

Abstract
Cancer stem cells (CSC) represent malignant cell subsets in hierarchically organized tumors, which are selectively capable of tumor initiation and self-renewal and give rise to bulk populations of non-tumorigenic cancer cell progeny through differentiation. Robust evidence for the existence of prospectively identifiable CSC among cancer bulk populations has been generated using marker-specific genetic lineage tracking of molecularly defined cancer subpopulations in competitive tumor development models. Moreover, novel mechanisms and relationships have been discovered that link CSC to cancer therapeutic resistance and clinical tumor progression. Importantly, proof-of-principle for the potential therapeutic utility of the CSC concept has recently been provided by demonstrating that selective killing of CSC through a prospective molecular marker can inhibit tumor growth. Herein, we review these novel and translationally relevant research developments and discuss potential strategies for CSC-targeted therapy in the context of resistance mechanisms and molecular pathways preferentially operative in CSC.
Keywords: ABCB5, cancer stem cells, genetic lineage tracking, melanoma, tumor hierarchy
Introduction
Over the past several decades, accelerating scientific and technological advances have enabled researchers to generate an abundance of knowledge in the realm of cancer biology. Numerous genes, mutant alleles, proteins, and signaling networks involved in the initiation and progression of cancer have been identified and many of the mechanisms conferring resistance to therapy have been characterized. Despite these groundbreaking advances and remarkable successes in the translation of this knowledge to the clinic, cancer remains a leading cause of human death and suffering, often due to the limited efficacy of currently available treatment modalities. The cancer stem cell (CSC) theory(1) has prompted some investigators to re-examine more established views of tumor initiation, cancer progression, and therapeutic resistance, with a view to develop novel CSC-directed therapeutics that might synergize with currently available treatments predominantly directed at cancer bulk populations, and that might hence serve to improve clinical cancer therapy.
Here, we review the experimental evidence for the existence of CSC in human cancers and discuss emerging novel links between CSC, neoplastic progression and cancer therapeutic resistance. Moreover, we review recent research findings that have provided initial proof-of-principle for the potential therapeutic utility of the CSC concept and discuss CSC-targeted treatment strategies that might synergize with more conventional therapeutic approaches directed at cancer bulk populations to effect tumor eradication.
Identification of cancer stem cells
Cancers, like physiologic tissues, are composed of morphologically and phenotypically heterogeneous cell populations.(2,3) Cancer cells may also exhibit functional heterogeneity, as evidenced by divergent in vitro clonogenic and proliferative capacities(4,5) and varying in vivo tumorigenic potentials.(6–8) Ongoing genetic mutations in the context of monoclonality of many cancers cannot fully explain this cellular heterogeneity. The CSC hypothesis of tumor initiation and growth provides an additional explanation for functional differences associated with cancer subpopulations.(1) It should be noted that the CSC model does not necessarily contradict the stochastic theory of tumor development, which postulates that cancers arise and expand through stochastic mutations and clonal selection processes.(9)
CSC, which are also referred to as tumor-initiating cells, are operationally defined by three distinct properties: (i) a selective capacity to initiate tumors and drive neoplastic proliferation, (ii) an ability to create endless copies of themselves through self-renewal, and (iii) the potential to give rise to more mature non-stem cell cancer progeny through a process termed differentiation(1,10) (Fig. 1). In order to experimentally verify the “stemness” of a given cancer cell subset, researchers until recently had predominantly relied on xenotransplantation assays in immunocompromised mice using human tumor biopsy-derived cancer subpopulations sorted for the presence or absence of a particular candidate CSC marker or set of markers. While established cancer cell lines may be useful for the validation of biological properties of tumorigenic subpopulations, they are, on their own, unlike primary tumor material, not sufficient for the identification of CSC. Using this experimental approach, tumorigenic populations that were capable of transferring human disease into immunodeficient murine hosts and that recapitulated the phenotype and morphology of the original patient tumors were characterized in diverse human malignancies, including leukemias(11–16) (Table 1), tumors of the breast,(17) central nervous system (CNS),(18) colon,(19–21) head and neck,(22) ovaries(23) and pancreas,(24,25) melanomas,(26) hepatocellular carcinomas,(27) and Ewing’s sarcomas(28) (Table 2).
Figure 1.
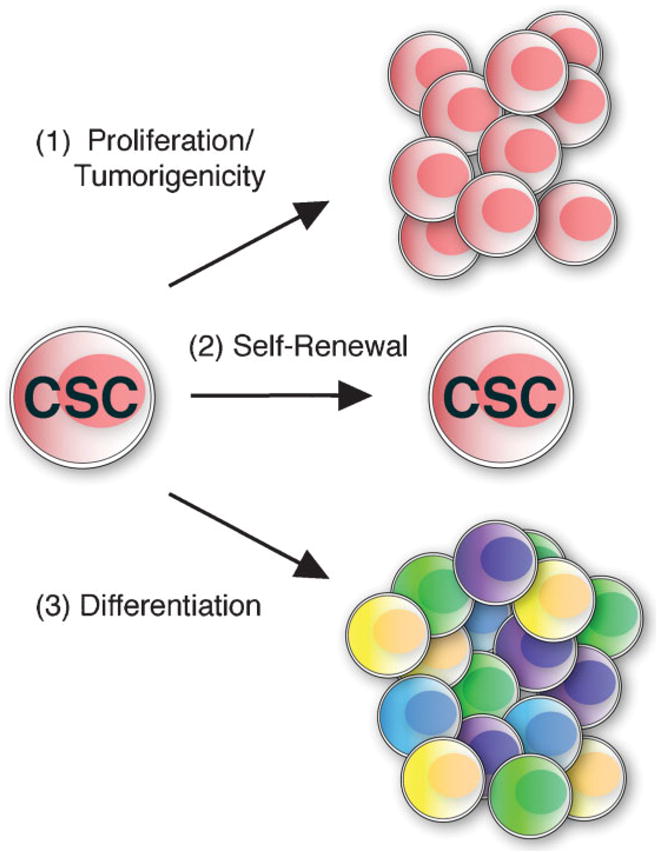
The cardinal features of CSC. CSC are characterized by three defining properties: (1) enhanced capacity for proliferation and tumorigenic growth, (2) long-term self-renewal ability, and (3) potential to give rise to more differentiated tumor bulk populations devoid of CSC characteristics. These cardinal CSC features can be attributed to molecularly defined cancer subpopulations using in vivo genetic lineage tracking in competitive tumor development models.
