colitis at all selected time points (Fig. 2). This was confirmed by histological analysis (Fig 1). RNA was isolated from colonic tissue at the 2, 5, and 8 month time points to foster a better understanding of inflammatory conditions in the FVB.mdr1a−/− large intestine as colitis progressed, and to enable us to better understand potential mitigating factors preventing colitis development in the B6.mdr1a−/− colons (for a complete list of genes selected and fold change see Supplemental Table I & Supplemental Table II). The 12 month time point was not included due to the severely reduced survival of the FVB.mdr1a−/− animals.
It has been previously been shown that colitis in the FVB.mdr1a−/− model is characterized by an increase in colonic expression of inflammatory cytokines and chemokines such as IFNγ, TNFα, and IL6, which correlates to changes seen in colitic human tissue.(22, 23, 25) Our data demonstrates that colonic tissue from FVB.mdr1a−/− animals not only display dramatically increased production of the inflammatory cytokine IFNγ, IL6, and TNFα, but also IL17, MIP-2, and IL1β (Fig. 3A/B and Supplemental Table II). Moreover, it further demonstrates that in the FVB.mdr1a−/− animals production of these cytokines increases with age and/or development of colitis. Interestingly, the expression of a key enzyme (COX-2) required for production of lipid mediators of inflammation, prostaglandins, remain unchanged in the FVB.mdr1a−/− colons (Fig 3D). Conversely, data from the B6.mdr1a−/− colons indicates that these animals, despite a moderate increase in the gene expression of the inflammatory cytokines and chemokines IFNγ, MIP-2, TNFα, and IL1β at 2 months of age, fail to demonstrate the increased production of inflammatory cytokines in later months as seen in the corresponding FVB.mdr1a−/− animals (Fig. 3 and Supplemental Table II). Furthermore, these animals display a moderate increase in Cox-2 production at early time points, indicating this may be a mitigating factor in strain linked pathology (Fig 3D). Given our observation that the B6.mdr1a−/− are resistant to the development of spontaneous colitis, we evaluated their resistance to the development of chemical- or bacterial-induced colitis.
FIGURE 3.
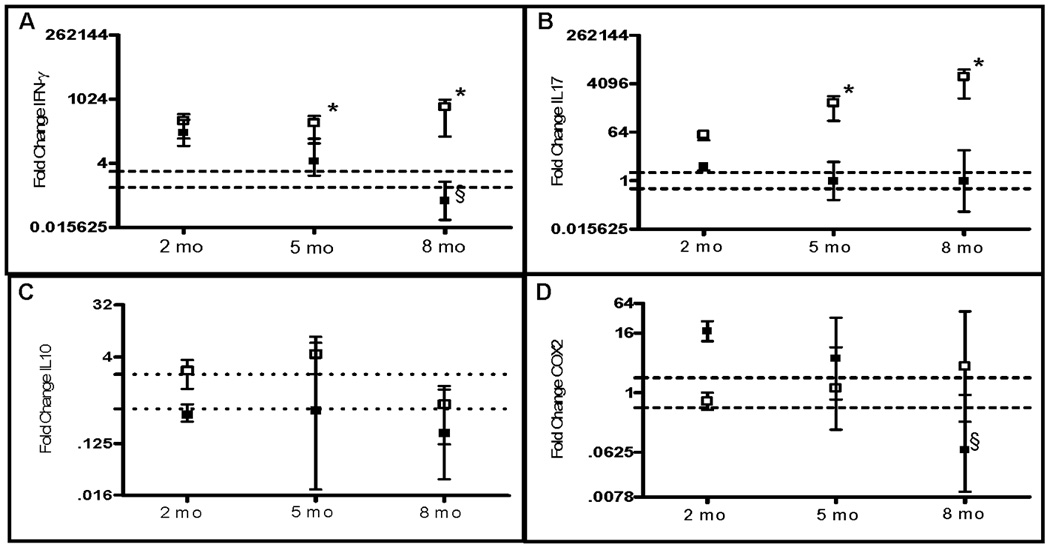
Colonic gene expression in B6 and FVB animals deficient in P-gp expression. Gene expression was derived from RNA isolated from whole colonic tissue. Target gene expression was normalized to expression of the 18S housekeeping gene, and the average fold change in gene expression from control strain values. Range is calculated from the standard deviation of the ΔΔCT value and the average gene expression is plotted with upper and lower limits of the range shown. Data is shown on a log 2 scale. Dotted lines demonstrate the range of physiological relevance, all values demonstrating a 2 fold alteration in gene expression from control values are considered physiologically different from control values. Gene expression of IFNγγ (A), IL17 (B), IL10 (C), and COX-2 (D) are represented graphically. Expression of other genes of interest can be found in Supplemental Table II. A minimum N of 2, 4, and 6 animals from control and mutant strains were analyzed for the 2, 5, and 8 month time points respectively. * Indicates statistical significance of P<.05 as determined by the Students T-test when the B6.mdr1a−/− is compared to the FVB.mdr1a−/−. § Indicates statistical significance of p<.05 between P-gp deficient and control animals as determined by Student’s T-Test
H. bilis infection does not induce colitis in B6.mdr1a−/− mice
It has been previously reported that inoculation of FVB.mdr1a−/− animals with H. bilis accelerates the development of colitis, corresponding with an increased inflammatory response in the mesenteric lymph node cells.(24) FVB.mdr1a−/− animals were shown to develop diarrhea and hunching within 3–4 weeks of inoculation (9–10 weeks of age), and severe colitis by 16 weeks post-inoculation (22 weeks of age).(24) Similar to previous reports we found that in our colony, FVB.mdr1a−/− but not FVB control strain animals, developed a significant colitis at 12 weeks post inoculation (18 weeks of age, data not shown). B6.mdr1a−/− and B6 controls were sacrificed at both 4 and 12 weeks post inoculation with H. bilis and examined for evidence of colitis. At 4 weeks post-inoculation B6 and B6.mdr1a−/− demonstrated no clinical or macroscopic indicators of colitis. Given the inherent resistance of the B6.mdr1a−/− strain, we went on to examine animals at 12 weeks post-inoculation. B6.mdr1a−/− animals demonstrated reduced weight gain over the course of the experiment when compared to inoculated wild type controls, however, at sacrifice they were shown to be free of diarrhea, fecal blood and alterations to intestinal permeability (Fig 4 and Table I). Serum and feces harvested at the time of sacrifice were analyzed for the presence of antibodies specific for H. bilis. B6 and B6.mdr1a−/− inoculated animals showed expression of serum IgG specific for H. bilis, while levels of H. bilis specific fecal IgA remained below quantifiable limit. There was no significant difference in the levels of anti-H. bilis IgG between H. bilis infected B6 and B6.mdr1a−/−, although control strain B6 animals had a tendency towards higher levels of antibody expression (Fig. 5). Additionally, antibody levels were not comparable to those seen in inoculated FVB.mdr1a−/− animals, which were shown to be in excess of 9000ng/ml. Colonic tissue appeared healthy at time of sacrifice and displayed no alterations in either length or weight (Fig 4D and data not shown). Histological examination confirmed these results (Fig 4B, C & E)).
