

Abstract
The interpretation of data-driven experiments in genomics often involves a search for biological categories that are enriched for the responder genes identified by the experiments. However, knowledge bases such as the Gene Ontology (GO) contain hundreds or thousands of categories with very high overlap between categories. Thus, enrichment analysis performed on one category at a time frequently returns large numbers of correlated categories, leaving the choice of the most relevant ones to the user's; interpretation.
Here we present model-based gene set analysis (MGSA) that analyzes all categories at once by embedding them in a Bayesian network, in which gene response is modeled as a function of the activation of biological categories. Probabilistic inference is used to identify the active categories. The Bayesian modeling approach naturally takes category overlap into account and avoids the need for multiple testing corrections met in single-category enrichment analysis. On simulated data, MGSA identifies active categories with up to 95% precision at a recall of 20% for moderate settings of noise, leading to a 10-fold precision improvement over single-category statistical enrichment analysis. Application to a gene expression data set in yeast demonstrates that the method provides high-level, summarized views of core biological processes and correctly eliminates confounding associations.
INTRODUCTION
Many studies in functional genomics follow a data-driven approach. Experiments such as transcriptional profiling with microarrays, ChIP-on-chip or gene knock-out screens are done at the scale of the whole genome without specifying a prior hypothesis. Instead, one seeks to discover new phenomena and generate new hypotheses from the data. Loosely formulated, the main question driving the analysis of data-driven experiments is: what is going on?
Although for technical and biological reasons the nature of the data differs between these types of experiments, they often can be summarized by a list of genes which responded to the experiment, e.g. genes found to be differentially expressed, bound by a particular transcription factor or whose knock-down elicits a phenotype of interest. However, extensive lists of responder genes are not per se useful to describe the experiment. A practical way to address the question of what is going on? is to perform a gene category analysis, i.e. to ask whether these responder genes (which we will refer to as the study set) share some biological features that distinguish them among the set of all genes tested in the experiment (which we will refer to as the population). Gene category analysis involves a list of gene categories, such as those provided by the Gene Ontology (GO) (1) or the pathways of the KEGG database (2), and a statistical method for identifying enriched categories such as overrepresentation analysis using Fisher's; exact test (3), gene set enrichment (4–7), logistic regression (8), random-set analysis (9) and Bayesian techniques for analyzing GO terms in a context where not all annotated genes are observed (10).
Gene category analyses that follow the mentioned approaches often return a large number of significant categories, which are related to one another and leave to the user the task of choosing the most meaningful categories at the risk of relying on biased judgments. The reason for the correlation is that genes can belong to multiple categories, so that if one category is significantly overrepresented in the study set, then it is more likely that other categories with many genes in common with it will also be significantly overrepresented. While these enrichments are correct in a statistical sense, long lists of results can make it difficult to focus on the most important ones in a biological sense.
With GO, annotations are understood to follow the true-path rule, meaning that whenever a gene is annotated to a term it is also implicitly associated with all parents of that term. This is an important cause of gene sharing, a phenomenon which we have termed the inheritance problem (11). A number of methods have been proposed to deal with these problems. The elim algorithm examines the GO graph in a bottom-up fashion (12). Once a GO term is found to be significantly overrepresented according to Fisher's; exact test, all genes annotated to it are removed from further analysis. A variant of this algorithm downweights the genes rather than eliminating them completely (12). The parent–child algorithm determines overrepresentation of a term in the context of annotations to the term's; parents by redefining the groups used to calculate the hypergeometric distribution (11).
All of the aforementioned procedures successively test overrepresentation for each of the categories. The above methods make use of the structure of GO to address statistical dependencies, but they are limited to ontologies and do not fundamentally differ from the original paradigm of term-for-term testing with the exact Fisher test. More recently, an approach called GenGO was presented, which fits a model on all the terms simultaneously. GenGO models the set of responder genes as members of a set of active GO terms. The objective of GenGO is to identify the most likely combination of active terms, while allowing for some false positive and some false negative responder genes. The fitting procedure optimizes an objective function that combines the likelihood of the model with a penalization that tends to limit the overall number of active terms. This procedure was shown to outperform other methods for detecting GO-term overrepresentation on simulated data and to identify concise yet biologically relevant sets of significantly overrepresented GO terms when applied to real data sets (13).
Here we present model-based gene set analysis (MGSA), a model-based approach that significantly improves over GenGO by scoring terms with their posterior probabilities, leading to more robust results and to improvements over standard methods across a broader range of sensitivity cutoffs. Our benchmark on simulated data confirm the drastic improvements of model-based approaches over term-by-term methods. MGSA uses a simple and adaptable Bayesian network that provides a great deal more flexibility as to the kinds of data that can be used in the analysis and to extensions of the model. MGSA is implemented as part of the free and open-source software Ontologizer (14), a Java application that implements a large number of GO enrichment methods and enables visual exploration of the results.
METHODS
Model
We model gene response in a genome-wide experiment as the result of an activation of a number of biological categories. These categories can be pathways as defined by the KEGG database (2), GO terms (15) or any other scheme (5,16) that associates genes to potentially overlapping biologically meaningful categories. Because we primarily work with GO, we call these categories terms. Our method does not make use of the graph structure of GO other than utilizing the true-path rule, which states that if a gene is associated to a term, then it is also associated to all of terms along the path up to the root of the ontology. Apart from that we make no explicit use of the structure.
We assume that the experiment attempts to detect genes that have a particular state (such as differential expression), which can be on or off. The true state of any gene is hidden. The experiment and its associated analysis provide observations of the gene states that are associated with unknown false positive (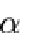 ) and false negative rates (
) and false negative rates (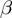 ), which we will assume to be identical and independent for all genes.