Table 1.
Cell surface phenotype of CSC identified in hematological malignancies
| Tumor type | Cell surface markers | Reference |
|---|---|---|
| AML | a CD34+CD38− | Lapidot et al.(46) |
| CD34+CD38− | Bonnet and Dick(11) | |
| CD34+CD38− | Ishikawa et al.(15) | |
| B-ALL | CD34+CD10−/CD34+CD19− | Cox et al.(13) |
| CD34+CD38−CD19+ | Castor et al.(12) | |
| Multiple Myeloma | a CD34−CD138− | Matsui et al.(16) |
| T-ALL | CD34+CD4−/CD34+CD7− | Cox et al.(14) |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia.
Serial xenotransplantation into secondary mouse recipients unsuccessful or not performed.
Table 2.
Cell surface phenotype of CSC identified in solid tumors
| Tumor type | Cell surface marker(s) | Reference |
|---|---|---|
| Breast | CD44+CD24−/low Lineage−ESA+ | Al-Hajj et al.(17) |
| CNS | CD133+ | Singh et al.(18) |
| Colon | CD133+ | O’Brien et al.(20) |
| CD133+ | Ricci-Vitiani et al.(21) | |
| ESAhighCD44+ Lineage− (CD166+) | Dalerba et al.(19) | |
| Ewing’s | CD133+ | Suva et al.(28) |
| Head and neck | CD44+Lineage− | Prince et al.(22) |
| Melanoma | ABCB5+ | Schatton et al.(26) |
| Liver | CD90+CD45− (CD44+) | Yang et al.(27) |
| Ovarian | CD44+CD117+ | Zhang et al.(23) |
| Pancreas | CD44+CD24+ESA+ | Li et al.(25) |
| CD133+ | Hermann et al.(24) |
It is important to recognize that CSC identification efforts that rely exclusively on tumorigenicity assays of sorted, untracked cancer cell subpopulations may have important potential limitations with respect to their ability to demonstrate hierarchical tumor organization. For example, conversion of marker phenotype among cancer subpopulations, as has been reported for the candidate CSC marker CD133,(29) could preclude unequivocal demonstration of hierarchical CSC-driven tumor organization in such studies. Furthermore, potential interactions between CSC and tumor bulk populations that may be operative in naturally occurring cancers(26) may not be taken into account in CSC investigations that rely exclusively on tumorigenicity assays of sorted, untracked cancer cell subpopulations. Therefore, marker-specific genetic lineage tracking of cancer subpopulations in competitive tumor development models enhances cell sorting-based xenotransplantation assays and can serve to confirm the existence of tumor hierarchies driven by molecularly defined CSC.(26)
In human acute myeloid leukemia (AML), tracking of unsegregated tumor cells in human to mouse xenotrans-plantation assays revealed heterogeneity of this malignancy with regard to CSC characteristics, but did not identify molecularly defined CSC.(30) In contrast, genetic lineage tracking of marker-sorted cancer subpopulations allowed the identification of molecularly defined CSC at the apex of hierarchically organized tumors in human malignant melanoma(26) and facilitated detection of cancer subpopulations of enhanced tumorigenicity in human breast and brain cancer cell lines.(31) In human melanomas, CSC responsible for tumor initiation and growth can be prospectively isolated based on expression of ABCB5 (ATP-binding cassette, subfamily B, member 5),(26) a mediator of cancer chemoresistance.(32–34) Previous studies had also suggested CD20 and CD133 as potential markers for CSC in melanoma, but serial xenotransplantation of prospectively isolated subpopulations was not performed in these experiments.(35,36)
In order to further examine and confirm the enhanced capacity of ABCB5+ CSC for tumorigenic growth, self-renewal, and differentiation,(26) genetic lineage tracking was performed in human to mouse xenotransplantation experiments using genetically encoded DsRed (red fluorescent protein) and EYFP (enhanced yellow–green fluorescent protein) fluorescent labeling of CSC and melanoma bulk populations, respectively.(26) Xenotransplantation of ABCB5+/DsRed+ melanoma subsets and ABCB5−/EYFP+ tumor bulk components reconstituted at naturally occurring ratios of ~1:10 resulted in significantly increased relative frequencies of DsRed+ cells of ABCB5+ origin in tumor xenografts up to a frequency of ~50% at the experimental endpoint of 6 weeks.(26) These finding established enhanced tumorigenicity of ABCB5+ melanoma subpopulations versus ABCB5− melanoma bulk populations in a competitive tumor development model.
When ABCB5+ cancer cells were re-isolated from the induced experimental tumors, fluorescent cells were of red-fluorescent phenotype of ABCB5+ origin, demonstrating self-renewal capacity of this cell subset.(26) EYFP+ cells were not found at significant levels among ABCB5+ isolates.(26) Thus, ABCB5+ tumor cells arose only from ABCB5+ inocula, and ABCB5− cells gave rise, at lower levels, exclusively to ABCB5− progeny. Fluorescent ABCB5− tumor cell isolates exhibited both DsRed and EYFP positivity, demonstrating that ABCB5+ cells also possess the capacity to differentiate into ABCB5− populations. These findings demonstrated through the use of genetic lineage tracking the existence of a tumor hierarchy, in which tumorigenic ABCB5+ melanoma cells, enriched for CSC, posses the exclusive capacity to self-renew and give rise to more differentiated, ABCB5− tumor progeny.(26) Moreover, the results showed that CSC may drive more differentiated and on their own non-tumorigenic cancer bulk populations to also contribute to a growing tumor mass, because EYFP+ cells of ABCB5− origin proliferated, albeit less efficiently, in the presence of ABCB5+/DsRed+ melanoma cells during the 6 weeks observation period.(26)
The existence of proliferating cancer bulk subsets in experimental tumors is consistent with clinical findings in human patients, where proliferating cancer bulk populations are susceptible to cell killing through cell-cycle-specific therapeutic agents. Further in vivo genetic lineage tracking experiments in human breast and brain cancer models utilized cell lines engineered to stably express the ZsGreen protein fused to the carboxyl-terminal degron of ornithine decarboxylase, which results in a fluorescent fusion protein that accumulates in cells with reduced proteasome activity.(31) These studies established CSC characteristics for cancer subpopulations characterized by low proteasome activity.(31) In addition, genetic lineage tracking was utilized to define self-renewal and differentiation capacity of murine Cd133+ or Lgr5+ (leucine-rich repeat-containing G protein-coupled receptor 5) cells of origin for colon cancer in inducible Cre knock-in mouse models.(37,38) These lineage tracking studies showed that long-lived intestinal stem cells are susceptible to cancer-causing mutations,(37,38) indicating that physiologic stem cells might represent the cell of origin for malignant transformation in colon cancer.