FIGURE 4.
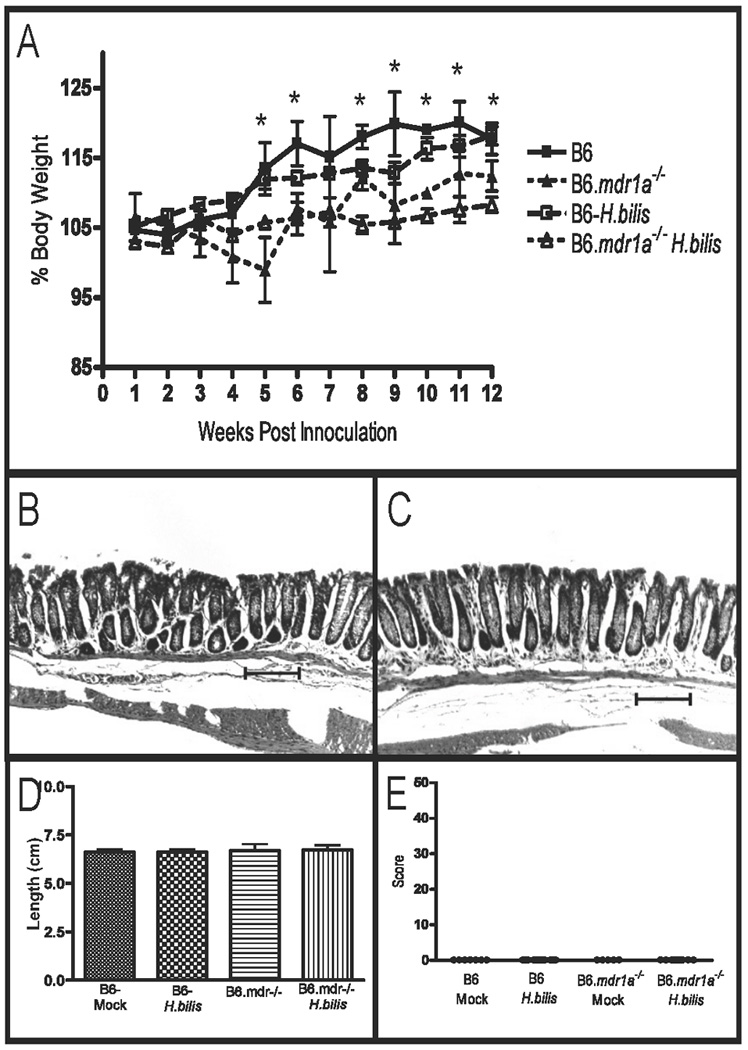
Evaluation of colitis in B6 and B6.mdr1a−/− mice following inoculation with H. bilis. Alterations in body weight in B6 and B6.mdr1a−/− following inoculation with H. bilis. (A) Colonic histology of B6 (B) and B6.mdr1a−/− (C) animals at sacrifice. Colonic length (D) and histological scoring of inoculated animals (E). Images captured at 100X magnification. Bar=100µm. * Indicates statistical significance of P<0.05 when comparing P-gp deficient and control animals inoculated with H.bilis as determined by a Student’s T-test.
TABLE 1.
Clinical colitis Observations from Treated B6.mdrla−/− Animals
| Diarrhea | Fecal Blood |
Death | Serum FITC- Dextran (ng/ml) |
||
|---|---|---|---|---|---|
| C57BL/6 | Mock | 0/7 | 0/7 | 0/7 | 0.12 ±0.27 |
| H.bilis | 0/11 | 0/11 | 0/11 | 0.37 ±0.47 | |
| Mock | 0/9 | 0/9 | 0/9 | 0.47 ±0.48 | |
| DSS | 2/10 | 0/10 | 0/10 | 0.66 ±0.79 | |
| Mock | 0/9 | 0/9 | 0/9 | 0.96 ±1.27 | |
| Piroxicam | 0/10 | 0/10 | 0/10 | 0.44 ±.66 | |
| C57BL/6mdr1a−/− | Mock | 0/5 | 0/5 | 0/5 | 1.96 ±2.74 |
| H.bilis | 0/9 | 0/9 | 0/9 | 0.83 ±2.13 | |
| Mock | 0/8 | 0/8 | 0/8 | 1.35 ±1.04 | |
| DSS | 1/9 | *6/9 | 0/9 | §*4.72 ±1.90 | |
| Mock | 0/15 | 0/15 | 0/15 | 1.00 ±1.31 | |
| Piroxicam | 0/10 | 3/10 | 3/10 | 0.49 ±0.38 | |
Indicates statistical significance of P<.05 as determined by the Students T-test when the B6.mdrla−/− is compared to the FVB.mdrla−/−.
Indicates statistical significance of p=.05 between P-gp deficient and control animals as determined by Student’s T-Test
FIGURE 5.