), which we will assume to be identical and independent for all genes.
For instance, in the setting of a microarray experiment, the on state would correspond to differential expression, and the off state would correspond to a lack of differential expression of a gene. Our model, hence, assumes that differential expression is the consequence of the annotation to some terms that are on.
An additional parameter 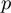 represents the prior probability of a term being in the on state. The probability
represents the prior probability of a term being in the on state. The probability 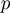 is typically low (<0.5), introducing an effective penalization for the number of active terms. This ingredient promotes parsimonious explanations of the data.
is typically low (<0.5), introducing an effective penalization for the number of active terms. This ingredient promotes parsimonious explanations of the data.
More formally, our model is a Bayesian network with three layers augmented with a set of parameters (Figure 1):
The term layer
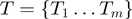 consists of Boolean nodes that represent the
consists of Boolean nodes that represent the 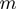 terms. There is a Boolean variable associated with each node that can have the state values on (1) or off (0).
terms. There is a Boolean variable associated with each node that can have the state values on (1) or off (0).The hidden layer
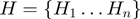 contains Boolean nodes that represent the
contains Boolean nodes that represent the 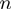 annotated genes. There are edges from the terms to their annotated genes. For instance, if gene
annotated genes. There are edges from the terms to their annotated genes. For instance, if gene 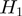 is annotated to terms
is annotated to terms 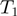 and
and 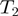 then there is an edge between
then there is an edge between 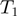 and
and 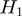 and another edge between
and another edge between 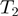 and
and 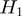 . The state of the nodes reflects the true activation pattern of the genes. Each node can have the state values on (1), or off (0).
. The state of the nodes reflects the true activation pattern of the genes. Each node can have the state values on (1), or off (0).The observed layer
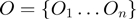 contains Boolean nodes reflecting the state of all observed genes. The observed gene state nodes are directly connected to the corresponding hidden gene state nodes in a one-to-one fashion.
contains Boolean nodes reflecting the state of all observed genes. The observed gene state nodes are directly connected to the corresponding hidden gene state nodes in a one-to-one fashion.The parameter set contains continuous nodes with values in
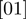 corresponding to the parameters of the model
corresponding to the parameters of the model 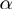 ,
, 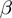 and
and 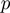 . These parameterize the distributions of the observed and the term layer as detailed below.
. These parameterize the distributions of the observed and the term layer as detailed below.
Figure 1.

A Bayesian network to model gene response with gene categories. Gene categories, or terms (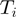 , ellipses) can be either on or off. Terms that are on activate the hidden state (
, ellipses) can be either on or off. Terms that are on activate the hidden state (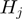 , rectangles) of all genes annotated to them, the other genes remain off. The observed states (
, rectangles) of all genes annotated to them, the other genes remain off. The observed states (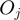 , diamonds) of the genes are noisy observations of their true hidden state. The parameters of the model (light gray nodes) are the prior probability of each term to be active,
, diamonds) of the genes are noisy observations of their true hidden state. The parameters of the model (light gray nodes) are the prior probability of each term to be active, 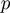 , the false positive rate,
, the false positive rate, 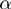 and the false negative rate,
and the false negative rate, 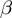 .
.
For didactic purposes, we will initially explain a simplified version of our procedure in which the parameters 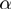 ,
, 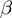 and
and 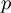 are considered to have known, fixed values. We will then show how the Bayesian network can be augmented to search for optimal values for
are considered to have known, fixed values. We will then show how the Bayesian network can be augmented to search for optimal values for 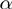 ,
, 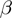 and
and 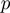 .
.
The state propagation of the nodes can be modeled using various local probability distributions (LPDs), denoted by 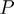 . The joint probability distribution for this Bayesian network can be written as
. The joint probability distribution for this Bayesian network can be written as
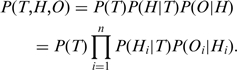 |
(1) |
We model the state of each 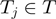 according to a Bernoulli distribution with hyperparameter
according to a Bernoulli distribution with hyperparameter 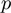 , i.e.
, i.e. 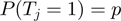 . Denote by
. Denote by 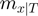 the number of terms that have state
the number of terms that have state 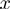 for a given
for a given 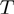 , i.e.
, i.e. 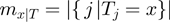 then
then
| (2) |
In the following, we denote by 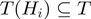 the set of terms to which gene
the set of terms to which gene 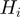 is annotated, i.e.
is annotated, i.e. 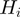 and the ancestors of
and the ancestors of 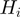 . For the
. For the 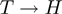 links, any node
links, any node 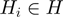 is on (
is on (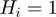 ) if at least one term in
) if at least one term in 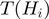 is on. Otherwise it is off:
is on. Otherwise it is off:
| (3) |
Note that this transition is deterministic. For the 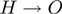 connection, we choose the following two Bernoulli distributions:
connection, we choose the following two Bernoulli distributions: 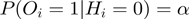 and
and 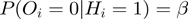 .
.
Therefore, 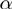 is the probability that a gene
is the probability that a gene 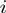 is observed to be on (i.e.
is observed to be on (i.e. 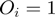 ), although its true hidden state is actually off (i.e.
), although its true hidden state is actually off (i.e. 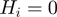 ) and thus, none of the terms which annotate the gene are on. Correspondingly,
) and thus, none of the terms which annotate the gene are on. Correspondingly, 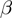 is the probability of a gene being observed to be off, although at least one term that annotates it is on.
is the probability of a gene being observed to be off, although at least one term that annotates it is on.
