

Abstract
Infection by human herpes virus 8 (HHV-8) in childhood is common in the Mediterranean basin; however, classic Kaposi’s sarcoma (KS) is exceedingly rare in children not infected with HIV and not receiving immunosuppression, with only 30 cases reported since 1960. We recently reported two children with autosomal and X-linked recessive primary immunodeficiencies underlying KS in a context of multiple clinical manifestations. These reports suggested that classic KS in otherwise healthy children might also result from inborn errors of immunity more specific to HHV-8. In this paper, we describe three unrelated Turkish children with classic KS born to first-cousin parents. The first patient, a girl, developed KS at two years of age with disseminated cutaneous and mucosal lesions. The clinical course progressed rapidly and the patient died within three months, despite treatment with vincristine. The other two children developed a milder form of KS at the age of nine years, with multiple cutaneous lesions. A boy treated with interferon alpha therapy for 12 months is now in full remission at age 14, two years after treatment. The second girl is currently stabilized with etoposide, which was begun four months ago. None of the three children had any relevant familial history or other clinical features. The occurrence of classic KS in three unrelated Turkish children, each born to consanguineous parents, strongly suggests that autosomal recessive predisposition may drive the rare occurrence of HHV-8-associated classic KS in children.
Introduction
Kaposi’s sarcoma (KS) is an angiogenic inflammatory neoplasm originating from vascular endothelial cells (1). The lesions can affect the skin and various internal organs. KS is actually divided into two entities, which correspond to the two main geographic areas affected: the African Sub-Saharan (endemic) form and the Mediterranean (classic) form (2). KS was first linked to infection by human herpes virus 8 (HHV-8) in 1994 (3). However, only a small minority of HHV-8-infected individuals develop KS. The human factors involved in the HHV-8-driven oncogenic transformation of endothelial cells have recently begun to be deciphered. Indeed, the development of KS may be favoured by acquired immuno-deficiencies, such as HIV co-infection (epidemic form) (4) or transplantation-related immuno-suppression (iatrogenic form) (5). This suggests that classic KS in other patients may occur due to predisposing impaired immunity, whether inherited or acquired.
In children, both epidemic and iatrogenic KS remain relatively rare (6, 7). Endemic and, even more so, classic KS, are exceedingly rare in children. This rarity contrasts with the high HHV-8 seroprevalence in children by 15 years of age. Only 30 children living around or originating from the Mediterranean basin have been reported with classic KS since 1960. Nothing was specified in these paediatric KS reports regarding other severe infections or any specific history of other diseases. Classic KS in childhood presents with a distinct and much more disseminated form than that which is seen in adults. In fact, childhood KS is often lethal within one or two years of evolution, especially when diagnosed in children under seven years of age. It therefore resembles, clinically, epidemic and iatrogenic forms of KS at all ages. None of the families with paediatric KS were reported to be multiplex, and information on parental consanguinity was not provided.
We hypothesize that classic KS of childhood may result from HHV-8-specific Mendelian inborn errors of immunity (8, 9). We reported the first two cases of KS in children with a primary immunodeficiency. The first child had autosomal recessive, complete IFN-γR1 deficiency, presented with chronic mycobacterial disease, and died of disseminated KS (10). The second child had X-linked recessive Wiskott-Aldrich syndrome (WAS) and presented with Epstein-Barr virus-driven B lymphocyte proliferation (EBV-BLP) and disseminated KS (11). He was cured of WAS, EBV-BLP and KS by hematopoietic stem cell transplantation. Although these two children, like those with iatrogenic and epidemic forms of KS, did not present with isolated KS, their cases suggested that isolated KS in other children may be favoured by monogenic inborn errors of immunity, specific to HHV-8. We here report three otherwise healthy, unrelated children with severe, classic KS, who were born to unrelated consanguineous families originating from Turkey.