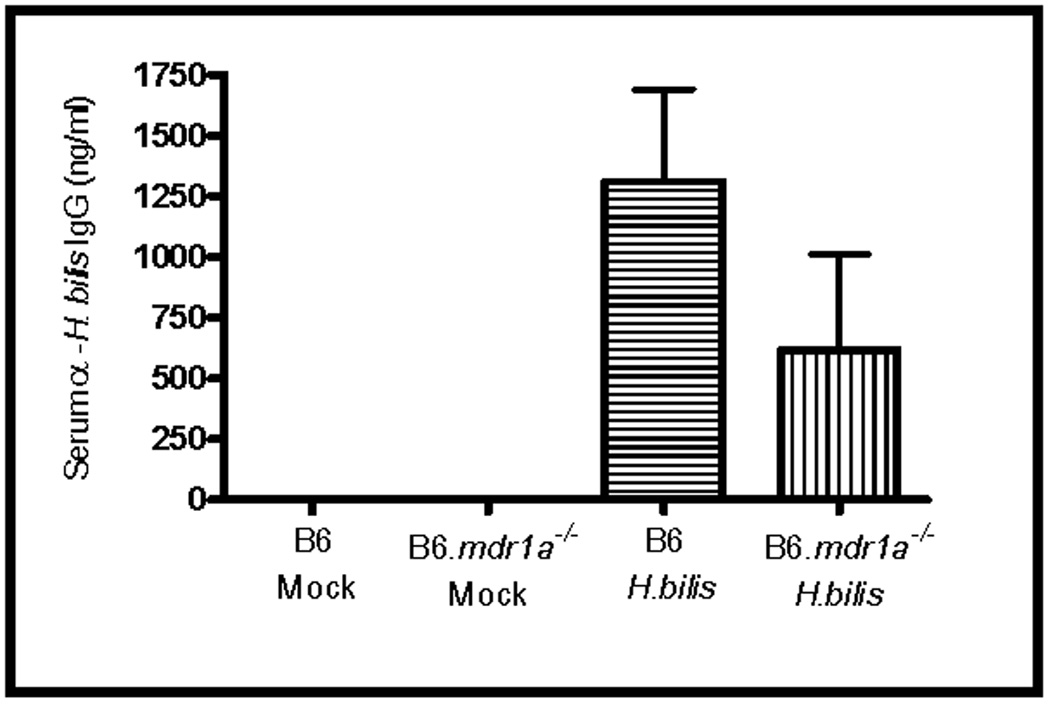
Levels of serum H. bilis antibodies in inoculated B6.mdr1a−/− and control B6 mice. Serum levels of anti-H. bilis IgG were determined using an H. bilis specific ELISA, and quantified using total IgG controls. N=5–12 animals per strain/treatment type.
B6.mdr1a−/− mice have increased susceptibility to DSS-induced colitis
P-gp deficiency has been shown to affect epithelial permeability, and is associated with altered phosphorylation of proteins involved in the formation of tight junctions, resulting in impaired intestinal integrity.(21) It has been previously reported that FVB.mdr1a−/− animals show an accelerated colitis development following exposure to DSS.(25) DSS serves to disrupt the intestinal epithelium, and has been shown to induce colitis in susceptible models. B6.mdr1a−/− and wild type B6 animals were given 2.25% DSS in drinking water for 7 days and sacrificed on day 11. B6.mdr1a−/− animals developed a more severe colitis in response to DSS treatment, as evidenced by a significant reduction in weight and an increased incidence of fecal blood and diarrhea (Fig 6A, and Table I). Colonic length was significantly reduced in B6.mdr1a−/− mice and they demonstrated macroscopic evidence of colitis at time of sacrifice (Fig 6C,D). Histological examination confirmed these findings and demonstrated that DSS-treated P-gp deficient animals had significantly higher colonic histology scores than did DSS-treated B6 control mice (Fig 6E). The DSS-treated B6.mdr1a−/− additionally demonstrated an increased colonic permeability to FITC-dextran as measured by serum levels following oral gavage (Table I). Results were mirrored in the long term DSS colitis models (Supplemental Fig. 1). Interestingly, it has been previously observed in the FVB.mdr1a−/− model that males demonstrate an increased incidence of colitis when compared to female animals. Our experiments contained mixed gender populations, which seems to be the source of the variance displayed in our histology scoring. B6.mdr1a−/− male animals treated with DSS typically had higher histological scores when compared to female animals (Fig. 6E and Supplemental Fig. 1).
FIGURE 6.
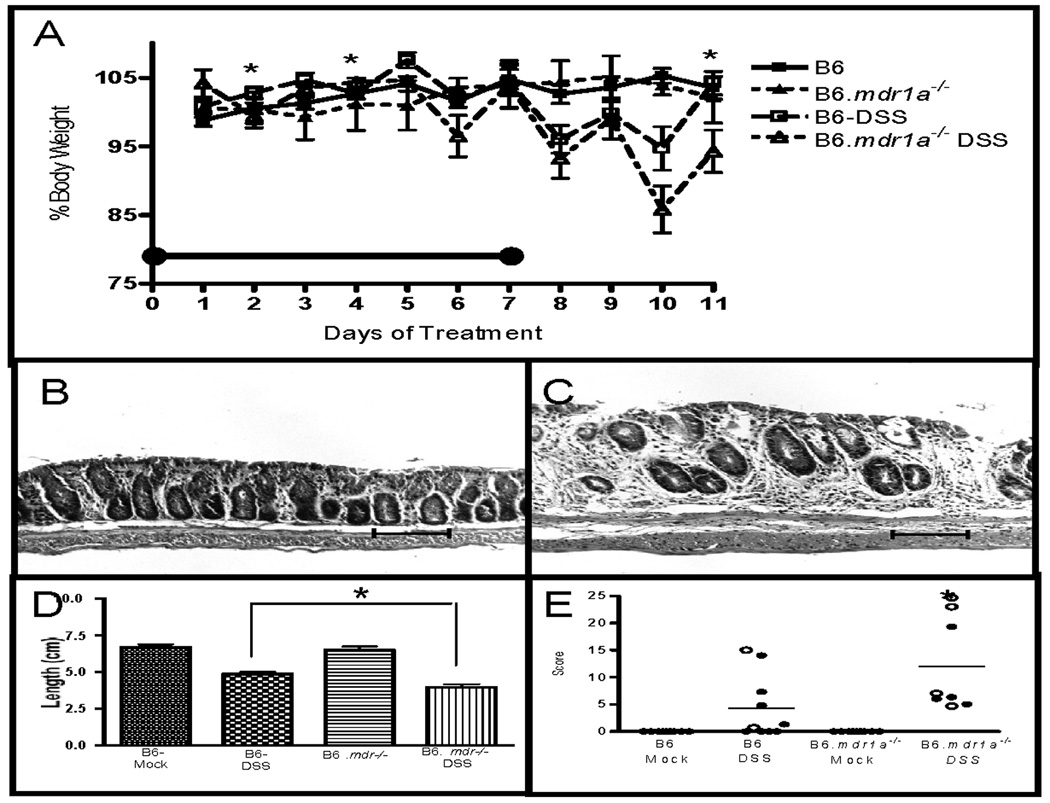
Evaluation of colitis in B6 and B6.mdr1a−/− mice following treatment with DSS. Reductions in body weight in B6 and B6.mdr1a−/− undergoing treatment with DSS (A). The black bar represents time period of DSS treatment. Colonic histology of B6 (B), and B6.mdr1a−/−(C) animals at sacrifice. Average colonic length (D) and histological scoring (E) of treated animals and controls. Closed circles represent female animals, open circles represent male animals. Images captured at 100X magnification. Bar=100µm. * Indicates statistical significance of P<0.05 as determined by Mann Whitney (A & C), or P<0.05 as determined by Student’s T-test (B) when comparing P-gp deficient and control animals treated with DSS.