It should be noted, however, that the commonly accepted definition of CSC(1) does not necessarily imply a specific relationship of CSC to physiological stem cells. Indeed, findings in other cancers show that committed progenitors, and even terminally differentiated cell types, can represent the cellular source of malignant transformation.(39–42) In summary, genetic lineage tracking experiments have identified molecularly defined cancer subpopulations in human malignancies that exhibit enhanced tumorigenicity, self-renewal, and differentiation capacity, and as a result have provided robust evidence for the existence of CSC in human cancers. Moreover, genetic lineage tracking has proven to be a useful technique to dissect further potential relationships of CSC to normal stem cells in physiological tissues.
Considerable variability has been observed in the course of CSC identification efforts with regard to estimated frequencies of CSC in human cancers. While CSC phenotype and function are independent of relative frequency in human malignancies,(1,43) it is nevertheless important to define the factors responsible for this variability, because such studies may help to identify additional CSC-specific functions. Such factors may relate, e.g., to immune and microenvironmental interactions.(10,44,45)
With regard to immunity, estimated CSC frequencies may vary with the immune status of tumor xenotransplantation recipients.(11,26,46,47) For example, in studies on human AML, a minimum of 2 × 105 CD34+CD38− patient-derived tumor cells were required to initiate leukemias in severe combined immunodeficient (SCID) mice(46) compared to a 40-fold reduced number of AML cells (5 × 103) of equal phenotype in more severely immunocompromised non-obese diabetic (NOD)/SCID mouse recipients.(11) In addition, while CD34+CD38− AML-cells could be serially passaged to form secondary neoplasms in NOD/SCID mice,(11) they failed to do so in SCID hosts,(46) indicating that CSC phenotype and function are largely dependent on recipient immune status in this malignancy.(48) Moreover, recent evidence indicates that the CD34+CD38+ fraction of certain AML may also contain cells capable of initiating leukemias, a function that may potentially be masked in more immunocompetent hosts through inhibitory effects of anti-CD38 antibodies used for leukemia cell isolation.(49)
Host immune status may also be a variable that might influence estimated CSC frequencies in human melanomas, as indicated by a study that examined cell numbers required for tumor formation in first-passage xenotransplantation experiments.(47) While estimated CSC frequencies in xenotransplantation models involving NOD/SCID murine recipients are low (~1 in 106 human melanoma cells when tumorigenicity is assessed 8 weeks post-tumor cell inoculation(26,47)), a higher frequency of cells capable of initiating experimental tumors is detected when utilizing more severely immunocompromised, interleukin-2 receptor gamma chain null (IL-2Rγ−/−) NOD/SCID xenograft recipients.(47) Based on calculations from the data provided, the findings by Quintana et al. revealed 18-fold enrichment of patient-derived tumorigenic cells from ~1 in 105 in NOD/SCID mice to 1 in 5.5 × 103 in IL-2Rγ−/− NOD/SCID recipients, when tumorigenicity was assessed 32 weeks following tumor xenotransplantation in those hosts that had not received matrigel injections.(47)
While the study did not directly address CSC functions such as self-renewal and differentiation capacity in serial xenotransplantation experiments, the results support the view that under conditions of relatively intact immunity, such as in human patients or lesser immunocompromised experimental hosts, only CSC might possess the phenotypic and functional characteristics to evade host immunosurveillance and initiate tumor growth. In contrast, tumor host environments characterized by absent antitumor immunity might permit tumor bulk populations, which do not normally initiate tumors and may not possess CSC-specific self-renewal and differentiation capacity to also cause experimental tumor growth. This view is consistent with findings of increased cancer incidence in immunocompromised human patients and experimental animals.(50) Furthermore, it is supported by clinical findings regarding circulating melanoma cells, which indicate that malignant cells detected in the peripheral blood of human melanoma patients frequently do not initiate tumors.(51)
The possibility of immunoevasive properties of CSC,(45,52,53) potentially resembling immunomodulatory functions of physiologic stem cells,(54,55) has important implications for the experimental design of studies aimed at characterizing clinically relevant CSC populations. These considerations are particularly relevant to cancers known to induce strong immunogenic responses in patients in which they arise, such as human malignant melanoma,(56) and indicate that experimental model systems that allow CSC/host immunity interactions to occur in a fashion more closely resembling the clinical patient scenario might be better suited for the identification and study of CSC than those employing more profoundly immunocompromised xenograft hosts that deviate further from the translational relevance of host tissues.
Additional microenvironmental, non-immune host factors might also influence CSC-driven tumorigenicity.(44) For example, Galli et al. found that intracranial orthotopic inoculation of stem-like glioblastoma cells consistently induced neoplastic growth, while significantly reduced rates of tumor formation were observed when cells were injected subcutaneously.(57) In a separate study, CD133+Nestin+ brain CSC were found to reside within a perivascular niche, where they interacted closely with endothelial cells,(58) and increasing the number of co-grafted endothelial cells in orthotopic human brain tumor xenografts led to expansion of the self-renewing CSC fraction and accelerated cancer initiation and growth.(58) CSC might also reciprocally modulate the surrounding niche through secretion of paracrine factors or direct cell–cell contact. For example, brain CSC were shown to promote tumor angiogenesis and, as a consequence, tumor xenograft growth through secretion of vascular endothelial growth factor (VEGF).(59) In addition, co-administration of tumor growth-promoting factors that may normally be produced by cancer cells, such as the extracellular matrix component laminin, can enable tumor cells, which in the absence of laminin do not initiate tumors, to also contribute to experimental tumor formation.(47) Thus, important interactions of CSC with the tumor host microenvironment exist, which are relevant to and should be considered for the design of effective and clinically relevant assays for CSC identification and targeting.