Case 1
The girl was born in 2001 to Turkish parents, who were first-cousins living in Istanbul, Turkey. Her parents and two brothers were healthy (Figure 1A). The patient had no particular medical history especially regarding vaccination. At the age of two years, a bleeding lesion of the upper lip began to enlarge, resulting in eating difficulties. Fifteen days later, she was examined by Drs Palanduz and Telhan, revealing low weight (<3rd percentile) and a height in the 10th-25th percentile. She had a fever (38° C) and was pale. There was a 10×10 mm hemorrhagic lesion with crusts on the upper lip. Mobile, painless bilateral cervical and sub-mandibular lymph nodes measuring 10×10 mm were present. Smaller axillary and inguinal lymphadenopathies were detected as well as hepatosplenomegaly. Eczematous lesions were present on fingers and feet. Laboratory examination measured hemoglobin at 8.7 g/dl, hematocrit at 26%. There was no laboratory evidence of known immunodeficiency, with normal immunoglobulin levels and negative HIV serology. During her hospitalization black-purple pigmented nodular lesions of a few mm to one cm appeared first around the lips and mouth and later on the ears spreading out to the whole body, arms and legs within a month. Worsening of the anemia, reticulocytosis, and a positive Coombs’ test with erythroid hyperplasia in the bone marrow led to a diagnosis of auto-immune hemolytic anemia. Treatment with steroids was started (2 mg/kg). Under this treatment, the lymph nodes enlarged (Figure 2). KS was diagnosed on the skin biopsies, which showed high numbers of spindle-shaped cells and positive in situ HHV-8 staining. She was treated by vincristine 1.5mg/m2 intravenously every week. Titration of the serum antibodies against latent HHV-8 antigens, performed under treatment, failed. The patient died of severe pulmonary infection three months after the beginning of the treatment and four months after the appearance of the first KS skin lesion.
Figure 1.

Pedigrees of the three unrelated consanguineous families with one child presenting KS (in black). Circles represent women, squares represent men. The child with psoriasis is marked with horizontal bars, the one with recurrent lower respiratory infections with vertical bars.
Figure 2.
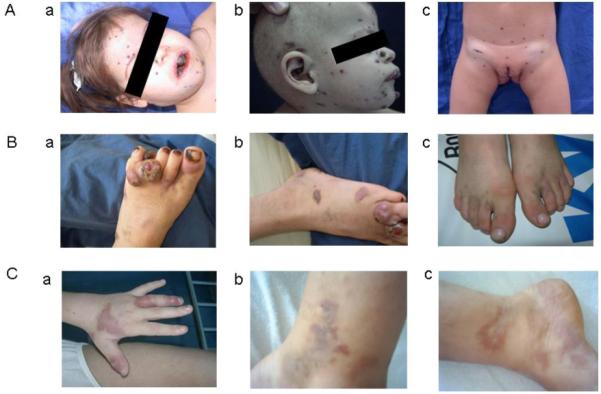
Kaposi’s sarcoma lesions in A. the two-year-old girl a. and b. around the lips, mouth and ears and c. the perinea and inguinal lymph nodes, B. the eleven-year-old boy on the foot a. and b. before and c. after treatment with interferon alpha-2a. C.a. presents the Kaposiform hemangioendothelioma lesions on the left hand in the nine-year-old girl reported in case 3, b. and c. present Kaposi’s sarcoma lesions on feet and thighs appearing 5 years later, all before treatment.
Case 2
The boy was born in 1995 to Turkish parents, first-cousins living in the region of Ankara, in Turkey. His father died in 1998 of pulmonary cancer. One of his brothers presents with psoriasis. His mother, his four other brothers and two sisters are healthy (Figure 1B). He presented no particular medical history, especially regarding vaccination. At the age of nine years, he developed multiple painless, small (10-20 mm), dark purple, cutaneous nodule lesions on the anterior surfaces of both feet. In the next two years, these lesions disseminated to the legs and groin. In 2006, after two years, the boy was examined by Drs Sahin and Ertem, revealing a weight in 10th-25th percentile and a height in the 50th percentile. He had no fever but presented enlarged bilateral cervical (15×9 mm), submandibular (10×10 mm), axillar (9×4 mm) and inguinal (25×8 mm) lymph nodes. There was no hepatosplenomegaly. Laboratory examination showed normal hemoglobin and hematocrit, a white blood count of 6.6 × 109/L (76% lymphocytes) and high immunoglobulin levels for IgG (13g/L) and IgE (890 IU/L). The PPD test was positive (18×21mm); HIV serology was negative. KS was diagnosed on skin lesions showing typical spindle cells and positive in situ HHV-8 staining. The patient had very high IgG titers against latent antigens (1/20,480). All family members except one of his healthy brothers presented IgG against latent antigens (titers from 1/160 to 1/5,120). He was first treated with interferon alpha-2a (IFN-α2a) therapy (4 million U/m2/dose, 3 times a week). Lesions disappeared within six months. IFN-α2a treatment was stopped after 12 months and the child is now in remission, 60 months after diagnosis of the first skin lesion and 24 months after IFN-α2a treatment was discontinued.
