

Abstract
Skeletal muscle contraction increases intracellular ATP turnover, calcium flux, and mechanical stress, initiating signal transduction pathways that modulate peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α)-dependent transcriptional programmes. The purpose of this study was to determine if the intensity of exercise regulates PGC-1α expression in human skeletal muscle, coincident with activation of signalling cascades known to regulate PGC-1α transcription. Eight sedentary males expended 400 kcal (1674 kj) during a single bout of cycle ergometer exercise on two separate occasions at either 40% (LO) or 80% (HI) of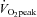 . Skeletal muscle biopsies from the m. vastus lateralis were taken at rest and at +0, +3 and +19 h after exercise. Energy expenditure during exercise was similar between trials, but the high intensity bout was shorter in duration (LO, 69.9 ± 4.0 min; HI, 36.0 ± 2.2 min, P < 0.05) and had a higher rate of glycogen utilization (P < 0.05). PGC-1α mRNA abundance increased in an intensity-dependent manner +3 h after exercise (LO, 3.8-fold; HI, 10.2-fold, P < 0.05). AMP-activated protein kinase (AMPK) (2.8-fold, P < 0.05) and calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylation (84%, P < 0.05) increased immediately after HI but not LO. p38 mitogen-activated protein kinase (MAPK) phosphorylation increased after both trials (∼2.0-fold, P < 0.05), but phosphorylation of the downstream transcription factor, activating transcription factor-2 (ATF-2), increased only after HI (2.4-fold, P < 0.05). Cyclic-AMP response element binding protein (CREB) phosphorylation was elevated at +3 h after both trials (∼80%, P < 0.05) and class IIa histone deacetylase (HDAC) phosphorylation increased only after HI (2.0-fold, P < 0.05). In conclusion, exercise intensity regulates PGC-1α mRNA abundance in human skeletal muscle in response to a single bout of exercise. This effect is mediated by differential activation of multiple signalling pathways, with ATF-2 and HDAC phosphorylation proposed as key intensity-dependent mediators.
. Skeletal muscle biopsies from the m. vastus lateralis were taken at rest and at +0, +3 and +19 h after exercise. Energy expenditure during exercise was similar between trials, but the high intensity bout was shorter in duration (LO, 69.9 ± 4.0 min; HI, 36.0 ± 2.2 min, P < 0.05) and had a higher rate of glycogen utilization (P < 0.05). PGC-1α mRNA abundance increased in an intensity-dependent manner +3 h after exercise (LO, 3.8-fold; HI, 10.2-fold, P < 0.05). AMP-activated protein kinase (AMPK) (2.8-fold, P < 0.05) and calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylation (84%, P < 0.05) increased immediately after HI but not LO. p38 mitogen-activated protein kinase (MAPK) phosphorylation increased after both trials (∼2.0-fold, P < 0.05), but phosphorylation of the downstream transcription factor, activating transcription factor-2 (ATF-2), increased only after HI (2.4-fold, P < 0.05). Cyclic-AMP response element binding protein (CREB) phosphorylation was elevated at +3 h after both trials (∼80%, P < 0.05) and class IIa histone deacetylase (HDAC) phosphorylation increased only after HI (2.0-fold, P < 0.05). In conclusion, exercise intensity regulates PGC-1α mRNA abundance in human skeletal muscle in response to a single bout of exercise. This effect is mediated by differential activation of multiple signalling pathways, with ATF-2 and HDAC phosphorylation proposed as key intensity-dependent mediators.
Introduction
The transcription factor coactivator peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is a critical regulator of mitochondrial biogenesis, cellular respiration and energy substrate utilization (Wu et al. 1999; St-Pierre et al. 2003; Wende et al. 2005; Rohas et al. 2007; Wende et al. 2007). The expression of PGC-1α is decreased in skeletal muscle of type 2 diabetic patients (Mootha et al. 2004) and with physical inactivity (Timmons et al. 2006). Conversely, acute skeletal muscle contraction increases PGC-1α mRNA abundance (Pilegaard et al. 2003; Russell et al. 2005; Vissing et al. 2005) and exercise training is associated with elevated PGC-1α protein content (Burgomaster et al. 2008). Nevertheless, relatively little is known about the intracellular mechanisms that regulate PGC-1α expression in human skeletal muscle.
Characterization of the promoter region of the PPARGC1A gene reveals conserved myocyte enhancer factor 2 (MEF2) and cyclic-AMP response element (CRE) sequences (Esterbauer et al. 1999; Irrcher et al. 2008), which regulate PGC-1α transcription in response to physiological stimuli (Czubryt et al. 2003; Handschin et al. 2003; Irrcher et al. 2008). Mutations of the MEF2 and CRE sites ablate the responsiveness of the PGC-1α promoter to motor nerve stimulation in rodent skeletal muscle (Akimoto et al. 2004) and this suggests that transcriptional activation of PGC-1α is dependent on functional interactions with regulatory factors on the MEF2 and CRE cis elements on the PGC-1α promoter (Akimoto et al. 2008). Several targets have been implicated as regulatory partners for MEF2 and CRE binding sites (Akimoto et al. 2005; Liu et al. 2005; Thomson et al. 2008; Wright et al. 2007a). In rodent muscle cells, the binding of activating transcription factor (ATF)-2, CRE binding protein (CREB) and class IIa histone deacetylases (HDACs) have been reported to modulate PGC-1α gene transcription (Akimoto et al. 2005, Akimoto et al. 2008; Irrcher et al. 2008; Wright et al. 2007a).
The activation of AMP-activated protein kinase (AMPK), calcium/calmodulin-dependent protein kinase (CaMK) II and p38 mitogen-activated protein kinase (MAPK) signalling cascades are well characterized upstream modulators of PGC-1α expression in skeletal muscle (Akimoto et al. 2005; Jager et al. 2007; Wright et al. 2007a). These cascades activate downstream regulatory factors (Akimoto et al. 2005; Liu et al. 2005; Thomson et al. 2008; Wright et al. 2007a), and in the case of AMPK (Jager et al. 2007) and p38 (Puigserver et al. 2001), also directly phosphorylate PGC-1α, thereby increasing transcriptional activation of the PGC-1α promoter through an auto-regulatory mechanism. Given that skeletal muscle energy flux during contraction is intensity dependent, it is unsurprising that signal transduction cascades are differentially regulated by the intensity of an acute exercise challenge (Widegren et al. 2000; Wojtaszewski et al. 2000; Rose et al. 2006). Whether differential activation of these signalling cascades could lead to an intensity-dependent regulation of downstream gene targets and transcriptional processes is unknown.