Piroxicam-diet does not induce colitis in B6.mdr1a−/− mice
Our initial colonic gene expression data suggested a potential role for elevated prostaglandin (PG) synthesis in modulating disease development in B6.mdr1a−/− mice, as gene expression was elevated in the colons isolated from the 2 month B6.mdr1a−/− mice. PGs are lipid mediators that have been shown to play a role in mediating inflammation in arthritis and inflammatory bowel disease.(34–36) PG synthesis is tightly regulated through expression of the COX enzymes. To better examine the potential role of COX in preventing or dampening intestinal inflammation in these animals, we treated B6.mdr1a−/− and wild type B6 mice with piroxicam, a NSAID that inhibits COX function.(27) B6.mdr1a−/− and B6 controls were initially treated for 7 days with piroxicam supplemented chow. Despite early clinical signs of disease including weight loss, and increased mortality, none of the animals treated showed any histological evidence of colitis (Supplemental Fig 2). We next looked at the effects of a long term piroxicam diet on P-gp deficient animals. Animals were treated with piroxicam supplemented chow for 2 weeks followed by a 4 week recovery period during which they returned to normal rodent diet. Animals were monitored on a weekly basis for weight loss and other physiologic evidence of colitis. B6.mdr1a−/− mice demonstrated increased weight loss during the period of piroxicam treatment, and additionally one animal had had transient fecal blood (Table I, Fig 7A). However, at sacrifice animals had an unaltered colonic phenotype including unperturbed colonic length, barrier permeability, and colonic histology (Table I, Fig 7). B6.mdr1a−/− animals displayed a slightly increased mortality in response to piroxicam treatment that was not seen in control strain animals, however, upon sacrifice this appeared to be associated with the development of liver inflammation (Table I, Fig 8). Liver inflammation was notably absent in both B6 controls, B6.mdr1a−/− controls, and B6 piroxicam treated animals (Fig 8).
FIGURE 7.
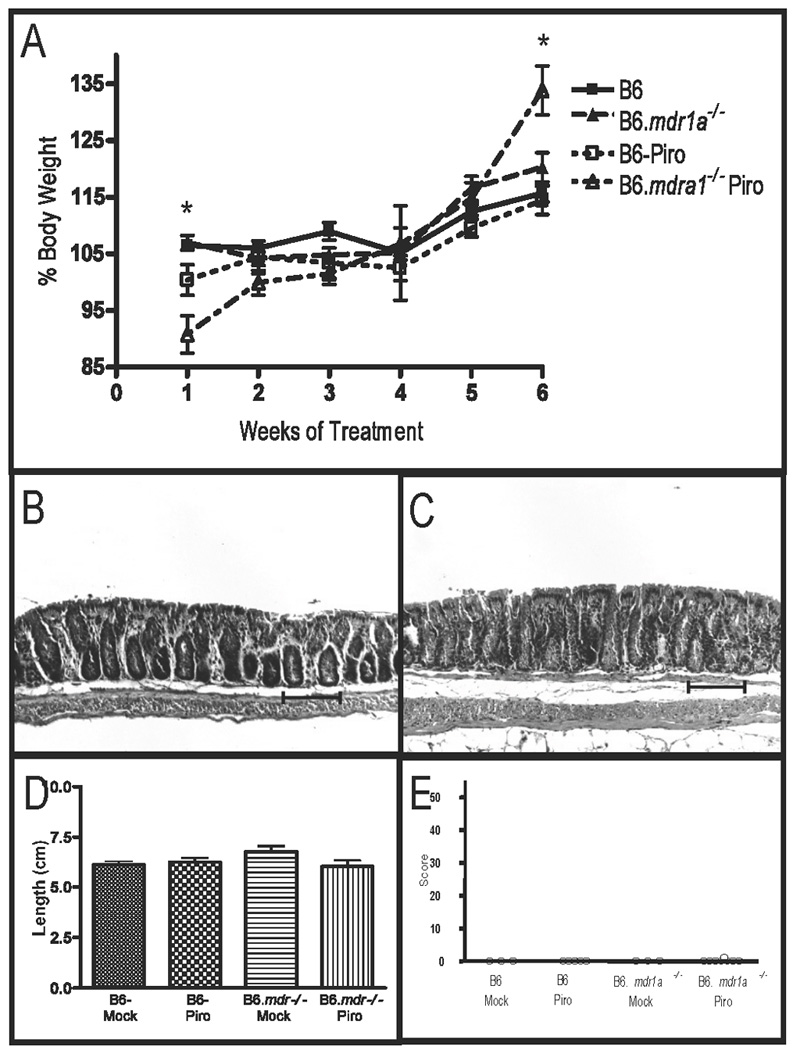
Evaluation of colitis in B6 and B6.mdr1a−/− mice following treatment with piroxicam. Body weight in B6 and B6.mdr1a−/− undergoing treatment with Piroxicam (A). Colonic histology of B6 (B), and B6.mdr1a−/− (C) animals at sacrifice. Colonic length (D) and histological scores of treated animals (E). Images captured at 100X magnification. Bar=100µm. * Indicates statistical significance of P<0.05 as determined by Mann Whitney analysis when comparing piroxicam treated P-gp deficient and control strain animals.