Cancer stem cell-mediated resistance to conventional tumor therapies
The effectiveness of cancer therapy as a strategy to cure metastatic or disseminated malignant disease is frequently impaired by either intrinsic or acquired tumor resistance to cytotoxic agents or ionizing radiation.(60) Cellular mechanisms of therapeutic resistance include increased DNA damage recognition and repair, alterations of cell cycle checkpoints, impairment of tumor apoptotic pathways, and reduced accumulation of cytotoxic chemotherapeutic agents through enhanced energy-dependent drug efflux.(60) As a corollary to observations of a drug-resistant phenotype of physiological stem cells,(61,62) it had been hypothesized that CSC might also represent a subpopulation within cancers, i.e., characterized by increased resistance to chemo- and radiotherapy.(63,64) Evidence for a preferential resistance of CSC to conventional cancer therapies (Fig. 2A) has recently been generated.
Figure 2.
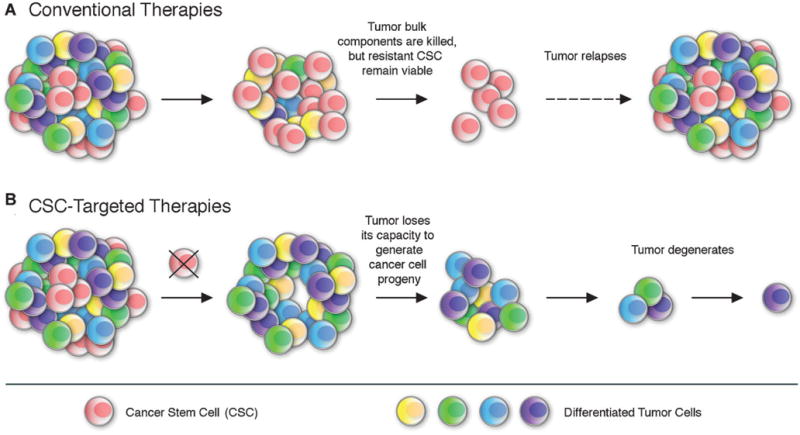
Conventional therapies versus CSC-targeted therapeutic strategies. A: conventional tumor therapies may initially shrink cancers by killing mainly tumor bulk populations with limited self-renewal and proliferative potential. Resistant CSC may remain viable after treatment and ultimately reestablish tumor growth, leading to relapse and neoplastic progression. B: In contrast, novel CSC-targeted approaches strip the tumor of its capacity to generate cancer cell progeny, which inhibits tumor growth and might ultimately result in tumor degeneration.
In hematological malignancies, leukemic stem cell (LSC) phenotype-expressing subpopulations were found to over-express several ABC drug efflux transporters implicated in drug resistance mechanisms.(65) In addition, LSC exhibited significantly higher daunorubicin and mitoxantrone efflux than tumor bulk components.(66) Moreover, Costello et al. demonstrated that both malignant and physiologic CD34+CD38− hematopoietic cells exhibit reduced in vitro sensitivity to daunorubicin.(52) The preferential chemoresistance was hereby associated with increased transcript levels of multi-drug resistance-associated proteins and reduced Fas-induced apoptosis following drug exposure.(52) In chronic myeloid leukemia (CML), resistance to the ABL tyrosine kinase inhibitor imatinib was linked to the failure of the chemotherapeutic agent to deplete the LSC compartment, particularly in CML patients in blast crisis.(39,67,68) Similarly, primary CD34+ CML cells also proved resistant to apoptosis induction by dasatinib(69) and nilotinib,(70) two alternative ABL tyrosine kinase inhibitors designed to circumvent imatinib resistance.(71) Furthermore, the dual SRC/ABL kinase inhibitor SKI-606 also failed to eradicate the CD34+ CML compartment in vitro.(72)
LSC isolated from human AML showed increased quiescence in vitro as indicated by a G0/G1 arrest of the cell cycle,(73,74) suggesting resistance to therapies targeted at proliferating cancer cells. Quiescence of human LSC was also demonstrated in human CML cell lines and primary patient material in vitro.(75) Moreover, resistance of LSC to cell cycle-dependent cytotoxic therapy was also shown in vivo in human AML to IL-2Rγ−/− NOD/SCID xenotransplantation experiments.(15) Specifically, cytosine arabinoside (Ara-C) treatment of human AML-bearing mice revealed that LSC had homed to the osteoblast niche within the bone marrow, protected from Ara-C-induced apoptosis.(15) Residual CD34+CD38− leukemic blasts were capable of stably engrafting secondary recipients, indicating that self-renewing LSC represent the Ara-C-resistant fraction in AML.(15) Insights into the mechanisms of CSC cell cycle arrest have been generated in model organisms.(76,77) During oncogene-induced acute myeloid leukemogenesis, a p21Cip1/Waf1-dependent cellular response is activated in LSC, which leads to reversible cell cycle arrest and DNA repair.(77) This mechanism prevents excessive DNA damage accumulation and functional exhaustion of hyper-proliferating LSC.(77) Furthermore, in a mouse model of CML, maintenance of LSC quiescence and resistance was functionally associated with the promyelocytic leukemia protein (PML) tumor suppressor gene.(76)
Evidence for CSC-associated resistance mechanisms has also been generated in solid tumors. For example, in human glioblastoma xenografts, the fraction of CD133+ CSC was found enriched after ionizing radiation.(78) The radioresistance of brain CSC was mechanistically linked to the preferential activation of the DNA damage repair pathway and to significantly reduced rates of apoptosis through the involvement of DNA checkpoint kinases.(78) In a separate study, cultured CD133+ glioblastoma cells exhibited lower rates of apoptosis in response to chemotherapeutic agents compared to their CD133− counterparts.(79)
Furthermore, in breast cancer, two independent studies found significantly increased levels of CD44+CD24−/low CSC phenotype-expressing cancer cells in tumor biopsies from patients who had received conventional chemotherapy compared to those evaluated prior to therapy.(80,81) Li et al. found that the frequency of CD44+CD24−/low tumor cells following therapeutic intervention correlated with the ability of treated breast cancer cells to form mammospheres and to initiate tumors in immunocompromised mice.(80) In a separate study, breast cancer cell line-derived spheroids enriched for CD44+CD24−/low subsets exhibited increased in vitro resistance to ionizing radiation as well as reduced levels of reactive oxygen species (ROS) and DNA double strand breaks after radiation exposure compared to adherent monolayer cultures.(82) Increased radiation resistance of stem-like cells in the human MCF-7 breast cancer cell line and murine mammary epithelium was subsequently also demonstrated by Woodward et al.(83) Consistent with these findings, mammary repopulating units as well as CSC phenotype-expressing murine and human patient-derived breast cancer cells displayed reduced levels of ROS and increased expression of free radical scavenging systems compared to tumor bulk components in a subsequent study by Diehn et al.(84)
In pancreatic cancer, CD133+tumor-initiating cells showed increased resistance to the chemotherapeutic agent gemcitabine compared to CD133− pancreatic cancer cells.(24) In colorectal cancer tumor xenografts, cells of CD44+ESA+ CSC phenotype were enriched following administration of chemotherapeutic agents involving cyclophosphamide or irrinotecan.(85) Moreover, the capacity to initiate tumors in xenotransplantation assays was preserved in the resistant colon cancer cell subset.(85)
In human malignant melanoma, where CSC can be prospectively isolated based on their expression of the ABCB5 drug efflux transporter,(26) the ABCB5+ cell subset was shown to possess increased resistance to the chemotherapeutic agent doxorubicin as a result of diminished drug accumulation.(32) Doxorubicin resistance could be reversed through monoclonal antibody-mediated inhibition of ABCB5-dependent cellular drug efflux, resulting in preferential chemosensitization of the more resistant CSC subset.(32) In addition, ABCB5 gene silencing through small interfering RNA treatment significantly increased the sensitivity of human melanoma cells to doxorubicin(33) and to 5-fluorouracil and camptothecin.(34) Furthermore, ABCB5 gene expression levels across a panel of human cancer cell lines used by the National Cancer Institute (NCI) for drug screening correlated significantly with chemoresistance to 45 out of 119 anticancer agents.(32)
The recent observation by Lehne et al. that development of drug resistance in a CML cell line results in ABCB5 gene amplification and mRNA overexpression and in the induction of stem cell genes raises the possibility that ABCB5 may serve similar chemoresistance functions in this malignancy, with potentially important implications should similar mechanisms be operative in drug-resistant clinical leukemias.(86,87) Since ABCB5+ CSC frequency also correlated significantly with clinical melanoma progression,(26) the ABCB5 marker identified a novel, direct link between CSC, cancer therapeutic resistance, and clinical neoplastic progression in a human malignancy.