Denote by 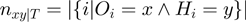 the number of genes having observed activation
the number of genes having observed activation 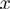 and true activation
and true activation 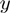 according to the states of
according to the states of 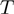 . For instance,
. For instance, 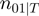 corresponds to the number of genes observed to be not differentially expressed but whose true activation state is on. Then, by considering the LPDs of nodes, we get the following product of Bernoulli distributions for
corresponds to the number of genes observed to be not differentially expressed but whose true activation state is on. Then, by considering the LPDs of nodes, we get the following product of Bernoulli distributions for 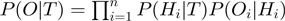 :
:
| (4) |
Markov Chain Monte Carlo algorithm for marginal probabilities inference with known parameters
As it is often the case, marginal posteriors for our network cannot be derived analytically. We estimate these values using a Metropolis–Hasting algorithm, which is a Markov chain Monte Carlo (MCMC) method (17–19). The MCMC algorithm performs a random walk over the term and parameter configurations, which asymptotically provides a random sampler according to the target distribution 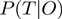 .
.
Given the current configuration of the terms denoted by 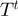 , the algorithm proposes a neighbor state
, the algorithm proposes a neighbor state 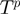 according to a proposal density function
according to a proposal density function 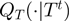 . We sample a value
. We sample a value  uniformly from the range (0,1). Then, if
uniformly from the range (0,1). Then, if
| (5) |
the proposal is accepted, i.e. 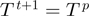 , otherwise it is rejected, i.e.
, otherwise it is rejected, i.e. 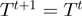 . Using Bayes' law, we have
. Using Bayes' law, we have
| (6) |
and similarly for 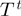 . Substituting these expressions for
. Substituting these expressions for 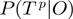 and
and 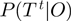 cancels out the normalization constant
cancels out the normalization constant 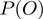 . The acceptance probability is then:
. The acceptance probability is then:
| (7) |
Equation (5) is used iteratively to define a random walk through the space of configurations. A burn-in period consisting of a certain number of iterations is used to initialize the MCMC chain (in our implementation, the default is 20 000 iterations). Following this, 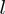 further iterations (default
further iterations (default 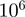 ) are performed. Let
) are performed. Let 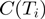 be the number of samples in which term
be the number of samples in which term 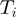 was on. Then
was on. Then
In order to finish the description of the algorithm, we need to define classes of operations of which a proposal is chosen, that is, we need to specify 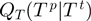 . We denote by
. We denote by 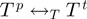 the binary relation that states that
the binary relation that states that 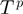 be constructed from
be constructed from 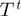 by either
by either
• toggling the on/off state of a single term, or by
• exchanging the state of a pair of terms that contains a single on term and a single off term.
We denote by 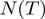 the neighborhood of a given configuration for
the neighborhood of a given configuration for 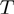 , that is, the number of different operations that can be applied once to
, that is, the number of different operations that can be applied once to 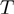 in order to get a new configuration. At first, there are
in order to get a new configuration. At first, there are 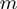 terms in total, each of which can be toggled. In addition, there are
terms in total, each of which can be toggled. In addition, there are 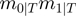 possibilities to combine terms that are on with terms that are off. Thus, there are a total of
possibilities to combine terms that are on with terms that are off. Thus, there are a total of 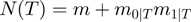 valid state transitions. We would like to sample the valid proposals with equal probability, therefore the proposal distribution
valid state transitions. We would like to sample the valid proposals with equal probability, therefore the proposal distribution 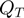 is determined by
is determined by
| (8) |
which we can use to rewrite Equation (7) to:
The procedure is shown in Algorithm 1. For simplicity, the burn-in period is omitted from the pseudocode. It is easy to see that all states of the chain are reachable from any state, as the Markov chain is finite and it is possible to reach an arbitrary state from any other state by a fixed number of operations. This accounts for the reducibility of the chain. Moreover, the chain is aperodic as it is always possible to stay in the same state, as any proposal can be rejected. Therefore, the resulting Markov chain is ergodic, which is a sufficient condition for a convergence to a stationary distribution, which matches the desired target distribution.

MCMC algorithm for marginal probabilities inferrence with unknown parameters
The estimation of the parameter 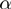 ,
, 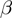 and
and 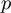 can be easily integrated directly into the MCMC algorithm. The parameters now must be explicitly considered in the joint probability distribution:
can be easily integrated directly into the MCMC algorithm. The parameters now must be explicitly considered in the joint probability distribution:
| (9) |
where 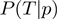 is given by Equation (2),
is given by Equation (2), 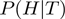 is given by Equation (3) and
is given by Equation (3) and 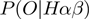 corresponds to
corresponds to 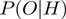 of the basic model. As
of the basic model. As 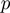 ,
, 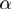 and
and 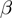 are now true random variables, we must define a prior distribution on them as well. Here, we have used uniform distributions to introduce as little bias as possible.
are now true random variables, we must define a prior distribution on them as well. Here, we have used uniform distributions to introduce as little bias as possible.
We are seeking for a scheme to sample from joint posterior distribution
In order to utilize the Metropolis–Hasting algorithm for this purpose, we are required to provide an efficient calculation for the numerator. This is straighforward, because the numerator factors to
| (10) |
and moreover, 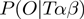 can be determined using Equation (4).
can be determined using Equation (4).
In addition to term state transitions, we also need to take parameter transitions within the proposal density into account. We define the new proposal density as a mixture of the state transition density 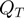 and a parameter transition density
and a parameter transition density 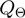 . We denote the current realization of the parameters by
. We denote the current realization of the parameters by 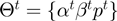 and by
and by 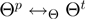 the relation of whether
the relation of whether 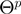 can be constructed from
can be constructed from 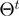 . The fully specified proposal density is then
. The fully specified proposal density is then
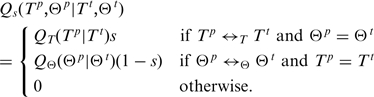 |
The parameter 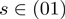 can be used to balance state transition proposals against parameter proposals. That is to say, depending on the outcome of a Bernoulli process with hyperparameter
can be used to balance state transition proposals against parameter proposals. That is to say, depending on the outcome of a Bernoulli process with hyperparameter  , we either propose a new state transition or a new parameter setting. For the experiments described in this article,
, we either propose a new state transition or a new parameter setting. For the experiments described in this article,  was set to 0.5.
was set to 0.5.