FIGURE 8.
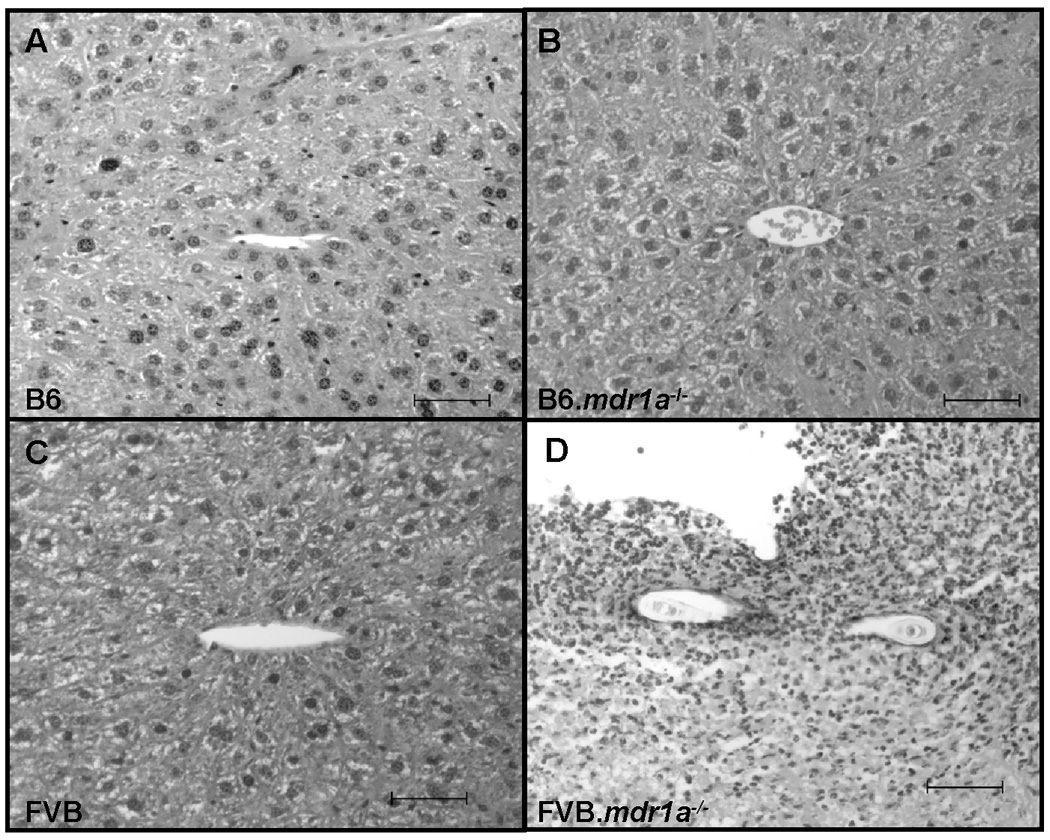
Liver histology from B6 control and P-gp deficient piroxicam treated animals. B6 (A), B6.mdr1a−/− (B) and piroxicam treated B6 wild type animals demonstrated no significant liver histological changes or inflammation, while B6.mdr1a−/− piroxicam treated animals showed severe liver inflammation. Images captured at 400X magnification. Bar=50µm
DISSCUSSION
The FVB.mdr1a deficient animal was originally derived for the purposes of drug extrusion studies at the blood brain barrier.(28) It was subsequently discovered that P-gp’s pumping capabilities were integral to the function of the intestinal epithelium, as indicated by the development of spontaneous colitis in these animals.(20) P-gp function is of particular importance with respect to interactions involving the intestinal microbiota, as it has been demonstrated that prophylactic treatment with antibiotics prevented colitis development for up to 16 weeks in P-gp deficient animals. Additionally, it was shown that 10 weeks of antibiotic treatment ameliorated colitis in FVB.mdr1a−/− mice.(20) Chimeric animals were generated to examine the respective roles P-gp deficiency in the intestinal epithelium, as compared to in immune effector cells with regards to colitis development. It was shown that P-gp deficiency in the intestinal epithelium was necessary and sufficient for the development of colitis FVB.mdr1a−/− animals.(20)
Subsequent work has focused on examining the effects of P-pg deficiency on the integrity of the intestinal epithelium. Evidence indicates that FVB.mdr1a−/− animals develop an altered permeability to molecularly tagged substrates at approximately 12 weeks of age, and prior to the development of any clinical or histological evidence of colitis.(21) These permeability changes correlate with altered phosphorylation of junctional proteins occludin and zonula occludin-1 (ZO-1).(21) These data clearly indicate that histological and phenotypic evidence of colitis occurs secondarily to the development of altered intestinal barrier function. Although the precise mechanism remains unclear it has also recently been demonstrated that FVB.mdr1a−/− demonstrate an increased expression of inflammatory cytokines, and an increased responsiveness to LPS prior to the development of intestinal inflammation.(22) It was our initial hypothesis that alterations in P-gp expression would affect the animal’s ability to respond to bacterial stimuli. In hopes of better understanding the affects of P-gp expression to individual bacterial ligands we re-derived the mdr1a mutation on the C57BL/6 background and began our efforts to characterize the development of colitis on this background.