In aggregate, several studies have provided experimental evidence for increased resistance of CSC to conventional cancer therapies directed at bulk populations of tumor cells (Fig. 2A). These findings underscore the need to dissect further the molecular pathways and mechanisms responsible for CSC-specific therapy resistance. Furthermore, they highlight the therapeutic promise of CSC-directed treatment strategies, which could facilitate eradication of tumors currently resistant to systemic therapy and thus potentially result in patient cures.
Cancer stem cell-directed targeting strategies
Cancer treatments that target CSC through specific markers or signaling pathways critically involved in CSC function could potentially increase the efficacy of current forms of therapy, by reducing the risk of relapse and dissemination (Fig. 2B), if CSC are indeed the major culprits of tumor initiation and progression in human patients. Several factors should be taken into account when designing CSC-directed treatment strategies: (i) given the similarities between CSC and physiologic stem cells,(37,38,88) CSC-targeted therapeutic agents could exert adverse effects on the renewal and maintenance of physiologic tissues due to potential toxic effects on a tumor host’s normal stem cell compartment. Therefore, preferred CSC targets would comprise those molecules or pathways that are preferentially induced or operative in malignant as opposed to physiological stem cells. (ii) CSC can represent heterogeneous cell populations that might differ in resistance profiles and might therefore not be efficiently targeted by a single therapeutic agent. (iii) CSC, like tumor bulk populations, might develop resistance to CSC-directed therapies. Furthermore, therapeutic efficacy will often depend also on the significant reduction of tumor bulk populations that may cause excessive tumor burden. Therefore, combination therapies that involve both CSC-directed agents as well as tumor bulk-targeted regimens would be predicted to prove most effective in improving clinical treatment responses and patient outcomes.
When examining the therapeutic efficacy of CSC-targeting strategies, it is important to recognize that such therapies might not necessarily reduce tumor burden in the short term. Longer observation intervals, and combination regimens, might be required to evaluate and discern the optimal therapeutic effects of CSC targeting. Therefore, in preclinical systems and ultimately in the clinic, experimental endpoints should include the status of CSC phenotype and function in addition to reduction of tumor burden. Several approaches might prove useful in enhancing responsiveness to systemic therapy in light of the CSC concept: (i) CSC ablation using antitumor agents, including monoclonal antibodies,(26,27) small molecules,(89–93) engineered oncolytic viruses,(94,95) or activated immune cells;(96) (ii) blockade of CSC function;(31,58,59,97,98) (iii) reversal of CSC resistance;(32,78,84,98–102) and (iv) CSC-directed differentiation therapy.(81,103–105)
Several studies have recently examined the potential therapeutic utility of the CSC concept. In human malignant melanoma, killing of CSC through their prospective identifier, ABCB5, could halt experimental tumor initiation and growth in vivo.(26) The tumor-inhibitory effects were shown to be mediated by immune effector populations induced to recognize and kill ABCB5+ CSC, as determined in antibody-dependent cell-mediated cytotoxicity (ADCC) assays in vitro and by histological analysis of ABCB5 monoclonal antibody-treated human melanoma xenografts in vivo.(26) These findings provided initial proof-of-principle that targeted ablation of CSC through a prospective marker can inhibit tumorigenesis and cancer growth, thereby validating for the first time the potential therapeutic utility of the CSC concept.(26) Also in melanoma, short hairpin RNA (shRNA) treatment directed at CD133, a marker preferentially coexpressed with ABCB5 in physiological skin progenitors(106) and a subset of melanomas,(32) was found to reduce melanoma cell clonogenicity and motility in vitro and melanoma metastatic potential in experimental model systems in vivo.(93)
Additional approaches to eliminate CSC might ultimately utilize oncolytic viruses or engineered immune cells. For example, Eriksson et al. used oncolytic adenoviruses to kill CD44+CD24−/low breast carcinoma cells.(94) Viral infection of CSC phenotype-expressing cancer cells could prevent the formation of tumor xenografts in immunodeficient mice in these studies, but adenoviral oncolytic effects were not tested on unsegregated populations or CSC-depleted subsets.(94) Therefore, it remained unclear whether selective CSC elimination or general antitumor effects were responsible for the observed inhibition of tumorigenicity. Similar caveats apply to a study that reported oncolytic virus-mediated killing of CD133+ glioblastoma cells.(95) In another approach, Mine et al. examined the antitumor effects of ex vivo primed T cells directed at immunogenic peptides present on breast CSC for cell killing.(96) This strategy proved useful in eliminating CD44+CD24−/low MCF7 breast cancer cells in vitro.(96) However, the immune cells in this study were not directed at CSC-specific markers and the effects were not examined in vivo or using clinical tumor specimens.