Many possibilities for the proposal density of the parameter transition 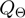 and for the relation
and for the relation 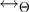 can be envisaged. We have considered transitions
can be envisaged. We have considered transitions 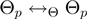 for which
for which 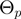 differs from
differs from 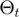 in the realization of not more than a single variable.
in the realization of not more than a single variable.
In contrast with the configuration space of the terms' activation state, the domain of these new variables is continuous. However, an internal study revealed that the algorithm is not overly sensitive to the exact parameter settings. Therefore, we can restrict the range of the variables to a set of discrete values. For the experiments described in this work, we used the restrictions 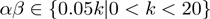 and
and 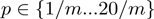 , where
, where 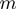 is the number of terms.
is the number of terms.
At last, we can state the proposal density function for parameter transitions
| (11) |
in which 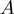 ,
, 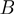 and
and 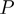 stand for the domain of the parameters
stand for the domain of the parameters 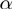 ,
, 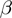 and
and 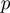 respectively. Note that
respectively. Note that 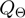 is symmetric, i.e.
is symmetric, i.e.
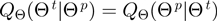 .
.
Simulation and evaluation of the performance
The simulations were based on revision 1.846 (dated October 21, 2009) of the GO term definition file. We restricted the entire simulation study to genes of Drosophila melanogaster. Annotations for this species were taken from revision 1.157 (dated October 19, 2009) of the gene association file provided by FlyBase (20), using all annotations regardless of their evidence code. This results in a total of 12 484 genes that are annotated directly or by inheritance to 7078 GO terms.
Study sets were generated according to our model as follows. One value for the false positive rate 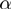 and one for the false negative rate
and one for the false negative rate 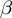 were set. A number (varying from one to five) of unrelated terms (i.e. pairs of terms related by parent–child relationships were avoided) are randomly picked to be in on state. In the remainder of this section, we denote by
were set. A number (varying from one to five) of unrelated terms (i.e. pairs of terms related by parent–child relationships were avoided) are randomly picked to be in on state. In the remainder of this section, we denote by 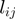 the state or label of term
the state or label of term  within study set
within study set 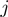 , i.e.
, i.e. 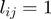 , if term
, if term 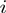 is on, or
is on, or 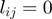 otherwise.
otherwise.
Each single study set 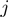 is then filled with all genes that are annotated to the term
is then filled with all genes that are annotated to the term 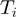 for all
for all 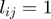 . Next, the noisy observations are simulated by removing each gene with a probability of
. Next, the noisy observations are simulated by removing each gene with a probability of 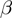 from the study set. Then, genes from the population not annotated to any of the active GO terms were added to the study set with a probability of
from the study set. Then, genes from the population not annotated to any of the active GO terms were added to the study set with a probability of 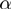 . The whole procedure was repeated 1500 times for each combination of
. The whole procedure was repeated 1500 times for each combination of 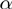 and
and 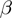 providing 1500 different study sets of varying sizes for that combination.
providing 1500 different study sets of varying sizes for that combination.
All tested algorithms were then applied to the simulated study sets. Note that the study set generation procedure controls merely the expected values of the proportion of false positive and false negative genes for the study sets, whereas the actual proportion of each individual study set may differ. MGSA and GenGO
and GenGO (which use fixed values of the parameters
(which use fixed values of the parameters 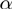 ,
, 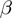 and
and 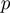 ) were supplied with the values of
) were supplied with the values of 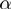 and
and 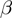 used for the simulation and
used for the simulation and 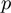 was set according to the number of GO terms that were set to on. The application of the algorithms results in prediction values (scores) for
was set according to the number of GO terms that were set to on. The application of the algorithms results in prediction values (scores) for 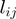 , denoted by
, denoted by 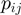 . We remark that for posterior marginal probabilities higher values (rather than lower as with
. We remark that for posterior marginal probabilities higher values (rather than lower as with 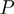 -values) indicate stronger support for the state on.
-values) indicate stronger support for the state on.
Benchmarking of the methods was done by using standard measures for the evaluation of discrimination procedures. We made use of receiver operating characteristic (ROC) curves and precisison/recall curves, pooling the results of all study sets with identical parameter combinations. In addition to the values of a ROC analysis, we calculated the 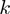 -truncated ROC value for each study set
-truncated ROC value for each study set 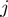 via
via
in which 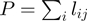 is the total number of positives and
is the total number of positives and 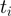 represents the number of true positives above the
represents the number of true positives above the 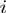 -th false positive (21,22). We reported the average over
-th false positive (21,22). We reported the average over 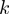 -truncated ROC values of all study sets for
-truncated ROC values of all study sets for 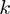 = 10.
= 10.
Analysis of yeast growth media
Raw tiling array data comparing yeast fermentative growth (YPD: Yeast extract Pepton Dextrose) and respiratory growth (YPE: Yeast extract Peptone Ethanol) (23) were processed to provide normalized intensity values for each probe in each hybridization. The expression level of each transcript in each growth condition was estimated by the midpoint of the shorth (shortest interval covering half of the values) of the probe intensities of the transcript across all arrays of the growth condition. Transcripts were called expressed if their expression level was above the threshold (24). Transcript expression levels of the two conditions ‘YPD’ and ‘YPE’ were normalized against each other using the vsn method (25) as differential expression at the transcript level appeared to still depend on average expression value. Next, transcripts were called differentially expressed if they showed at least 2-fold change between the two conditions. We then compared a study set of 510 differentially expressed genes to the population of 5308 genes (23) (Supplementary Table 1). We used GO annotations obtained from the Saccharomyces Genome Database (26) as of October 22, 2009 and restricted our analysis to the biological process ontology.