P-gp deficient mice on the B6 and FVB backgrounds were first analyzed at 2 months of age. Although neither strain demonstrated histological colitis, both the B6.mdr1a−/− and the FVB.mdr1a−/− colons had increased expression of the inflammatory cytokines and chemokines IFNγ, MIP2, TNFα and IL1β. The strains differed in their expression of colonic IL17 (high in FVB.mdr1a−/−) and COX-2 (high in B6.mdr1a−/−), therefore providing the first clue that genetic strain background was modifying the intestinal effects of P-gp deficiency. This genetic background effect was even stronger at the later times examined, as the FVB.mdr1a−/− continued to have increased inflammatory cytokine and chemokine expression and significant histological colitis, while the B6.mdr1a−/− was similar to its B6 wildtype control in both colonic cytokine levels and histology. This modulation of colitis incidence by genetic background may help to explain the conflicting data on the association of P-gp mutations and colitis incidence in human populations. Due to this genetic resistance to colitis in the absence of P-gp, we investigated other environmental insults known to induce colitis in murine models.
H. bilis is a microaerophilic gram-negative organism which colonizes the liver and lower GI tract of susceptible hosts.(37) It has been shown to induce colitis in a number of immune-dysregulated animal models including immunodeficient rats, SCID mice, and B6 mice deficient in IL10 gene expression.(38–40) It has previously been shown that bacterial colonization of the FVB.mdr1a−/− mice with Helicobacter bilis induces a more rapid, and phenotypically distinct colitis.(24) FVB.mdr1a−/− animals display a significant alterations intestinal permeability as early as 4–6 weeks of age, prior to the development of colitic phenotype.(21, 22) It may be that the accelerated colitic phenotype seen in this model system is a result of the increased immune access resulting from the impaired intestinal integrity. To determine whether H. bilis would serve as an adequate environmental factor to initiate inflammatory bowel disease we inoculated our newly derived B6.mdr1a−/− mice. Our results demonstrated that contrary to the colitis induced in FVB.mdr1a−/− mice, inoculation with H. bilis was not a sufficient to induce colitis in B6.mdr1a−/−. It should be noted that the B6 and B6.mdr1a−/− mice did recognize the H. bilis innoculum, as demonstrated by the serum IgG response; however, this immune response was not associated with colitis and did not extend to a mucosal fecal IgA response.
As the FVB.mdr1a−/− disease model has aberrant functioning of the intestinal epithelium,, we hypothesized that chemical disruption of the intestinal epithelium may increase colitis susceptibility in B6.mdr1a−/− deficient animals. Additional support for this theory comes from the recent report that FVB.mdr1a−/− showed accelerated colitis development following DSS treatment.(25) Furthermore, mice deficient in expression of the multi-drug resistant protein-1, a transport protein of similar function and substrate specificity to mdr1a, although resistant to spontaneous colitis show an increased susceptibility to both DSS and TNBS induced colitis.(41) Interestingly, the mrp-1 deficient mouse was also derived on the FVB background, which has subsequently been shown to be innately more resistant to DSS colitis, requiring far higher doses DSS than those reported those previously reported for the C57BL/6 animal.(25, 42) This indicates both an increasingly important role for P-gp in mitigating DSS colitis, and implied that colitis induction in our strain would take place at much lower doses than those described for FVB.mdr1a deficient animals.
DSS is a sulfated polysaccharide, and although the exact mechanism of colitis induction is unknown; it has been shown to have toxic affects on the intestinal epithelium and cause stripping of epithelial cells. It was our hypothesis that animals deficient in P-gp would be more susceptible to damage induced at the epithelial level. To investigate the effects of epithelial insult as a mitigating factor in disease development, B6.mdr1a−/− and control strain mice were treated with DSS. B6.mdr1a−/− animals were shown to develop a significant colitis in response to treatment with DSS in both long term and short term treatment models. It has been suggested that P-gp expression plays a role in cellular apoptosis, and regulation of cell cycle.(43) It may be that the increased susceptibility to DSS-induced colitis in the P-gp deficient animals is directly linked to impaired cell cycling, thus affecting wound repair. Further studies will need to be conducted to determine whether this plays a factor.
Our initial studies done to characterize strain specific disease development revealed that B6.mdra1a−/− mice demonstrated altered colonic gene expression at 2 months of age, when compared to B6 controls, even though they never developed spontaneous colitis. B6.mdr1a−/− animals expressed elevated levels of IFNγ, MIP2, TNFα, IL1β, and COX-2 at 2-months of age. This increased gene expression returned to normal by 5-months of age. The COX enzymes tightly regulate the synthesis of the lipid inflammatory mediators prostaglandins (PG), thromboxanes and prostacyclins.(44) PGs have been shown to have strong immunomodulatory effects and can inhibit LPS-induced inflammation.(45, 46) COX-1 expression is considered constitutive and has been associated with intestinal maintenance and homeostatic mechanisms, while COX-2 expression is induced in response to inflammatory stimuli and cytokines.(47–49) It has been previously shown that intestinal inflammation can be initiated through inhibition of COX-2, and is thought to be a function of disregulation of its biosynthetic bi-products.(50) COX-2 has been previously shown to be integral in regulating the immune response in IL10 deficient animals on the resistant C57BL/6 background, perhaps serving as a potential immune modulator in the absence of IL10.(27) In keeping with this hypothesis, treatment of B6.IL10 deficient animals with the COX-inhibitor, piroxicam, has been shown to induce an acceleration of colitis onset.(27) We hypothesized that in our model system early upregulation of COX-2 gene expression might be acting to modulate the immune response therefore preventing the development of colitis. To clarify the potential role of COX-2 gene expression in mitigating colitis development, B6.mdr1a−/− animals were treated with piroxicam. We show that COX inhibition has no affect on the colitis resistance seen in B6.mdr1a−/− mice. This is perhaps not surprising as it has also been previously shown that increased COX-2 expression reduces expression of P-gp, and that P-gp expression is decreased in IL10 deficient animals.(51) It may be that the diminished or even absent P-gp expression enhances the affect of COX-2 products, such as prostaglandins, on the intestinal epithelium, and that the initiation of inflammation after inhibition of COX function requires additional inhibition of IL10 expression.