Further preclinical in vivo evidence for the potential therapeutic utility of CSC-targeting strategies was provided in hepatocellular carcinoma.(27) Using a human liver cancer to nude mouse xenotransplantation model, Yang et al. characterized a subpopulation of CD90+ CSC that coexpressed CD44 and exhibited an increased capacity to initiate experimental tumors in comparison to CD90− human liver cancer cells. Systemic administration of a human CD44 antibody at the time of subcutaneous tumor cell inoculation of CD90+ carcinoma cells significantly inhibited tumor initiation and growth compared to no treatment.(27) Anti-CD44 antibody-induced apoptosis was implicated as the mechanism of inhibited tumor xenograft formation, based on flow-cytometric quantification of cell death among CD90+ tumor cell fractions in response to antibody treatment in vitro.(27) Whether murine immune effector responses were also responsible for inhibited in vivo tumor growth was not examined in this study. While the results by Yang et al. provide further evidence for the potential therapeutic utility of CSC targeting, it is important to note that CD44 was not always found differentially expressed in CD90+ versus CD90− hepatocellular carcinoma cells in this study,(27) leaving uncertain its role as a CSC-specific target in this malignancy.
In human gliomas, CD133+ CSC were found to express up to 42-fold higher levels of the neural cell adhesion molecule L1 cell adhesion molecule (L1CAM) compared to CD133− tumor bulk populations,(89) and targeting of L1CAM using lentivirus-mediated shRNA interference significantly decreased the ability of CD133+ glioblastoma subpopulations to form neurospheres compared to shRNA controls. Moreover, L1CAM shRNA treatment induced apoptosis in CD133+ glioblastoma cells but had no significant effect on apoptosis in CD133− tumor populations in vitro.(89) Knockdown of L1CAM expression in CD133+ glioma cells before xenotransplantation to immunocompromised mice significantly decreased their in vivo tumorigenic capacity and resulted in increased survival of murine recipients. In order to determine the effect of L1CAM targeting on established tumors, immunocompromised mice were intracranially injected 5 days post-CD133+ glioma cell inoculation with lentiviral preparations expressing L1CAM shRNA. This treatment regimen significantly suppressed tumor growth and prolonged the lifespan of tumor-bearing mice compared to recipients of control shRNA,(89) indicating that targeting L1CAM may be useful as a CSC-directed therapy.
A recent report by Vlashi et al. further highlights the potential promise of targeting molecular mechanisms preferentially operative in CSC.(31) Human glioma and breast cancer cell lines with CSC characteristics were found to exhibit reduced 26S proteasome activity compared to tumor bulk components and selective targeting of this cell subset via a proteasome-dependent thymidine kinase suicide gene resulted in experimental tumor regression,(31) indicating that differences in proteasome activity might also serve as potential targets for CSC-directed cancer therapy. Additional studies have targeted CSC in preclinical model systems, but mostly through targets that were less specifically expressed by these critical subpopulations. Therefore, inhibition of cancer cell proliferation or tumor growth in these studies cannot always be attributed with certainty to specific inhibitory effects on CSC.
In AML, administration of an anti-human CD44 antibody significantly delayed malignant growth and prolonged survival of human leukemia-engrafted mice.(97) Mechanistically, CD44 antibody treatment was shown to inhibit the interaction of CD44-expressing leukemia cells with their niche and as a consequence migration and engraftment of these cells to the bone marrow and spleen of immunodeficient mice.(97) In addition, antibody treatment was shown to induce differentiation to more mature cancer cell progeny that was unable to establish robust leukemia upon xenotrans-plantation.(97) These data indicated a potential role of the microenvironment in regulating CSC function, with important implications for the development of CSC-directed treatment strategies.(44) In brain cancer, Calabrese et al. demonstrated that depletion of vascular endothelial cells, shown capable of enhancing in vivo self-renewal capacity of CD133+ brain CSC, using inhibitors of ERBB2 (erythroblastic leukemia viral homolog 2, also known as human epidermal growth factor receptor 2) or VEGF signaling, caused a reduction in CSC numbers and significantly inhibited in vivo tumor xenograft growth.(58) In an additional study, anti-VEGF neutralizing monoclonal antibody treatment was also found to suppress the growth of CD133+ human glioma cell-derived xenografts.(59)
A further CSC-directed therapeutic strategy might be the targeting of signals that regulate CSC resistance to chemo- or radiotherapy. Bao et al. generated experimental support for the potential promise of such approaches in human gliomas, where CSC subpopulations could be sensitized to ionizing radiation via pharmacological inhibition of DNA checkpoint kinases.(78) Similarly, in breast cancers, pharmacological depletion of ROS scavengers decreased CSC clonogenicity and resulted in radiosensitization.(84) Chemoresistance reversal in CSC populations may be achieved through specific blockade of ABC transporters, as shown in human melanoma.(32) The potential therapeutic utility of CSC-chemosensitizing agents is further supported by recent findings in colon cancer;(101) Todaro et al. demonstrated that treatment of CD133+ colon cancer cells with a neutralizing antibody to IL-4 prior to treatment with agents such as 5-fluorouracil and oxiplatin resulted in increased apoptosis of CSC through selective sensitization in vitro and in vivo.(101) Furthermore, CSC sensitization to immunotherapy might improve cancer therapy, because CSC, for example in melanoma, can possess an immunoevasive phenotype.(45)
The effects of various small molecules on CSC have been examined in AML(91,92,107) and breast cancer.(90) For example, inhibitors of nuclear factor kappa B (NF-κB) induced apoptosis in CD34+CD38− leukemic cells in vitro and inhibited tumor growth in experimental animal models in vivo, while sparing the physiologic hematopoietic stem cell compartment.(91) Similarly, in additional studies conducted by the same group, in vitro treatment of AML and CML cultures with MG-132 and TDZD-8, respectively, induced programmed cell death in CD34+CD38− leukemia cells.(91,92) Stem-like cancer cells have also been targeted through modulation of the phosphoinositide 3-kinase/phosphatase and tensin homolog (PI3K/PTEN) pathway in a mouse model of leukemia(108) as well as in prostate cancer cultures maintained under sphere-forming conditions.(109)
In breast cancer patients, treatment with the HER2 pathway inhibitor lapatinib resulted in a decrease in the relative frequency of CD44+CD24−/low cells after 12 weeks of treatment whereas conventional chemotherapy increased the percentage of CSC phenotype-expressing cells.(80) In a separate study, treatment of human patient-derived breast cancer cells with DPPE resulted in apoptosis among the CD44+CD24−/low CSC subset.(90) Eradication of tumorigenic cells was facilitated by concurrent treatment with doxorubicin,(90) pointing to a potential promise of combination therapy.