Devising exercise modalities that are especially metabolically effective and time efficient in upregulating PGC-1α, which should then optimally stimulate gene expression to increase insulin sensitivity and fatty acid oxidation, has been proposed as a key treatment of metabolic disease (Benton et al. 2008). Thus, we focused on the modulation of PGC-1α mRNA abundance by the intensity of exercise, which in itself is a key determinant in the nature of adaptation to regular physical activity (Dudley et al. 1982). The purpose of this study was to determine if the intensity of a single bout of exercise differentially regulates the expression of PGC-1α and its regulatory signalling cascades in human skeletal muscle. Having observed an intensity-dependent increase in PGC-1α mRNA abundance during recovery from a single bout of isocaloric exercise, we focused on the regulatory factors known to regulate the PGC-1α promoter, namely ATF-2, CREB and class IIa HDACs and their upstream kinases. We hypothesized that the intensity-dependent effect of exercise on PGC-1α gene expression was mediated by differential activation of signal cascades purported to regulate transcription through PGC-1α promoter activity, thereby forming part of a signal transduction network that regulates PGC-1α transcription (see Fig. 6).
Figure 6. Proposed model of PGC-1α gene activation in human skeletal muscle by a single bout of exercise.

Acute myofibrillar contraction results in the phosphorylation (p) and activation of AMPK, CaMKII and p38 MAPK through molecular sensing of increased ATP turnover, increased calcium release from the sarcoplasmic reticulum, and mechanical stress, respectively. AMPK and CaMKII phosphorylate class IIa HDACs, leading to their nuclear exclusion and relieving the inhibitory effect of HDACs on MEF2 transcriptional activity at the MEF2 binding sequence on the PGC-1α promoter. AMPK and CaMKII also phosphorylate and activate CREB, resulting in an activating effect on the CRE sequence of the PGC-1α promoter. p38 MAPK phosphorylates and activates ATF-2, that in turn acts on the same CRE site resulting in transcriptional activation. These combined effects on the MEF2 and CRE sequences result in increased PGC-1α promoter activity and increased PGC-1α gene transcription. The intensity of the exercise bout may modulate the magnitude of this response with the phosphorylation of class IIa HDACs and ATF-2 being sensitive to the intensity of contraction, analogous to high intensity exercise (HI).
Methods
Participants and ethical approval
Eight healthy, sedentary males volunteered to participate in the study (24 ± 1 years, 1.79 ± 0.02 m, 80.3 ± 2.2 kg, 25.1 ± 1.2 kg m−2, 16.0 ± 3.3% body fat). All experimental procedures were approved by the Dublin City University Research Ethics Committee in accordance with the Declaration of Helsinki. Each participant underwent a thorough medical screening and provided written informed consent prior to participation. Subjects were physically inactive for at least 6 months and peak oxygen uptake (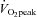 , 3.23 ± 0.18 l min−1) was determined by indirect calorimetry (Vmax 29C, SensorMedics, Yorba Linda, CA, USA) using an incremental protocol on an electronically braked stationary cycle ergometer (Ergoline 900, SensorMedics).
, 3.23 ± 0.18 l min−1) was determined by indirect calorimetry (Vmax 29C, SensorMedics, Yorba Linda, CA, USA) using an incremental protocol on an electronically braked stationary cycle ergometer (Ergoline 900, SensorMedics).
Experimental design
Subjects were required to complete two isocaloric acute exercise trials at (i) 40% (low intensity; LO) and (ii) 80% (high intensity; HI) 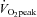 , on separate occasions in random order separated by at least 1 week. Seven days before the first experimental trial, the power outputs required to elicit 40% and 80%
, on separate occasions in random order separated by at least 1 week. Seven days before the first experimental trial, the power outputs required to elicit 40% and 80%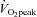 were verified. For the main experimental trials, subjects reported to the Metabolic Physiology Research Unit after an overnight fast and had a resting muscle biopsy taken (Pre). Subjects then consumed a high carbohydrate breakfast and remained in the laboratory for 4 h, at which point they started the exercise bout. Participants began cycling on a stationary ergometer (cadence at 70–75 r.p.m.) and continued until 400 kcal (1674 kj) were expended, as determined by indirect calorimetry monitored on a minute-by-minute basis (Weir, 1949). A muscle biopsy was taken immediately (+0 h) and 3 h after the cessation of exercise (+3 h). During this 3 h of recovery, subjects remained in the laboratory and were permitted to consume only water ad libitum. After the third biopsy (+3 h), subjects were provided with a standard meal, after which they were free to leave the laboratory. Another meal and snack were provided to eat later that evening and water was allowed ad libitum. No other food or beverages were allowed. The following morning, subjects returned to the laboratory at the same time after an overnight fast for a final muscle biopsy at 19 h after cessation of exercise (+19 h).
were verified. For the main experimental trials, subjects reported to the Metabolic Physiology Research Unit after an overnight fast and had a resting muscle biopsy taken (Pre). Subjects then consumed a high carbohydrate breakfast and remained in the laboratory for 4 h, at which point they started the exercise bout. Participants began cycling on a stationary ergometer (cadence at 70–75 r.p.m.) and continued until 400 kcal (1674 kj) were expended, as determined by indirect calorimetry monitored on a minute-by-minute basis (Weir, 1949). A muscle biopsy was taken immediately (+0 h) and 3 h after the cessation of exercise (+3 h). During this 3 h of recovery, subjects remained in the laboratory and were permitted to consume only water ad libitum. After the third biopsy (+3 h), subjects were provided with a standard meal, after which they were free to leave the laboratory. Another meal and snack were provided to eat later that evening and water was allowed ad libitum. No other food or beverages were allowed. The following morning, subjects returned to the laboratory at the same time after an overnight fast for a final muscle biopsy at 19 h after cessation of exercise (+19 h).