RESULTS
Bayesian networks to model experimental observations
In order to summarize the meaning of a long list of genes by naming biologically meaningful categories or terms, we propose a knowledge-based system in form of a Bayesian network. We model the state of the genes as a function of the activity of the associated terms. The true state of the genes is hidden and propagated to corresponding entities at the observation layer, by which we reflect the noisy nature of the data. The errors between the observation and the hidden states are assumed to be independent and to occur with a potentially unknown false positive (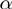 ) and false negative rate (
) and false negative rate (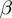 ), identical for all genes. Furthermore, an additional parameter
), identical for all genes. Furthermore, an additional parameter 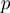 represents the prior probability of a term being in the on state (Figure 1). The purpose of this model is to infer the marginal posterior probability of each term
represents the prior probability of a term being in the on state (Figure 1). The purpose of this model is to infer the marginal posterior probability of each term 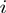 being active, i.e.
being active, i.e. 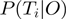 , given the observations of the experiment. See the ‘Methods’ section for a formal introduction of the model and the inference process.
, given the observations of the experiment. See the ‘Methods’ section for a formal introduction of the model and the inference process.
In practice, the parameters 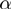 ,
, 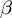 and
and 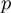 are not known. While the estimation is taken into account by the specification of the algorithm, we also run instances in which these parameters are fixed a priori to the true ones. This provides an upper bound for the parameter estimation. In the following, we refer to the general case as MGSA and to the case with known parameters as MGSA
are not known. While the estimation is taken into account by the specification of the algorithm, we also run instances in which these parameters are fixed a priori to the true ones. This provides an upper bound for the parameter estimation. In the following, we refer to the general case as MGSA and to the case with known parameters as MGSA .
.
Performance on simulated data
We simulated 1500 study sets in which the number of active terms varied from one to five (‘Methods’ section). The simulations were performed with 12 484 Drosophila genes that are annotated directly or indirectly via to parent–child relationships to 7078 GO terms. We followed this approach for each combination of 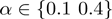 and
and 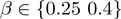 , resulting in a total of 6000 simulated study sets. We then compared MGSA and MGSA
, resulting in a total of 6000 simulated study sets. We then compared MGSA and MGSA against three single-term association procedures: the standard term-for-term (TfT) GO overrepresentation analysis by Fisher's; exact test (3), Parent–Child union (PCU) analysis (11) and topological weight (TopW) analysis (12), and against the other global model approach, GenGO (13). Similar to our approach, GenGO has two parameters that are intended to capture false positive and false negative responders and an additional parameter that penalizes superfluous terms. In the original implementation of GenGO (13), a heuristic procedure was used to search for the best values of these parameters. Unfortunately, the full GenGO software is not applicable for batched runs. We have implemented the algorithm denoted as GenGO
against three single-term association procedures: the standard term-for-term (TfT) GO overrepresentation analysis by Fisher's; exact test (3), Parent–Child union (PCU) analysis (11) and topological weight (TopW) analysis (12), and against the other global model approach, GenGO (13). Similar to our approach, GenGO has two parameters that are intended to capture false positive and false negative responders and an additional parameter that penalizes superfluous terms. In the original implementation of GenGO (13), a heuristic procedure was used to search for the best values of these parameters. Unfortunately, the full GenGO software is not applicable for batched runs. We have implemented the algorithm denoted as GenGO in the simple case where the parameters are known. For the simulations described here, we follow the authors' recommendation to set the penalty parameter to 3, while the remaining parameters were set to the optimal values. This provides an upper bound on the performance of the GenGO procedure with unknown parameters.
in the simple case where the parameters are known. For the simulations described here, we follow the authors' recommendation to set the penalty parameter to 3, while the remaining parameters were set to the optimal values. This provides an upper bound on the performance of the GenGO procedure with unknown parameters.
GO analyses typically contain a very large number of terms. Therefore, an important issue is whether a GO analysis method inflates the number of terms reported as significant. The most critical measure is therefore the precision, i.e. the proportion of true positives among the true and false positives. Comparing the precision of the different methods as a function of the recall, which is the proportion of true positives among all positive terms, demonstrates the drastic improvement of global model approaches. Both global model methods, GenGO and MGSA, dominate all three single-term association approaches by a factor of at least 3 (5 for MGSA) in precision at 20% recall (Figure 2A, B) across all investigated parameter settings. For a false positive rate
and MGSA, dominate all three single-term association approaches by a factor of at least 3 (5 for MGSA) in precision at 20% recall (Figure 2A, B) across all investigated parameter settings. For a false positive rate 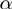 of 0.1, the improvement reaches even 8- to 10-fold. Moreover, MGSA largely outperforms GenGO' in all settings, for example, with a precision of ∼95% versus ∼80% for GenGO' in the case of
of 0.1, the improvement reaches even 8- to 10-fold. Moreover, MGSA largely outperforms GenGO' in all settings, for example, with a precision of ∼95% versus ∼80% for GenGO' in the case of 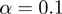 and
and 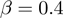 . Values of
. Values of 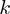 -truncated ROC scores (‘Methods’ section and Table 1) confirm the ranking of these methods when focusing on stringent cutoffs. Moreover, these improvements of MGSA are seen at any cutoff. MGSA outperform all other methods for the whole range of recall cutoffs and with all investigated parameter settings (Figure 2C, D).
-truncated ROC scores (‘Methods’ section and Table 1) confirm the ranking of these methods when focusing on stringent cutoffs. Moreover, these improvements of MGSA are seen at any cutoff. MGSA outperform all other methods for the whole range of recall cutoffs and with all investigated parameter settings (Figure 2C, D).
Figure 2.