One possible explanation for the lack of colitis development in our B6.mdr1a−/− animals could be linked to unaltered barrier permeability. Initial studies utilizing this animal model indicate that these animals show no evidence of alteration in intestinal barrier function, as quantified by permeability to FITC-Dextran. This theory also is supported by the fact that we see no evidence of colitis in our H. bilis inoculated animals. It may be that the lack of invasive properties of this bacterium, in conjunction with unaltered barrier function in our animals, prevented the bacterium from initiating a significant mucosal immune response. This would the complete lack of fecal antibodies seen in our inoculated animals. It may be that an organism more capable of penetrating the intestinal epithelium would be better suited to initiate colitis in the B6.mdr1a−/− model. Furthermore we have shown that an insult to the intestinal epithelium serves as an initiating factor for colitis development in our animals, arguing that upon epithelial insult these animals develop colitis similar to that seen in the FVB.mdr1a−/− animal model.
Supplementary Material
Colitis in B6 and B6.mdr1a−/− mice following long term treatment with DSS. Average colonic length (A) and histological scoring (B) of B6.mdr1a−/− and wild type B6 controls. Clinical measures of colitis are assessed in panel C, data is given for diarrhea, presence of fecal blood, increased mortality following treatment, and increased intestinal permeability to FITC-Dextran.* Indicates statistical significance of P<.05 as determined by Mann Whitney (A, and C) when comparing P-gp deficient and control animals treated with alternating cycles of DSS infused drinking water.
Evaluation of colitis in B6 and B6.mdr1a−/− mice following short term treatment with piroxicam. Average colonic length (A) and histological scoring (B) of B6.mdr1a−/− and wild type B6 controls. Clinical measures of colitis are assessed in panel C. Data is given for diarrhea, presence of fecal blood, mortality following treatment, and intestinal permeability to FITC-Dextran.
ACKNOWLEDGEMENTS
We would like to thank Peggy R. McKie-Bell and Jamie L. McNaught for their technical assistance and members of the Lorenz for valuable advice. We thank Dr. Chuck O. Elson for use of the Synergy Microplate Reader and Wayne Duck for experimental input and comments. We would also like to thank the Gnotobiotic and Genetically Engineered Mouse Core Facility for technical assistance.
Footnotes
This work was supported in part by the NIH grants P01 DK071176, T32A107051 and the University of Alabama at Birmingham Digestive Diseases Research Development Center Grant #P30 DK064400.
REFERENCES
- 1.Pena AS. Contribution of genetics to a new vision in the understanding of inflammatory bowel disease. World J Gastroenterol. 2006;12:4784–4787. doi: 10.3748/wjg.v12.i30.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kucharzik T, Maaser C, Lugering A, et al. Recent understanding of IBD pathogenesis: implications for future therapies. Inflamm Bowel Dis. 2006;12:1068–1083. doi: 10.1097/01.mib.0000235827.21778.d5. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 4.Satsangi J, Parkes M, Louis E, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 5.Ho GT, Soranzo N, Nimmo ER, et al. ABCB1/MDR1 gene determines susceptibility and phenotype in ulcerative colitis: discrimination of critical variants using a gene-wide haplotype tagging approach. Hum Mol Genet. 2006;15:797–805. doi: 10.1093/hmg/ddi494. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–3478. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onnie CM, Fisher SA, Pattni R, et al. Associations of allelic variants of the multidrug resistance gene (ABCB1 or MDR1) and inflammatory bowel disease and their effects on disease behavior: a case-control and meta-analysis study. Inflamm Bowel Dis. 2006;12:263–271. doi: 10.1097/01.MIB.0000209791.98866.ba. [DOI] [PubMed] [Google Scholar]
- 8.Yacyshyn B, Maksymowych W, Bowen-Yacyshyn MB. Differences in P-glycoprotein-170 expression and activity between Crohn's disease and ulcerative colitis. Hum Immunol. 1999;60:677–687. doi: 10.1016/s0198-8859(99)00036-1. [DOI] [PubMed] [Google Scholar]
- 9.Fiedler T, Buning C, Reuter W, et al. Possible role of MDR1 two-locus genotypes for young-age onset ulcerative colitis but not Crohn's disease. Eur J Clin Pharmacol. 2007;63:917–925. doi: 10.1007/s00228-007-0334-0. [DOI] [PubMed] [Google Scholar]
- 10.Brant SR, Panhuysen CI, Nicolae D, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–1292. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glas J, Torok HP, Schiemann U, et al. MDR1 gene polymorphism in ulcerative colitis. Gastroenterology. 2004;126:367. doi: 10.1053/j.gastro.2003.08.045. [DOI] [PubMed] [Google Scholar]
- 12.Fromm MF. P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int J Clin Pharmacol Ther. 2000;38:69–74. doi: 10.5414/cpp38069. [DOI] [PubMed] [Google Scholar]
- 13.Croop JM, Raymond M, Haber D, et al. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu SI, Lothstein L, Horwitz SB. Differential overexpression of three mdr gene family members in multidrug-resistant J774.2 mouse cells. Evidence that distinct P-glycoprotein precursors are encoded by unique mdr genes. J Biol Chem. 1989;264:12053–12062. [PubMed] [Google Scholar]
- 15.Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 16.Cordon-Cardo C, O'Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klimecki WT, Futscher BW, Grogan TM, et al. P-glycoprotein expression and function in circulating blood cells from normal volunteers. Blood. 1994;83:2451–2458. [PubMed] [Google Scholar]
- 18.Thiebaut F, Tsuruo T, Hamada H, et al. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho GT, Moodie FM, Satsangi J. Multidrug resistance 1 gene (P-glycoprotein 170): an important determinant in gastrointestinal disease? Gut. 2003;52:759–766. doi: 10.1136/gut.52.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 21.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–G162. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 22.Collett A, Higgs NB, Gironella M, et al. Early molecular and functional changes in colonic epithelium that precede increased gut permeability during colitis development in mdr1a(−/−) mice. Inflamm Bowel Dis. 2008;14:620–631. doi: 10.1002/ibd.20375. [DOI] [PubMed] [Google Scholar]