Muscle biopsies
Each muscle biopsy was taken from the m. vastus lateralis under local anaesthesia. An area of skin was anaesthetized with 2% lidocaine and a small (0.5 cm) incision made. The biopsy needle was inserted into the muscle and, with suction applied, ∼100 mg of tissue was removed. A fresh incision was made for each of the eight biopsies, at least 2 cm from a previous biopsy site. Muscle samples were snap-frozen in liquid nitrogen and stored at −80°C until analysis.
Dietary control
Pre-exercise preparation was the same for each exercise trial. Subjects were asked to abstain from caffeine and alcohol and refrain from physical activity of any kind for 24 h prior to testing. Subjects were asked to keep a one-day food diary on the day prior to the first experimental trial and asked to repeat the content and pattern of dietary intake on the day preceding the second experimental trial. The dietary intake during each experimental trial was standardized, in terms of food type, total energy intake and macronutrient composition, for each participant. Total daily energy expenditure was estimated by the Harris–Benedict equation (Harris & Benedict, 1919), multiplied by a physical activity factor (1.4) with 400 kcal added to account for the exercise trial (Durnin, 1996). Three meals, each with 30% of predicted total energy expenditure, were provided during the day, with the remaining 10% energy provided with an evening snack. The daily energy intake (36 kcal kg−1; 151 kj kg−1) was composed of 6.0 g kg−1 carbohydrate, 0.8 g kg−1 fat, and 1.2 g kg−1 protein. Hence, the percentage contribution of each macronutrient to total energy intake was 67% carbohydrate, 20% fat and 13% protein.
Muscle glycogen
Frozen muscle samples (∼10 mg) were lyophilized, dissected free of connective tissue, weighed and hydrolysed with 1 m HCl by incubation at 100°C for 2 h and then neutralized with 0.67 m NaOH. Glycogen concentrations were determined by a standard enzymatic technique with fluorometric detection (Passonneau & Lauderdale, 1974).
mRNA abundance
Total RNA was isolated from ∼20 mg crude tissue using TRI reagent (Sigma-Aldrich, UK) as per the manufacturer's instructions. Total RNA concentration was quantified spectrophotometrically at an absorbance of 260 nm (NanoDrop ND-1000 Spectrophotometer, ThermoFisher Scientific, Waltham, MA, USA). The integrity and purity of each RNA sample was verified by gel electrophoresis (RNA 6000 Nano Lab Chip and 2100 Bioanalyzer, Agilent Technologies, Palo Alto, CA, USA) and by measuring the spectrophotometric A260/A280 (>1.8) and A260/A230 (>1.5) ratios. RNA (1 μg) was reverse transcribed to cDNA using the Reverse Transcription System (Promega, Madison, WI, USA) primed with oligo-dT(15) as per the manufacturer's instructions. The cDNA template was stored at −20°C until subsequent analysis. mRNA abundance (30 ng cDNA template per reaction) was determined using quantitative real-time PCR (ABI Prism 7500, Applied Biosystems, Foster City, CA, USA) using Assay-On-Demand primer pairs and probes (P/N 4331182, Assay IDs PGC-1α Hs00173304_m1, PDK4 Hs00176875_m1; Taqman Gene Expression Assays, Applied Biosystems) and Taqman Universal PCR Master Mix (Applied Biosystems). GAPDH mRNA (4333764F, Applied Biosystems) was stable across all time points and used as the housekeeping gene to which target mRNA expression was normalized. The average CT values of the unknown samples were converted to relative expression data using an appropriate standard curve.
Protein quantification
Approximately 25 mg of crude muscle was homogenized in 1 ml of ice-cold homogenization buffer (20 mm Tris (pH 7.8), 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 1% Triton X-100, 10% (w/v) glycerol, 10 mm NaF, 1 mm EDTA, 5 mm sodium pyrophosphate, 0.5 mm Na3VO4, 1 μg ml−1 leupeptin, 0.2 mm phenylmethyl sulfonyl fluoride, 1 μg ml−1 aprotinin, 1 mm dithiothreitol, 1 mm benzamidine, 1 μm microcystin) using a motorized pestle. Homogenates were rotated end-over-end for 60 min at 4°C and centrifuged (12,000 g for 15 min at 4°C), and the protein content of the supernatant was determined by a commercially available detergent-compatible colorimetric assay (Bio-Rad Laboratories, Hercules, CA, USA). An aliquot of muscle homogenate (50 μg protein) was mixed with Laemmli buffer (20% glycerol, 62.5 mmol l−1 Tris-HCl, 2% SDS, 0.00125% bromophenol blue, 2%β-mercaptoethanol). The samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane. Non-specific binding was blocked in a 5% milk/TBS-t (10 mmol l−1 Tris pH 7.5, 100 mmol l−1 NaCl, 0.1% Tween 20) for 2 h at room temperature. Membranes were incubated overnight with primary antibodies directed towards phospho-AMPKα Thr172 (1:1000; Cell Signaling Technology, Beverly, MA; no. 2531), AMPKα (1:1000; Cell Signaling; no. 2532), phospho-acetyl-CoA carboxylase (ACC) Ser79 (1:500; Cell Signaling; no. 3661), ACC (1:1000; Millipore, Billerica, MA, USA; 07-439), phospho-ATF-2 Thr71 (1:500; Cell Signaling; no. 9221), ATF-2 (1:1000; Millipore; 06-326), phospho-CaMKII Thr286 (1:1000; Cell Signaling; no. 3361), CaMKII (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-13082), phospho-CREB Ser133 (1:1000; Cell Signaling; no. 9191), CREB (1:1000; Cell Signaling; no. 9197), phospho-HDAC4/5/7 Ser632/Ser498/Ser486 (1:1000; Cell Signaling; no. 3424), HDAC4 (1:1000; Cell Signaling; no. 2072), HDAC5 (1:1000; Cell Signaling; no. 2082), phospho-p38 MAPK Thr180/Tyr182 (1:1000; Cell Signaling; no. 9211), p38 MAPK (1:1000; Cell Signaling; no. 9212), and GAPDH (1:4000; Santa Cruz; sc-25778). Membranes were washed in TBS-t and incubated with appropriate secondary horseradish peroxidase-conjugated antibodies (1:20000; Bio-Rad), visualized by enhanced chemiluminescence (ECL; GE Healthcare, Arlington Heights, IL, USA) and quantified by densitometry (GS800 Calibrated Imaging Densitometer, Bio-Rad). Samples for each subject from the respective exercise trials were compared in parallel on the same gel, and representative blots are included. GAPDH and total protein abundance of each phosphorylated protein were used for normalization where appropriate. A representative blot for each protein analysed is presented in Fig. 1.