Benchmarking on simulated data set. Performance of the TfT, PCU, TopW, GenGO , MGSA
, MGSA and MGSA algorithms on simulated data set with different settings of false positive (
and MGSA algorithms on simulated data set with different settings of false positive (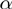 ) and false negative (
) and false negative (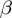 ) rates. In each row, the leftmost panel shows the precision for a recall of 0.2 (A, B), the middle panel precision as a function of recall (C, D) and the rightmost panel the ROC curve (E, F).
) rates. In each row, the leftmost panel shows the precision for a recall of 0.2 (A, B), the middle panel precision as a function of recall (C, D) and the rightmost panel the ROC curve (E, F).
Table 1.
ROC10 Analysis
| α | β | GenGO′ | MGSA′ | MGSA | TfT | PCU | TopW |
|---|---|---|---|---|---|---|---|
| 0.1 | 0.4 | 0.35 | 0.44 | 0.41 | 0.31 | 0.24 | 0.26 |
| 0.4 | 0.25 | 0.22 | 0.28 | 0.25 | 0.22 | 0.17 | 0.15 |
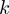 -truncated ROC curves were generated for the simulated data shown in Figure 2 for
-truncated ROC curves were generated for the simulated data shown in Figure 2 for 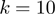 . The ROC10 score is the area under the ROC curve up to the tenth false positive.
. The ROC10 score is the area under the ROC curve up to the tenth false positive. 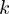 -truncated ROC scores range from 0 to 1, with 1 corresponding to the most sensitive and selective result.
-truncated ROC scores range from 0 to 1, with 1 corresponding to the most sensitive and selective result.
Notably, the performance of GenGO′, which reports only a single maximum likelihood solution and discards any alternative solution, even if it is almost as likely, drops off much earlier than MGSA (Figure 2C, D). This behavior is more apparent in Receiver Operating Characteristic curves (ROC curves, Figure 2E, F), which plot the true positive rate (or recall) as a function of the false positive rate (proportion of false positives among all negative terms). Indeed, away from the most stringent zone, GenGO appears as the least accurate of all tested methods. See also Supplementary Figures S1–S12 for results on other parameter combinations
Together these results on simulation confirm the drastic improvement of global model approaches and demonstrate that our marginal posterior method, MGSA, largely outperforms GenGO by showing an accurate behavior on the whole range of cutoffs.
Dealing with unknown values of the parameters 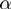 ,
, 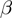 and
and 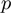 had required a significant extension of our basic algorithm (‘Methods’ section). The simulation data allowed us to investigate the ability of the full MGSA algorithm to cope with unknown parameter values. We ran the basic version of the algorithm, MGSA
had required a significant extension of our basic algorithm (‘Methods’ section). The simulation data allowed us to investigate the ability of the full MGSA algorithm to cope with unknown parameter values. We ran the basic version of the algorithm, MGSA , in which the parameters are known and fixed a priori. Performance of MGSA
, in which the parameters are known and fixed a priori. Performance of MGSA are displayed in the precision–recall and the ROC curves (Figure 2) and represent what MGSA could reach if the parameters were known. As Figure 2 shows, the performance of the algorithm is not drastically affected by this, showing that the full MGSA algorithm performs reasonably well when dealing with unknown parameter values.
are displayed in the precision–recall and the ROC curves (Figure 2) and represent what MGSA could reach if the parameters were known. As Figure 2 shows, the performance of the algorithm is not drastically affected by this, showing that the full MGSA algorithm performs reasonably well when dealing with unknown parameter values.
Analysis of expression data from fermentative and respiratory respiration in yeast
We applied our method to 510 yeast genes found to be differentially expressed in fermentative growth compared with respiratory growth (‘Methods’ section). Respiration and fermentation are two well-studied growth modes of yeast, thus facilitating the interpretation of the results. We ran TfT, PCU and MGSA on the biological process ontology of GO. Using Benjamini–Hochberg correction for multiple testing and a cutoff of false discovery rate of 0.1, the term-for-term method returns 193 significantly enriched terms, many of which are highly related (Supplementary Table 1). We investigated how the results of MGSA fluctuate by running 20 independent Markov chains, each of length 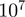 , using a cutoff of 0.5 on the posterior probability to call a term on, i.e. a level at which a term is estimated to be more likely to be on than to be off. MGSA reports only seven terms with a marginal posterior probability >0.5 (in at least one of the 20 runs). Out of these 7 terms, 6 showed a posterior probability >0.5 consistently across the 20 chains (Figure 3 and Supplementary Table 1), while the seventh one, monocarboxylic acid metabolic process, had estimated posterior probability between 0.410 and 0.502. Hence, results for the most likely terms were reproducible between runs. We checked the robustness of these results against variations in the study set by creating 2 000 random subsamples of the study set containing 90% of the original genes. The terms identified by the original analysis were consistently identified in the subsamples (Supplementary Figure 13).
, using a cutoff of 0.5 on the posterior probability to call a term on, i.e. a level at which a term is estimated to be more likely to be on than to be off. MGSA reports only seven terms with a marginal posterior probability >0.5 (in at least one of the 20 runs). Out of these 7 terms, 6 showed a posterior probability >0.5 consistently across the 20 chains (Figure 3 and Supplementary Table 1), while the seventh one, monocarboxylic acid metabolic process, had estimated posterior probability between 0.410 and 0.502. Hence, results for the most likely terms were reproducible between runs. We checked the robustness of these results against variations in the study set by creating 2 000 random subsamples of the study set containing 90% of the original genes. The terms identified by the original analysis were consistently identified in the subsamples (Supplementary Figure 13).
Figure 3.