A number of combinatorial approaches have been shown to sensitize the chemorefractory LSC compartment(52,67–70,72) in AML and CML to chemotherapeutic drugs in vitro.(98–100,102) Specifically, pharmacologic inhibition of essential autophagy genes,(99) the CXCR4 pathway,(102) farnesyltransferase,(100) or of the hedgehog signaling network(98) increased the in vitro sensitivity of quiescent leukemic cells to the apoptogenic effects of tyrosine kinase inhibitors, including imatinib, dasatinib, or nilotinib. Characterization of gene expression profiles of CSC compared to physiological stem cells(110–112) may also serve to identify additional small molecules that selectively target CSC. However, further studies, including additional in vivo validation strategies, are needed in this regard. Ultimately, a more refined characterization of signaling networks specifically operative in CSC, especially vis-à-vis cancer bulk populations, may lead to effective CSC-targeted therapies.
Differentiation therapy represents an additional promising approach to target CSC. Differentiation strategies might induce quiescent CSC to differentiate into more mature tumor cell types, either through activation of distinct signaling pathways,(105) through altering gene expression profiles using micro-RNAs (miRNAs),(81) or through epigenetic therapy.(103) For example, Piccirillo et al. used BMP signaling to induce differentiation of CSC in human brain cancer models.(105) Specifically, administration of BMP4 either in vitro to glioblastoma cultures or in vivo to human brain cancer-bearing mice induced differentiation of glioblastomas and significantly reduced CD133+ cell frequency.(105) Moreover, mice implanted with tumor cells that had been transiently exposed to BMP4 survived significantly longer and showed smaller tumor lesions compared to recipients of untreated glioblastoma cells.(105) Interestingly, the BMP4 receptor BMPR1A is preferentially expressed on ABCB5+ CSC in human melanomas,(26) suggesting that a similar strategy could also be promising in this malignancy. In a separate study, administration of Notch pathway inhibitors resulted in depletion of medulloblastoma stem-like cells.(104)
miRNAs represent an additional class of molecules that could sensitize CSC to conventional anticancer therapies through differentiation.(81) This is indicated by findings in breast cancer, where differentiation of CD44+CD24−/low cells was induced through enforced expression of let-7 miRNA.(81) Finally, epigenetic therapy could also potentially be employed to induce CSC to differentiate and thus to render these aggressive cells more susceptible to conventional cytotoxic treatment.(103) This possibility is supported by clinical findings in patients with locally advanced breast cancer who were treated with the demethylating agent hydralazine and the histone deacetylase inhibitor magnesium valproate prior to doxorubicin and cyclophosphamide therapy, with a trend toward improved clinical outcome compared to doxorubicin and cyclophosphamide therapy alone.(103) The CSC marker and doxorubicin chemoresistance mediator ABCB5(26,32,106) were significantly downregulated following differentiation therapy in these studies,(103) raising the possibility that loss of more primitive and doxorubicin-resistant ABCB5-expressing cancer cells through differentiation might have contributed to the observed results.
In summary, several approaches can be envisioned to therapeutically target CSC to render tumor therapy more effective. Importantly, proof-of-principle has been established for the potential therapeutic utility of the CSC concept, and an increasing number of studies suggest that current cancer therapy could be enhanced through CSC eradication, blockade of CSC functions, reversal of CSC resistance, or induction of CSC differentiation. Therefore, it is likely that more effective cancer therapies will ultimately emerge as a result of addition of CSC-targeted strategies to more conventional treatments directed at tumor bulk populations.
Conclusions
Robust evidence has been generated for the existence of molecularly defined CSC in human cancer through in vivo genetic lineage tracking in experimental model systems. Furthermore, critical links between CSC, tumor progression, and therapy resistance have emerged that underscore the need for novel therapeutic strategies that target these aggressive cancer subpopulations. Multiple lines of investigation indicate that several approaches might prove useful in enhancing clinical responses to systemic therapy through CSC targeting, including CSC ablation through monoclonal antibodies, blockade of CSC functions, reversal of CSC-associated resistance mechanisms, or induction of CSC differentiation through epigenetic differentiation therapy. Therefore, combination therapies targeting CSC and tumor bulk populations are most likely to lead to optimized cancer treatments and to further reduce cancer morbidity and mortality in human patients.