Figure 1. Representative immunoblots.

Representative immunoblots corresponding to phosphorylated protein, total protein expression and loading control (GAPDH) measured before (Pre), immediately after (+0 h) and after 3 (+3 h) and 19 h (+19 h) of recovery from isocaloric (400 kcal) stationary cycle ergometer exercise at either 40% (low intensity, LO) or 80% (high intensity, HI) 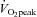 . See text for abbreviations and antibody descriptions.
. See text for abbreviations and antibody descriptions.
Statistical analysis
Experimental data are presented as means ±s.e.m. Data were evaluated using the SigmaStat for Windows v3.11 software package (Systat Software, Inc, San Jose, CA, USA). Two-way (trial × time) repeated measures ANOVA, with Student–Neuman–Keuls post-hoc pair-wise comparisons was performed to identify differences between the two intensities of exercise for variables with serial measurements. Student's paired t test was performed to identify differences between trials for variables with single measurements. The significance level was set at α= 0.05 for all statistical tests.
Results
Energy expenditure and substrate utilization
Total energy expenditure was similar between trials, despite the difference in exercise intensity and total exercise time (P < 0.05) (Table 1). The difference in the rate of energy expenditure (P < 0.05) resulted in a greater reliance on the relative (P < 0.05) and absolute (P < 0.05) contribution of carbohydrate oxidation during the high intensity trial. As expected, the contribution of fat oxidation to total energy expenditure was lower during the high intensity trial (P < 0.05).
Table 1.
Energy expenditure and metabolic responses during isocaloric low and high intensity exercise trials
| LO | HI | |
|---|---|---|
| Total EE (kcal) | 412 ± 11 | 403 ± 1 |
| Rate of EE (kcal min−1) | 6.0 ± 0.3 | 11.5 ± 0.7* |
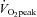 |
38.8 ± 0.4 | 79.4 ± 1.5* |
| Exercise time (min) | 69.9 ± 4.0 | 36.0 ± 2.2* |
| RER | 0.90 ± 0.01 | 0.98 ± 0.01* |
| CHO oxidation rate (g min−1) | 0.9 ± 0.1 | 2.5 ± 0.2* |
| Total carbohydrate oxidized (g) | 64 ± 2 | 89 ± 3* |
| Rate of fat oxidation (g min−1) | 0.23 ± 0.02 | 0.12 ± 0.04* |
| Total fat oxidized (g) | 15 ± 1 | 4 ± 1* |
| Rate of glycogen utilization (mmol (kg dw)−1 min−1) | 1.3 ± 0.2 | 3.1 ± 1.0* |
| Plasma lactate at rest (mm) | 1.12 ± 0.16 | 0.99 ± 0.14 |
| Plasma lactate at termination (mm) | 1.22 ± 0.11 | 7.23 ± 1.07* |
Values are mean ±s.e.m.
Significantly different from low intensity trial (P < 0.05). EE, energy expenditure; CHO, carbohydrate; RER, respiratory exchange ratio.
Muscle glycogen content was similar at baseline (259 ± 17 vs. 249 ± 19 mmol (kg dw)−1 for the LO and HI trials, respectively) and decreased following both exercise trials (176 ± 22 vs. 128 ± 34 mmol (kg dw)−1 for the LO and HI trials, respectively; P < 0.05), but returned to baseline the following morning. The net rate of glycogen utilization was higher during the high intensity trial (1.3 ± 0.2 vs. 3.1 ± 1.0 mmol (kg dw)−1 min−1, for the LO and HI trials respectively, P < 0.05). Plasma lactate concentration was similar at rest (LO, 1.12 ± 0.16 mm; HI, 0.99 ± 0.14 mm), and was unchanged from baseline during LO, but increased to 7.23 ± 1.07 mm at the end of HI (P < 0.05, compared to both baseline and LO).
PGC-1α mRNA abundance
PGC-1α mRNA abundance was elevated 3.8- and 10.2-fold 3 h after LO and HI, respectively (P < 0.05), with an effect of exercise intensity observed between trials (P < 0.05) (Fig. 2). PDK4 mRNA abundance was elevated 6.8–7.2-fold at +3 h in both trials (data not shown), suggesting that intensity-dependent effects on mRNA abundance are gene-specific rather than a generalized exercise effect.
Figure 2. Intensity-dependent regulation of PGC-1α gene expression by a single bout of exercise.

The effect of exercise intensity on PGC-1α mRNA abundance immediately after (+0 h) and during recovery (+3 h and +19 h) from isocaloric (400 kcal) exercise bouts. Open bars represent low intensity trial, LO; filled bars represent high intensity trial, HI. PGC-1α mRNA was normalized using the housekeeping gene GAPDH. Values are means ±s.e.m., n= 8. *Significantly different from baseline within same trial (P < 0.05); †significantly different from LO at same time point (P < 0.05).
Contraction-activated signal cascades
AMPK phosphorylation (Fig. 3A) was similar at baseline, but increased 2.8-fold at +0 h following exercise in the high (P < 0.05) but not low intensity trial, resulting in a difference between trials (P < 0.05). Acetyl-CoA carboxylase β (ACCβ) phosphorylation (Fig. 3B) increased 2.3- and 5.2-fold immediately following exercise for LO and HI, respectively (P < 0.05), resulting in a difference between trials at this time point (P < 0.05).
Figure 3. Greater activation of AMPK signalling by a single bout of high intensity exercise.