Application on a respiratory versus fermentative growth expression dataset in yeast. (A) Ranked list of the 192 overrepresented terms using a term-for-term Fisher's; test with Benjamini–Hochberg correction for multiple testing. Many of the top terms are redundant and relate to similar functions. The term cell death (highlighted in blue) is a spurious association (see text). (B) Ranked list of the top 10 terms identified by a single run of MGSA (six of them with a posterior >0.5 in green). (C) Error bars (95% confidence intervals) obtained with 20 runs of MGSA. Each of the seven terms was identified with a posterior >0.5 in at least one of the 20 runs.
Among the seven terms, oxidation reduction summarizes the main biological process that distinguishes growth in these two different media, namely the use of oxidation phosphorylation during respiration to regenerate ATP. The other terms, such as, carbohydrate transport, or monocarboxylic acid metabolic process capture processes that are linked to the change of carbon source but not directly involved in the oxidation reduction reactions. Hence, MGSA provides a high-level, summarized view of the core biological process, respiration, avoiding redundant results while still keeping the necessary level of granularity in other branches of the ontology.
The term cell death illustrates very well the difference between single-term association approaches and global model approaches. Both tested enrichment-based approaches, TfT and PCU, report cell death as an enriched term whereas MGSA does not. It happens that mitochondria are implicated both in cell death and in respiration (27). The differentially expressed genes annotated to cell death encode mitochondrial proteins and are also involved in respiration. Hence, it is correct to report cell death enriched for differentially expressed genes. However, cells are not dying in any of these two conditions. The enrichment is due to the sharing of genes with respiration, a process which is genuinely differentially activated. In this study set, 113 genes are annotated to oxidation reduction including 25 out of 28 genes annotated to cell death. MGSA, which infers the terms that are on and not simply enriched, does not report cell death. One should also note that cell death is not a type of respiratory pathway or vice versa. Methods such as TfT that examine the statistical significance of each term separately cannot compensate for correlations between terms due to gene sharing. Although methods such as PCU and TopW can compensate for some kinds of statistical correlations that arise because of the inheritance of annotations from descendent nodes in the GO graph (11,12), they fail in situations such as the one described here because, oxidation reduction and cell death share some annotated genes but are not directly connected to one another by the graph structure of GO.
DISCUSSION
Of trees and forests
Data-driven molecular biology experiments can be used to identify a list of genes that respond in the context of a given experiment. With the advent of technologies, such as microarray hybridization and next-generation sequencing, which enable biologists to generate data reflecting the response profiles of thousands of genes or proteins, gene category analysis has become ever more important as a means of understanding the salient features of such experiments and for generating new hypotheses. By using the knowledge as it is provided by GO, KEGG or other similar systems of categorization, these analyses have become a de facto standard for molecular biological research. Almost all previous methods are based on algorithms that analyze each term in isolation. For each term under consideration, the methods consider whether the study set is significantly enriched in genes annotated to the term compared with what one would expect based on the frequency of annotations to the term in the entire population of genes or using other related statistical models (28).
We suggest that single-term association methods that determine the significance of each term in isolation essentially do ‘not see the forest for the trees’, by which we mean that they tend to return many related terms which are statistically significant if considered individually, but they are not designed to return a set of core terms that together best explain the set of genes in the study set. Although some methods have been developed that partially compensate for statistical dependencies in GO (11,12), the work of Lu and colleagues (13) addressed the problem by modeling the gene responses using all categories together.
Modeling requires formulating a generative process of the data. We, and Lu and colleagues (13), considered the categories as the potential cause of the gene responses. Fitting the model then enables distinguishing between the causal categories (according to the model) from the categories merely associated with gene response. Although one cannot conclude that the identified categories are causal in reality (this is only a model and one only has observational data), this feature of model-fitting explains why it provides a better answer to the question what is going on? than testing for associations on a term for term basis.
Searching for an optimal set of terms that together explain a biological observation is a more difficult problem than examining each term for enrichment one at a time. In particular, the model used to find term sets must specify how the terms interact with one another. This imposes assumptions on the model that must be kept in mind when the results are interpreted. For instance, the model presented in this work assumes that activation of a single term suffices to activate genes, and does not require that a certain minimum number of genes are annotated by the term set. The Bayesian framework we present can easily be adapted for different kinds of models encoding different biological assumptions by choosing different priors or distributions. This will be the subject of future research.
A Bayesian framework for inference
In contrast to the method of Lu and coworkers (13), we embed the model into a Bayesian Network, to which we apply standard methods of probabilistic inference. This not only leads to an intuitive derivation of the score, but also increases the versatility of the framework. That is, although we have demonstrated our method with simple classes of LPDs for the nodes, one can easily use more involved distributions. Moreover, we show that by augmenting the Bayesian network, we get a streamlined approach that includes both the inference of the states and of the parameters. This in principle also enables the inclusion of prior knowledge, be it parameters that are known or estimated in form of 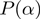 and
and 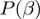 , or a specific term configuration in form of
, or a specific term configuration in form of 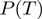 or
or 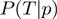 .
.
Instead of scoring terms by finding the maximum a posteriori as in the GenGO method (13), we use marginal posterior probabilities. Finding the maximum a posteriori provides a single combination of active terms and is not informative about alternative solutions. If several solutions show near-maximal likelihood then it is implausible that the single one with the largest likelihood is always the right solution. In contrast, marginal posterior as in MGSA associates a natural weight to each term that reflects a measure of certainty of its involvement in the process. Importantly, using marginal posterior probabilities increases the robustness and lowers the sensitivity of the procedure to high variance related to the multimodality of the problem, i.e. the existence of local maxima of the likelihood function. As we demonstrated using simulations, MGSA is indeed more robust than GenGO.