Acknowledgments
Our work is supported by funds provided by the NIH/NCI (grants 1RO1CA113796-01A1, 1R01CA138231-01, and 2P50CA093683-06A20006 to M. H. F.). T. S. is the recipient of a Postdoctoral Fellowship Award from the American Heart Association Founders Affiliate.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science. 1982;217:998–1003. doi: 10.1126/science.7112116. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Griffin JD, Lowenberg B. Clonogenic cells in acute myeloblastic leukemia. Blood. 1986;68:1185–1195. [PubMed] [Google Scholar]
- 5.Sabbath KD, Ball ED, Larcom P, Davis RB, Griffin JD. Heterogeneity of clonogenic cells in acute myeloblastic leukemia. J Clin Invest. 1985;75:746–753. doi: 10.1172/JCI111756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199:79–80. doi: 10.1038/199079a0. [DOI] [PubMed] [Google Scholar]
- 7.Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461–463. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 8.Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46:411–422. [PubMed] [Google Scholar]
- 9.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 10.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008;21:39–55. doi: 10.1111/j.1755-148X.2007.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 12.Castor A, Nilsson L, Astrand-Grundstrom I, Buitenhuis M, Ramirez C, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
- 13.Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104:2919–2925. doi: 10.1182/blood-2004-03-0901. [DOI] [PubMed] [Google Scholar]
- 14.Cox CV, Martin HM, Kearns PR, Virgo P, Evely RS, Blair A. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood. 2007;109:674–682. doi: 10.1182/blood-2006-06-030445. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 16.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–2336. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 19.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 21.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 22.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Balch C, Chan MW, Lai HC, Matei D, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 26.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, et al. Significance of CD90(+) cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 28.Suva ML, Riggi N, Stehle JC, Baumer K, Tercier S, et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009;69:1776–1781. doi: 10.1158/0008-5472.CAN-08-2242. [DOI] [PubMed] [Google Scholar]
- 29.Jaksch M, Munera J, Bajpai R, Terskikh A, Oshima RG. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 2008;68:7882–7886. doi: 10.1158/0008-5472.CAN-08-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 31.Vlashi E, Kim K, Lagadec C, Donna LD, McDonald JT, et al. In vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst. 2009;101:350–359. doi: 10.1093/jnci/djn509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 33.Elliott AM, Al-Hajj MA. ABCB8 mediates doxorubicin resistance in melanoma cells by protecting the mitochondrial genome. Mol Cancer Res. 2009;7:79–87. doi: 10.1158/1541-7786.MCR-08-0235. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y, Anderle P, Bussey KJ, Barbacioru C, Shankavaram U, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004;64:4294–4301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- 35.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 36.Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 38.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 40.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 41.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 42.Sun B, Chen M, Hawks CL, Pereira-Smith OM, Hornsby PJ. The minimal set of genetic alterations required for conversion of primary human fibroblasts to cancer cells in the subrenal capsule assay. Neoplasia. 2005;7:585–593. doi: 10.1593/neo.05172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennedy JA, Barabe F, Poeppl AG, Wang JC, Dick JE. Comment on “Tumor growth need not be driven by rare cancer stem cells”. Science. 2007;318:1722. doi: 10.1126/science.1149590. [DOI] [PubMed] [Google Scholar]
- 44.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 45.Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04568.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 47.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 49.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 50.Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22:1136–1151. doi: 10.1200/JCO.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 51.Medic S, Pearce RL, Heenan PJ, Ziman M. Molecular markers of circulating melanoma cells. Pigm Cell Res. 2007;20:80–91. doi: 10.1111/j.1600-0749.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- 52.Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, et al. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–4411. [PubMed] [Google Scholar]
- 53.Kawasaki BT, Farrar WL. Cancer stem cells, CD200 and immunoevasion. Trends Immunol. 2008;29:464–468. doi: 10.1016/j.it.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Frank MH, Sayegh MH. Immunomodulatory functions of mesenchymal stem cells. Lancet. 2004;363:1411–1412. doi: 10.1016/S0140-6736(04)16134-5. [DOI] [PubMed] [Google Scholar]
- 55.Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 56.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 58.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 59.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 60.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 61.Peters R, Leyvraz S, Perey L. Apoptotic regulation in primitive hematopoietic precursors. Blood. 1998;92:2041–2052. [PubMed] [Google Scholar]
- 62.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 63.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 64.Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene. 2004;23:7178–7187. doi: 10.1038/sj.onc.1207935. [DOI] [PubMed] [Google Scholar]
- 65.de Grouw EP, Raaijmakers MH, Boezeman JB, van der Reijden BA, van de Locht LT, et al. Preferential expression of a high number of ATP binding cassette transporters in both normal and leukemic CD34+CD38-cells. Leukemia. 2006;20:750–754. doi: 10.1038/sj.leu.2404131. [DOI] [PubMed] [Google Scholar]
- 66.Wulf GG, Wang RY, Kuehnle I, Weidner D, Marini F, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–1173. doi: 10.1182/blood.v98.4.1166. [DOI] [PubMed] [Google Scholar]
- 67.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 68.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 69.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 70.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109:4016–4019. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 71.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–2596. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 72.Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood. 2008;111:2329–2338. doi: 10.1182/blood-2007-05-092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML) Blood. 2003;101:3142–3149. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 74.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 75.Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94:2056–2064. [PubMed] [Google Scholar]
- 76.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- 78.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 79.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 81.Yu F, Yao H, Zhu P, Zhang X, Pan Q, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 82.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 83.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009 doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lehne G, Grasmo-Wendler UH, Berner JM, Meza-Zepeda LA, Adamsen BL, et al. Upregulation of stem cell genes in multidrug resistant K562 leukemia cells. Leuk Res. 2009 doi: 10.1016/j.leukres.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 87.Frank NY, Frank MH. ABCB5 gene amplification in human leukemia cells. Leuk Res. 2009 doi: 10.1016/j.leukres.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 89.Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng T, Liu JC, Pritchard KI, Eisen A, Zacksenhaus E. Preferential killing of breast tumor initiating cells by N,N-diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine/tesmilifene. Clin Cancer Res. 2009;15:119–130. doi: 10.1158/1078-0432.CCR-08-1708. [DOI] [PubMed] [Google Scholar]
- 91.Guzman ML, Li X, Corbett CA, Rossi RM, Bushnell T, et al. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8) Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guzman ML, Swiderski CF, Howard DS, Grimes BA, Rossi RM, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220–16225. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rappa G, Fodstad O, Lorico A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for meta-static melanoma. Stem Cells. 2008;26:3008–3017. doi: 10.1634/stemcells.2008-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eriksson M, Guse K, Bauerschmitz G, Virkkunen P, Tarkkanen M, et al. Oncolytic adenoviruses kill breast cancer initiating CD44(+)CD24(−/low) cells. Mol Ther. 2007;15:2088–2093. doi: 10.1038/sj.mt.6300300. [DOI] [PubMed] [Google Scholar]
- 95.Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 96.Mine T, Matsueda S, Li Y, Tokumitsu H, Gao H, et al. Breast cancer cells expressing stem cell markers CD44(+) CD24 (lo) are eliminated by Numb-1 peptide-activated T cells. Cancer Immunol Immunother. 2008 doi: 10.1007/s00262-008-0623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 98.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–1123. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Copland M, Pellicano F, Richmond L, Allan EK, Hamilton A, et al. BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood. 2008;111:2843–2853. doi: 10.1182/blood-2007-09-112573. [DOI] [PubMed] [Google Scholar]
- 101.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 102.Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2008 doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arce C, Perez-Plasencia C, Gonzalez-Fierro A, de la Cruz-Hernandez E, Revilla-Vazquez A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant Doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:e98. doi: 10.1371/journal.pone.0000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fan X, Matsui W, Khaki L, Stearns D, Chun J, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 105.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 106.Frank NY, Pendse SS, Lapchak PH, Margaryan A, Shlain D, et al. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem. 2003;278:47156–47165. doi: 10.1074/jbc.M308700200. [DOI] [PubMed] [Google Scholar]
- 107.Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 109.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106:268–273. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 111.Majeti R, Becker MW, Tian Q, Lee TL, Yan X, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U S A. 2009;106:3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]