The effect of exercise intensity on phosphorylation of AMPK (A) and ACCβ (B) protein immediately after (+0 h) and during recovery (+3 h and +19 h) from isocaloric (400 kcal) exercise bouts. Open bars represent low intensity trial, LO; filled bars represent high intensity trial, HI. Representative immunoblots are shown in Fig. 1. Phosphorylated protein is normalised to total protein content (con) of the respective protein. Values are means ±s.e.m., n= 8. *Significantly different from baseline within same trial (P < 0.05); †significantly different from LO at same time point (P < 0.05).
p38 MAPK phosphorylation (Fig. 4A) was increased at +0 h in both exercise trials (∼2.0-fold, P < 0.05), but the phosphorylation of a downstream transcription factor, ATF-2, a regulator of PGC-1α expression, was increased only in HI (2.4-fold, P < 0.05; Fig. 4B), indicating intensity-dependent regulation of this pathway.
Figure 4. Similar activation of p38 MAPK, but greater activation of ATF-2, by a single bout of high intensity compared to low intensity exercise.

The effect of exercise intensity on phosphorylation of p38 MAPK (A) and ATF-2 (B) protein immediately after (+0 h) and during recovery (+3 h and +19 h) from isocaloric (400 kcal) exercise bouts. Open bars represent low intensity trial, LO; filled bars represent high intensity trial, HI. Representative immunoblots are shown in Fig. 1. Phosphorylated protein is normalised to total protein content (con) of the respective protein. Values are means ±s.e.m., n= 8. *Significantly different from baseline within same trial (P < 0.05); †significantly different from LO at same time point (P < 0.05).
Total CaMKII phosphorylation (summation of βM and γ/δ isoforms; Rose et al. 2006) increased immediately following the high (84%, P < 0.05), but not low intensity trial, with a difference between trials (Fig. 5A; P < 0.05). Phosphorylation of the calcium-dependent transcription factor CREB was reduced at +0 h in HI (−64%, P < 0.05), but also tended to be reduced in LO (−36%, P= 0.104). However, CREB phosphorylation was subsequently elevated at +3 h after both trials (∼80%, P < 0.05; Fig. 5B). Phosphorylation of class IIa HDACs, purported downstream targets of both AMPK and CaMKII signalling and negative regulators of gene transcription, was measured by HDAC4/5/7 phosphorylation (Fig. 5C) and also increased only in HI at +0 h (2.0-fold, P < 0.05), consistent with the intensity-dependent activation of these upstream kinases.
Figure 5. Greater activation of CaMKII and HDACs, but not CREB, by a single bout of high intensity exercise.

The effect of exercise intensity on phosphorylation of CaMKII (A), CREB (B) and class IIa HDAC (C) protein immediately after (+0 h) and during recovery (+3 h and +19 h) from isocaloric (400 kcal) exercise bouts. Open bars represent low intensity trial, LO; filled bars represent high intensity trial, HI. Representative immunoblots are shown in Fig. 1. Phosphorylated protein is normalised to total protein content (con) of the respective protein. Phosphorylated HDAC4/5/7 is normalised to the summed total protein content of HDAC4 and 5. Values are means ±s.e.m., n= 8. *Significantly different from baseline within same trial (P < 0.05); †significantly different from LO at same time point (P < 0.05).
Discussion
In the present study, differential expression of PGC-1α mRNA following an acute bout of isocaloric exercise was associated with intensity-dependent regulation of intracellular signalling cascades. Skeletal muscle contraction activates AMPK, CaMKII and p38 MAPK signalling cascades in an intensity-dependent manner as a result of increased bioenergetic processes, calcium flux and cellular stress (Widegren et al. 2000; Wojtaszewski et al. 2000; Rose et al. 2006). These cascades also directly and indirectly regulate PGC-1α transcriptional activity (Puigserver et al. 2001; Akimoto et al. 2005; Jager et al. 2007; Wright et al. 2007a). Surprisingly, the effect of exercise intensity on skeletal muscle gene expression is poorly described. We hypothesized that altering the metabolic stress (intensity) of an acute bout of exercise would differentially modulate skeletal muscle PGC-1α gene expression, coincident with differential activation of signalling cascades known to regulate the PPARGC1A promoter. PGC-1α mRNA was elevated during recovery from both exercise bouts, with a greater elevation (10.2- vs. 3.8-fold) after high compared to isocaloric low intensity exercise. Phosphorylation of p38 MAPK and CREB was increased to the same extent by both low and high intensity exercise, but the differential expression of PGC-1α mRNA coincided with greater activation of AMPK and CaMKII signalling, activation of ATF-2 and repression of class IIa HDAC activity associated with high intensity exercise (Fig. 6).
Low intensity exercise increases ATP turnover, but not necessarily the AMP/ATP ratio (Howlett et al. 1998), unlike isocaloric high intensity exercise where the rates of energy expenditure and glycogen utilization are higher (Table 1). However, AMPK activity may still be increased during low intensity exercise as ACCβ phosphorylation, a sensitive indicator of skeletal muscle AMPK activity (Chen et al. 2003; Park et al. 2002), was increased following both exercise trials (Fig. 3B). An increase in ACCβ phosphorylation at low intensities of exercise in the absence of an increase in AMPK phosphorylation, as seen in the present study, has been previously reported (Chen et al. 2003; Sriwijitkamol et al. 2007). Our results also demonstrate a similar intensity-dependent effect on CaMKII phosphorylation (Fig. 5A), which is consistent with changes in the amplitude and frequency of calcium oscillations during high intensity contraction (Baylor & Hollingworth, 2003; Rose et al. 2006). However, p38 MAPK phosphorylation was not influenced by the intensity of the exercise bout (Fig. 4A). Next, we characterized the downstream targets of signalling cascades that target the PPARGC1A promoter to investigate the potential mechanisms regulating the observed differential effect on PGC-1α mRNA abundance.