The Java implementation that we provide in the Ontologizer estimates the marginal posteriors with a MCMC algorithm and is fast enough to perform 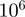 steps in <3 s on a standard 2.5 GHz PC. Since runs of MCMC are not guaranteed to converge in any a priori defined number of steps, we suggest that users repeat the analysis in order to see how the reported marginal probabilities of the top terms fluctuate. If fluctuations are too large, the number of MCMC steps should be increased.
steps in <3 s on a standard 2.5 GHz PC. Since runs of MCMC are not guaranteed to converge in any a priori defined number of steps, we suggest that users repeat the analysis in order to see how the reported marginal probabilities of the top terms fluctuate. If fluctuations are too large, the number of MCMC steps should be increased.
Using a Bayesian approach that models the data with all categories simultaneously, rather than using hypothesis testing on each category, avoids the issue of multiple testing. Moreover, the interpretation of the score, which is simply the probability of a category to be active, might appear more natural than a P-value. Finally, one should note that the ranking of the scores is reversed to values given by hypothesis-based procedures, i.e. high marginals give high support in the Bayesian setting, while high confidence in the hypothesis-based approach is indicated by low 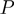 -values.
-values.
CONCLUSION
We have addressed the question of gene category analysis using a model-based approach, MGSA. In this Bayesian model, the genes responding to the experiment are assumed to belong to a small number of ‘active’ categories. Therefore, to answer the question of what is going on in an experiment, MGSA infers the ‘active’ categories, among all considered categories, given the actual gene state observations. We have shown that under the assumptions of our model our approach is better in identifying the causal sets than other procedures. We suggest that considering the forest instead of the trees is an advantageous strategy for gene category analysis, and that global model procedures such as the one presented in this article may be better able to describe the biological meaning of high-throughput data sets than are procedures that examine associations of categories one at a time. We note that the Bayesian network analyzed in this article is but one of many potential network structures that are made possible with our framework.
Our methods have been integrated into the Ontologizer project, an easy-to-use Java Webstart application for performing analysis of overrepresentation. The Ontologizer as well as the implementation of the described benchmark procedure have been released under the terms of the modified BSD licence. The application is available from http://compbio.charite.de/index.php/ontologizer2.html. The source code is available from http://sourceforge.net/projects/ontologizer/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG SFB 760 and RO 2005/4-1). Additional support was provided by the Lab of Lars Steinmetz. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Emilie Fritsch for insightful discussions about the yeast gene expression dataset analysis and to Simon Anders for critical reading of the manuscript.
REFERENCES
- 1.The Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhee SY, Wood V, Dolinski K, Draghici S. Use and misuse of the Gene Ontology annotations. Nat. Rev. Genet. 2008;9:509–515. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Z, Gentleman R. Extensions to gene set enrichment. Bioinformatics. 2007;23:306–313. doi: 10.1093/bioinformatics/btl599. [DOI] [PubMed] [Google Scholar]
- 5.Mootha VK, Lindgren CM, Eriksson K.-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrle M, Laurila E, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 6.Nam D, Kim S.-Y. Gene-set approach for expression pattern analysis. Brief. Bioinform. 2008;9:189–197. doi: 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- 7.Oron AP, Jiang Z, Gentleman R. Gene set enrichment analysis using linear models and diagnostics. Bioinformatics. 2008;24:2586–2591. doi: 10.1093/bioinformatics/btn465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics. 2009;25:211–217. doi: 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton MA, Quintana FA, den Boon JA, Sengupta S, Ahlquist P. Random-set methods identify distinct aspects of the enrichment signal in gene-set analysis. Ann. Appl. Stat. 2007;1:85–106. [Google Scholar]
- 10.Vêncio RZN, Koide T, Gomes SL, Pereira C.AB. BayGO: Bayesian analysis of ontology term enrichment in microarray data. BMC Bioinformatics. 2006;7:86. doi: 10.1186/1471-2105-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grossmann S, Bauer S, Robinson PN, Vingron M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- 12.Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Rosenfeld R, Simon I, Nau GJ, Bar-Joseph Z. A probabilistic generative model for GO enrichment analysis. Nucleic Acids Res. 2008;36:e109. doi: 10.1093/nar/gkn434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer S, Grossmann S, Vingron M, Robinson PN. Ontologizer 2.0—a multifunctional, tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008;24:1650–1651. doi: 10.1093/bioinformatics/btn250. [DOI] [PubMed] [Google Scholar]
- 15.Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009 – an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37:D396–D403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrieu C, De Freitas N, Doucet A, Jordan MI. An introduction to MCMC for machine learning. Mach. Learn. 2003;50:5–43. [Google Scholar]
- 18.Diaconis P. The Markov chain Monte Carlo revolution. Bull. Am. Math. Soc. 2009;46:179–205. [Google Scholar]
- 19.Diaconis P, Saloff-Coste L. ACM. 1995. What do we know about the Metropolis algorithm?. STOC’95: Proceedings of the Twenty-Seventh Annual ACM Symposium on Theory of Computing; pp. 112–129. [Google Scholar]
- 20.Tweedie S, Ashburner M, Falls K, Leyland P, Mcquilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, et al. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37(Suppl. 1):D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gribskov M, Robinson NL. Use of receiver operating characteristic (ROC) analysis to evaluate sequence matching. Comput. Chem. 1996;20:25–33. doi: 10.1016/s0097-8485(96)80004-0. [DOI] [PubMed] [Google Scholar]
- 22.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM. A high-resolution map of transcription in the yeast genome. Proc. Natl Acad. Sci. USA. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl. 1):S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 26.Hong EL, Balakrishnan R, Dong Q, Christie KR, Park J, Binkley G, Costanzo MC, Dwight SS, Engel SR, Fisk DG, et al. Gene Ontology annotations at SGD: new data sources and annotation methods. Nucleic Acids Res. 2008;36:D577. doi: 10.1093/nar/gkm909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 28.Goeman JJ, Bühlmann PP. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–987. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.