- 23.Banner KH, Cattaneo C, Le Net JL, et al. Macroscopic, microscopic and biochemical characterisation of spontaneous colitis in a transgenic mouse, deficient in the multiple drug resistance 1a gene. Br J Pharmacol. 2004;143:590–598. doi: 10.1038/sj.bjp.0705982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maggio-Price L, Shows D, Waggie K, et al. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a−/−) mice. Am J Pathol. 2002;160:739–751. doi: 10.1016/S0002-9440(10)64894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilk JN, Bilsborough J, Viney JL. The mdr1a−/− mouse model of spontaneous colitis: a relevant and appropriate animal model to study inflammatory bowel disease. Immunol Res. 2005;31:151–159. doi: 10.1385/IR:31:2:151. [DOI] [PubMed] [Google Scholar]
- 26.Iizasa H, Genda N, Kitano T, et al. Altered expression and function of P-glycoprotein in dextran sodium sulfate-induced colitis in mice. J Pharm Sci. 2003;92:569–576. doi: 10.1002/jps.10326. [DOI] [PubMed] [Google Scholar]
- 27.Berg DJ, Zhang J, Weinstock JV, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 28.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 29.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 30.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 31.Bas A, Forsberg G, Hammarstrom S, et al. Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol. 2004;59:566–573. doi: 10.1111/j.0300-9475.2004.01440.x. [DOI] [PubMed] [Google Scholar]
- 32.Ropenga A, Chapel A, Vandamme M, et al. Use of reference gene expression in rat distal colon after radiation exposure: a caveat. Radiat Res. 2004;161:597–602. doi: 10.1667/rr3173. [DOI] [PubMed] [Google Scholar]
- 33.Rubie C, Kempf K, Hans J, et al. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes. 2005;19:101–109. doi: 10.1016/j.mcp.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Amin AR, Attur M, Abramson SB. Nitric oxide synthase and cyclooxygenases: distribution, regulation, and intervention in arthritis. Curr Opin Rheumatol. 1999;11:202–209. doi: 10.1097/00002281-199905000-00009. [DOI] [PubMed] [Google Scholar]
- 35.MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn's disease. Med Clin North Am. 1994;78:1207–1231. doi: 10.1016/s0025-7125(16)30096-7. [DOI] [PubMed] [Google Scholar]
- 36.Portanova JP, Zhang Y, Anderson GD, et al. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J Exp Med. 1996;184:883–891. doi: 10.1084/jem.184.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox JG, Yan LL, Dewhirst FE, et al. Helicobacter bilis sp. nov., a novel Helicobacter species isolated from bile, livers, and intestines of aged, inbred mice. J Clin Microbiol. 1995;33:445–454. doi: 10.1128/jcm.33.2.445-454.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burich A, Hershberg R, Waggie K, et al. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G764–G778. doi: 10.1152/ajpgi.2001.281.3.G764. [DOI] [PubMed] [Google Scholar]
- 39.Haines DC, Gorelick PL, Battles JK, et al. Inflammatory large bowel disease in immunodeficient rats naturally and experimentally infected with Helicobacter bilis. Vet Pathol. 1998;35:202–208. doi: 10.1177/030098589803500305. [DOI] [PubMed] [Google Scholar]
- 40.Shomer NH, Dangler CA, Schrenzel MD, et al. Helicobacter bilis-induced inflammatory bowel disease in scid mice with defined flora. Infect Immun. 1997;65:4858–4864. doi: 10.1128/iai.65.11.4858-4864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.ten Hove T, Drillenburg P, Wijnholds J, et al. Differential susceptibility of multidrug resistance protein-1 deficient mice to DSS and TNBS-induced colitis. Dig Dis Sci. 2002;47:2056–2063. doi: 10.1023/a:1019629013945. [DOI] [PubMed] [Google Scholar]
- 42.Mahler M, Bristol IJ, Leiter EH, et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 43.Pallis M, Turzanski J, Higashi Y, et al. P-glycoprotein in acute myeloid leukaemia: therapeutic implications of its association with both a multidrug-resistant and an apoptosis-resistant phenotype. Leuk Lymphoma. 2002;43:1221–1228. doi: 10.1080/10428190290026277. [DOI] [PubMed] [Google Scholar]
- 44.van der Heijde DM. The continuing challenge of predictive factors in rheumatoid arthritis: prediction or association? J Rheumatol. 1997;24:6–8. [PubMed] [Google Scholar]
- 45.Kunkel SL, Spengler M, May MA, et al. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem. 1988;263:5380–5384. [PubMed] [Google Scholar]
- 46.Strassmann G, Patil-Koota V, Finkelman F, et al. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J Exp Med. 1994;180:2365–2370. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrios-Rodiles M, Chadee K. Novel regulation of cyclooxygenase-2 expression and prostaglandin E2 production by IFN-gamma in human macrophages. J Immunol. 1998;161:2441–2448. [PubMed] [Google Scholar]
- 48.Endo T, Ogushi F, Sone S. LPS-dependent cyclooxygenase-2 induction in human monocytes is down-regulated by IL-13, but not by IFN-gamma. J Immunol. 1996;156:2240–2246. [PubMed] [Google Scholar]
- 49.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)−1 and −2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 50.Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-dependent arachidonic acid metabolites are essential modulators of the intestinal immune response to dietary antigen. Nat Med. 1999;5:900–906. doi: 10.1038/11341. [DOI] [PubMed] [Google Scholar]
- 51.Buyse M, Radeva G, Bado A, et al. Intestinal inflammation induces adaptation of P-glycoprotein expression and activity. Biochem Pharmacol. 2005;69:1745–1754. doi: 10.1016/j.bcp.2005.03.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Colitis in B6 and B6.mdr1a−/− mice following long term treatment with DSS. Average colonic length (A) and histological scoring (B) of B6.mdr1a−/− and wild type B6 controls. Clinical measures of colitis are assessed in panel C, data is given for diarrhea, presence of fecal blood, increased mortality following treatment, and increased intestinal permeability to FITC-Dextran.* Indicates statistical significance of P<.05 as determined by Mann Whitney (A, and C) when comparing P-gp deficient and control animals treated with alternating cycles of DSS infused drinking water.
Evaluation of colitis in B6 and B6.mdr1a−/− mice following short term treatment with piroxicam. Average colonic length (A) and histological scoring (B) of B6.mdr1a−/− and wild type B6 controls. Clinical measures of colitis are assessed in panel C. Data is given for diarrhea, presence of fecal blood, mortality following treatment, and intestinal permeability to FITC-Dextran.