The PPARGC1A promoter contains conserved MEF2 and CRE sequences (Esterbauer et al. 1999; Irrcher et al. 2008) that regulate PGC-1α transcription in response to physiological stimuli (Czubryt et al. 2003; Handschin et al. 2003; Irrcher et al. 2008). MEF2 binds to the PGC-1α promoter on at least two binding sites and modulates PGC-1α expression through MEF2 interaction with HDACs (Czubryt et al. 2003). Association with class IIa HDACs (HDAC 4, 5, 7 and 9) in the basal state represses MEF2 transcriptional activity (Lu et al. 2000) and removal of HDAC repression on MEF2 via HDAC phosphorylation allows MEF2 to act on its binding site in the promoter region of a target gene (Lu et al. 2000; McKinsey et al. 2000). The CRE family of transcription factors includes CREB and ATF-2, both of which regulate PGC-1α expression (Handschin et al. 2003; Akimoto et al. 2005, Akimoto et al. 2008; Wu et al. 2006; Wright et al. 2007a). Differential regulation of PGC-1α mRNA induction in skeletal muscle by the intensity of exercise may be mediated in vivo by these regulatory factors as our data demonstrate that ATF-2 and class IIa HDAC phosphorylation is greater after high intensity exercise, whereas CREB phosphorylation is similarly increased during recovery at both intensities.
AMPK and CaMKII can phosphorylate HDACs in skeletal muscle (McKinsey et al. 2000; Backs et al. 2008; McGee et al. 2008). Both HDAC4 and 5 regulate transcriptional processes in response to phosphorylation by AMPK (McGee et al. 2008) and CaMKII (Backs et al. 2008), and via the same pathways, in association with MEF2, in response to contractile activity (McGee & Hargreaves, 2004; Liu et al. 2005). A causal link exists between the activation of CaMKII by electrical stimulation of cultured muscle cells, its phosphorylation and subsequent nuclear exclusion of HDAC4, thereby relieving repression of MEF2 transcriptional activity (Liu et al. 2005), and in addition, acute exercise increases the association of MEF2 and PGC-1α (McGee & Hargreaves, 2004). This collectively suggests that the activation of AMPK and CaMKII signalling cascades removes HDAC inhibition of MEF2 and promotes PGC-1α transcription. This supports recent evidence for class IIa HDACs export from the nucleus during exercise, thereby removing their repressive action on transcription (McGee et al. 2009). Evidence in the present study suggests that this signalling pathway is intensity-dependent and may partly explain the intensity-related induction of the PGC-1α transcript.
With respect to the CRE motif and regulation of PGC-1α, we observed an increase in CREB phosphorylation in both trials after 3 h of recovery (Fig. 5B), but only activation of ATF-2 after the high intensity trial (Fig. 4B). Activation of CREB via phosphorylation at Ser133 is necessary, but not always sufficient, for stimulus-induced activation of CREB and consequent gene transcription (Mayr & Montminy, 2001). Phosphorylation of CREB has been demonstrated in response to a variety of protein kinases including AMPK, CaMKII, PKA, Akt/PKB and p38 MAPK (Shaywitz & Greenberg, 1999). Therefore, CREB-related transcription factors potentially integrate a number of upstream regulatory signals (Thomson et al. 2008). However, an exercise-dependent decrease in CREB Ser133 phosphorylation was observed immediately after exercise, prior to the increase in CREB phosphorylation during recovery (Fig. 5B). The response of CREB to exercise in human skeletal muscle is not well characterized, but may be either unchanged (Widegren et al. 1998) or repressed (Widegren et al. 2000) immediately after exercise. In rat brain a similar decrease, but subsequent increase, in CREB Ser133 phosphorylation occurs in response to exercise (Ploughman et al. 2007). The biphasic response and delayed increase in CREB activation that we observed may be permissive in sustaining the elevation in PGC-1α mRNA that has been observed up to 8 h after exercise cessation (Vissing et al. 2005).
ATF-2 is a nuclear CRE motif-binding transcription factor that is activated by p38 MAPK and binds to the CRE binding site on the PGC-1α promoter enhancing PGC-1α transcription (Cao et al. 2004). This pathway is critical for the exercise-induced increase in PGC-1α mRNA (Akimoto et al. 2005; Wright et al. 2007b). Contractile activity has been associated with p38 MAPK and ATF-2 phosphorylation, coincident with an increase in PGC-1α mRNA (Akimoto et al. 2005, 2008; Wright et al. 2007b), although the present study is the first to demonstrate this pathway in human skeletal muscle. The acute increase in PGC-1α mRNA induced by p38 MAPK activation or exercise is ATF-2 dependent (Akimoto et al. 2005, 2008), whereas inhibition of CaMKII or p38 MAPK prevents caffeine-induced PGC-1α mRNA induction in an ATF-2-dependent manner (Wright et al. 2007a). In the present study, we observed a similar increase in p38 MAPK phosphorylation in both trials, but a marked increase in ATF-2 phosphorylation after high intensity exercise only. Therefore, other as yet unidentified factors may be involved in the activation of ATF-2 during high intensity exercise.
The complexity of the signalling response in the present study is consistent with a framework wherein multiple cellular signals converge to control PGC-1α expression. In addition, these pathways broadly demonstrate some degree of dependence, cross-talk and redundancy in the regulation of metabolism (Hurley et al. 2005; Murgia et al. 2009; Wright et al. 2007a). A multiple signal/transcription control system would allow for fine-tuning of the adaptive response (Akimoto et al. 2008), as illustrated in this context by the exercise intensity-dependent differential regulation of PGC-1α mRNA induction. The complexity of this regulation is further illustrated by the observation that posttranslational modification of existing PGC-1α protein also regulates PGC-1α mRNA abundance through autoregulatory co-activation of its own promoter (Puigserver et al. 2001; Jager et al. 2007; Canto et al. 2009). PGC-1α is directly phosphorylated by AMPK (Jager et al. 2007) and p38 MAPK (Puigserver et al. 2001), and the exercise-induced activation of these kinases observed in the present study may contribute to the induction of PGC-1α mRNA. This would explain the induction of PGC-1α mRNA during low intensity exercise, where p38 MAPK phosphorylation was similar to that observed during high intensity exercise, despite little AMPK or CaMKII activation. Hence, this mechanism may be permissive for the regulation of PGC-1α mRNA during low intensity exercise, whereas the modulation of regulatory factors of the PGC-1α promoter may be more permissive with higher intensity exercise.
Further work is required to determine if the marked difference in mRNA induction during the present protocols would be manifested as divergent increases in PGC-1α protein content after a period of exercise training at the respective intensities. Exercise at higher exercise intensities is speculated to be necessary to improve indices of metabolic function and gene expression, especially in insulin-resistant and aged muscle (Dipietro et al. 2006; Sriwijitkamol et al. 2007; De Filippis et al. 2008). In addition, the relative, rather than absolute, intensity of exercise is a strong determinant of the PGC-1α response to a single bout, even in trained individuals (Nordsborg et al. 2010). Extrapolation of our single bout effects supports the contention that reduced duration, high intensity exercise is as effective as traditional training (Coyle, 2005; Baar, 2006). The stimulus for adaptation, particularly in terms of substrate metabolism and mitochondrial biogenesis, may be greater when the intensity of the exercise training is higher. This study may provide a molecular insight into findings that first established this principle over a quarter of a century ago (Dudley et al. 1982): ‘for the same adaptive response, the length of daily exercise necessary to bring about the change becomes less as the intensity of training is increased.’ Finally, we chose to limit the caloric expenditure of the exercise bout within a physiologically relevant range consistent with industry exercise guidelines (ACSM, 1998). High intensity repeated sprint training activates similar signalling pathways and acutely increases PGC-1α mRNA abundance (Gibala et al. 2009), but does not utilize as many calories as longer duration endurance exercise (Gibala & McGee, 2008). This means that the efficacy of this type of training is reduced if the goal of training is weight loss. However, high intensity endurance exercise may expend sufficient calories, require less time commitment and result in more pronounced cellular adaptations than low intensity recreational exercise.
In conclusion, a single bout of exercise at high intensity resulted in a greater elevation of PGC-1α mRNA abundance during recovery compared to isocaloric low intensity exercise. We propose that this response was mediated by intensity-dependent regulation of ATF-2 and class IIa HDAC activity, and downstream of activation of the established contraction-induced signalling kinases AMPK and CaMKII, but independent of the activation of p38 MAPK, which was similarly activated by both intensities. Furthermore, CREB phosphorylation may prolong the activation of PGC-1α transcription in the hours after exercise.
Acknowledgments
This study was supported by an internal grant from Dublin City University, the Irish Research Council for Science, Engineering and Technology, the European Foundation for the Study of Diabetes, the European Research Council (Advance Grant to J.R.Z.), the Swedish Research Council, the Swedish Diabetes Association, the Foundation for Scientific Studies of Diabetology, the Strategic Research Foundation, and the Commission of the European Communities (Contract No LSHM-CT-2004-005272 EXGENESIS). The authors would like to acknowledge the excellent technical assistance provided by Paul O’Connor and Javier Monedero, Dr Gavin McHugh for performing the muscle biopsies and the research participants who volunteered for this demanding study.
Glossary
Abbreviations
- ACC
acetyl-CoA carboxylase
- Akt/PKB
v-akt murine thymoma viral oncogene homolog 1/protein kinase B
- AMPK
AMP-activated protein kinase
- ATF-2
activating transcription factor 2
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CRE
cyclic-AMP response element
- CREB
cyclic-AMP response element binding protein
- HDAC
histone deacetylase
- MAPK
mitogen-activated protein kinase
- MEF2
myocyte enhancer factor 2
- PDK4
pyruvate dehydrogenase kinase 4
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator-1α
- PKA
protein kinase A
- PPAR
peroxisome proliferator-activated receptor
Author contributions
The exercise testing and main experimental trials were carried out at the School of Health and Human Performance, Dublin City University, Ireland. Analysis of muscle samples were performed at Dublin City University and the Department of Molecular Medicine and Surgery, Section of Integrative Physiology, Karolinska Institute, Stockholm, Sweden. B.E., N.M.M., J.R.Z. and D.J.O’G. conceived, designed and planned the study; B.E., B.P.C., F.M.S., N.McC. and D.J.O’G. performed the exercise testing and main experimental trials; B.E. B.P.C. P.M.G.-R., A.V.C. and N.B. performed the data collection and analysis. All authors contributed to the interpretation and drafting of the manuscript. All authors approved the final manuscript for submission and publication.
Author's present address
B. Egan: Department of Molecular Medicine and Surgery, Section of Integrative Physiology, Karolinska Institute, SE-171 77 Stockholm, Sweden.
References
- ACSM. American College of Sports Medicine Position Stand. The recommended quantity and quality of exercise for developing and maintaining cardiorespiratory and muscular fitness, and flexibility in healthy adults. Med Sci Sports Exerc. 1998;30:975–991. doi: 10.1097/00005768-199806000-00032. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Li P, Yan Z. Functional interaction of regulatory factors with the Pgc-1α promoter in response to exercise by in vivo imaging. Am J Physiol Cell Physiol. 2008;295:C288–C292. doi: 10.1152/ajpcell.00104.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587–19593. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-γ coactivator-1α promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. 2004;287:C790–C796. doi: 10.1152/ajpcell.00425.2003. [DOI] [PubMed] [Google Scholar]
- Baar K. To perform your best: work hard not long. J Physiol. 2006;575:690. doi: 10.1113/jphysiol.2006.117317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol. 2008;28:3437–3445. doi: 10.1128/MCB.01611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J Physiol. 2003;551:125–138. doi: 10.1113/jphysiol.2003.041608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton CR, Wright DC, Bonen A. PGC-1α-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions. Appl Physiol Nutr Metab. 2008;33:843–862. doi: 10.1139/H08-074. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL, Gibala MJ. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol. 2008;586:151–160. doi: 10.1113/jphysiol.2007.142109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signalling in humans. Diabetes. 2003;52:2205–2212. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- Coyle EF. Very intense exercise-training is extremely potent and time efficient: a reminder. J Appl Physiol. 2005;98:1983–1984. doi: 10.1152/japplphysiol.00215.2005. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC-1 α) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis E, Alvarez G, Berria R, Cusi K, Everman S, Meyer C, Mandarino LJ. Insulin-resistant muscle is exercise resistant: evidence for reduced response of nuclear-encoded mitochondrial genes to exercise. Am J Physiol Endocrinol Metab. 2008;294:E607–E614. doi: 10.1152/ajpendo.00729.2007. [DOI] [PubMed] [Google Scholar]
- Dipietro L, Dziura J, Yeckel CW, Neufer PD. Exercise and improved insulin sensitivity in older women: evidence of the enduring benefits of higher intensity training. J Appl Physiol. 2006;100:142–149. doi: 10.1152/japplphysiol.00474.2005. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Abraham WM, Terjung RL. Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol. 1982;53:844–850. doi: 10.1152/jappl.1982.53.4.844. [DOI] [PubMed] [Google Scholar]
- Durnin JV. Energy requirements: general principles. Eur J Clin Nutr. 1996;50(Suppl 1):S2–S9. [PubMed] [Google Scholar]
- Esterbauer H, Oberkofler H, Krempler F, Patsch W. Human peroxisome proliferator activated receptor γ coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics. 1999;62:98–102. doi: 10.1006/geno.1999.5977. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, McGee SL. Metabolic adaptations to short-term high-intensity interval training: a little pain for a lot of gain? Exerc Sport Sci Rev. 2008;36:58–63. doi: 10.1097/JES.0b013e318168ec1f. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, McGee SL, Garnham AP, Howlett KF, Snow RJ, Hargreaves M. Brief intense interval exercise activates AMPK and p38 MAPK signalling and increases the expression of PGC-1α in human skeletal muscle. J Appl Physiol. 2009;106:929–934. doi: 10.1152/japplphysiol.90880.2008. [DOI] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Benedict FG. A Biometric Study of Basal Metabolism in Man. Washington, DC: Carnegie Institution of Washington; 1919. [Google Scholar]
- Howlett RA, Parolin ML, Dyck DJ, Hultman E, Jones NL, Heigenhauser GJ, Spriet LL. Regulation of skeletal muscle glycogen phosphorylase and PDH at varying exercise power outputs. Am J Physiol Regul Integr Comp Physiol. 1998;275:R418–R425. doi: 10.1152/ajpregu.1998.275.2.R418. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Kirwan AF, Hood DA. AMP-activated protein kinase-regulated activation of the PGC-1α promoter in skeletal muscle cells. PLoS ONE. 2008;3:e3614. doi: 10.1371/journal.pone.0003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Randall WR, Schneider MF. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J Cell Biol. 2005;168:887–897. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009;587:5951–5958. doi: 10.1113/jphysiol.2009.181065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SL, Hargreaves M. Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes. 2004;53:1208–1214. doi: 10.2337/diabetes.53.5.1208. [DOI] [PubMed] [Google Scholar]
- McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci U S A. 2000;97:14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St PJ, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia M, Jensen TE, Cusinato M, Garcia M, Richter EA, Schiaffino S. Multiple signalling pathways redundantly control glucose transporter GLUT4 gene transcription in skeletal muscle. J Physiol. 2009;587:4319–4327. doi: 10.1113/jphysiol.2009.174888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordsborg NB, Lundby C, Leick L, Pilegaard H. Relative workload determines exercise induced increases in PGC-1α mRNA. Med Sci Sports Exerc. 2010 doi: 10.1249/MSS.0b013e3181d2d21c. in press doi 10.1249/MSS.0b013e3181d2d21c. [DOI] [PubMed] [Google Scholar]
- Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol. 2002;92:2475–2482. doi: 10.1152/japplphysiol.00071.2002. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement in tissues. Anal Biochem. 1974;60:405–412. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploughman M, Granter-Button S, Chernenko G, Attwood Z, Tucker BA, Mearow KM, Corbett D. Exercise intensity influences the temporal profile of growth factors involved in neuronal plasticity following focal ischemia. Brain Res. 2007;1150:207–216. doi: 10.1016/j.brainres.2007.02.065. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1α. Proc Natl Acad Sci U S A. 2007;104:7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Kiens B, Richter EA. Ca2+–calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol. 2006;574:889–903. doi: 10.1113/jphysiol.2006.111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Hesselink MK, Lo SK, Schrauwen P. Regulation of metabolic transcriptional co-activators and transcription factors with acute exercise. FASEB J. 2005;19:986–988. doi: 10.1096/fj.04-3168fje. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, Winder WW. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–438. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Norrbom J, Scheele C, Thonberg H, Wahlestedt C, Tesch P. Expression profiling following local muscle inactivity in humans provides new perspective on diabetes-related genes. Genomics. 2006;87:165–172. doi: 10.1016/j.ygeno.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Vissing K, Andersen JL, Schjerling P. Are exercise-induced genes induced by exercise? FASEB J. 2005;19:94–96. doi: 10.1096/fj.04-2084fje. [DOI] [PubMed] [Google Scholar]
- Weir J. New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol. 1949;109:1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP. PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol. 2005;25:10684–10694. doi: 10.1128/MCB.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Schaeffer PJ, Parker GJ, Zechner C, Han DH, Chen MM, Hancock CR, Lehman JJ, Huss JM, McClain DA, Holloszy JO, Kelly DP. A role for the transcriptional coactivator PGC-1α in muscle refueling. J Biol Chem. 2007;282:36642–36651. doi: 10.1074/jbc.M707006200. [DOI] [PubMed] [Google Scholar]
- Widegren U, Jiang XJ, Krook A, Chibalin AV, Bjornholm M, Tally M, Roth RA, Henriksson J, Wallberg-Henriksson H, Zierath JR. Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB J. 1998;12:1379–1389. doi: 10.1096/fasebj.12.13.1379. [DOI] [PubMed] [Google Scholar]
- Widegren U, Wretman C, Lionikas A, Hedin G, Henriksson J. Influence of exercise intensity on ERK/MAP kinase signalling in human skeletal muscle. Pflugers Arch. 2000;441:317–322. doi: 10.1007/s004240000417. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol. 2000;528:221–226. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor γ coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007a;282:18793–18799. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007b;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci U S A. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]