Case 3
The girl was born in 1994 to Turkish parents, first-cousins living in Istanbul, Turkey. Her parents and younger sister are healthy, and her younger brother has a history of recurrent lower respiratory infections (Figure 1C). She had no particular medical history, especially regarding vaccination. At the age of nine years, she presented with a massive splenomegaly (10 cm under the left costal margin), pink rashes on the left hand and nodular lesions (3-4 cm) on the right shoulder and thigh (Figure 2C) associated with pancytopenia. She was negative for HIV serology. A skin biopsy showed kaposiform hemangioendeothelioma. Leishmaniosis was diagnosed on bone marrow biopsy, culture and specific IgG (Leishmania infantum) Treatment with liposomal amphotericin B for three weeks resulted in regression of the skin lesions, splenomegaly, pancytopenia and bone marrow abnormality.
Five years later in 2007, she suffered from similar hyperpigmented pink to purple eruptions on bilateral lower extremities (Figure 2C), multiple warts on both hands and splenomegaly (3 cm under the left costal margin). She was examined by Drs Aydogan and Canpolat revealing height and weight in the 25th-50th percentile. A relapse of leishmaniasis was excluded by biopsy and culture. There was no laboratory evidence of known immunodeficiency, with normal immunoglobulin levels, negative HIV serology and a negative PPD test. KS was diagnosed on the skin biopsy showing typical spindle cells and positive in situ HHV-8 staining. The patient had 1/320 titers of IgG against HHV-8 latent antigens. Her two siblings were negative. IFN-α2a therapy (3 million units/week for 5 weeks then 3 million units 3 times a week) was started. After six months, the lesions had not regressed and new lesions appeared. Chemotherapy with vinblastine was then started (6 mg/m2 a week for one month, then every two weeks for five months). The initial lesions regressed in the first three months but new ones appeared. The treatment was switched to etoposide (50 mg/m2/day for 15 consecutive days; then stopped for 15 days). After four rounds, the lesions started to regress.
Discussion
To our knowledge, we have reported on the first three children with classic KS not infected by HIV who have not received any immunosuppressive treatment, born to consanguineous parents. Among the thirty children with classic KS previously described in the literature (12-19), a familial clustering was specified only once (19) in a family from Italy with two cases of KS in an 18 month interval in two generations, suggesting autosomal dominant predisposition. Among the other 24 children, out of the 30 with classic KS for whom the country of origin was available, 10 were Italian, 3 Turkish, 1 Israeli, 1 Tunisian, 1 Egyptian, 1 Lebanese, and 7 were white Americans of unknown descent (12-19). Whether the families were consanguineous or not was not reported, despite the high consanguinity rates within some of those regions (up to 24% in Turkey and Tunisia). The rare occurrence of classic KS in HHV-8-infected children from the Mediterranean basin, the known impact of HIV and immunosuppression on the development of KS, and the two cases of primary immunodeficiency associated with KS previously described (10, 11), strongly suggested that classic KS in childhood may result from HHV-8-specific inborn errors of immunity. These three unrelated cases born to consanguineous Turkish parents add further weight to this hypothesis and suggest that predisposition may be autosomal recessive, at least in a fraction of children.
Acknowledgments
We thank all members of the laboratory of Human Genetics of Infectious Diseases for helpful discussions. We are particularly grateful to the patients and their families for their participation in this study. Sabine Plancoulaine is supported in part by Assistance Publique-Hôpitaux de Paris. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from INSERM, University Paris Descartes, the Agence Nationale pour la Recherche, the BNP Paribas Foundation, the March of Dimes and the Dana Foundation. The Laboratory is also supported by grants from The Rockefeller University Center for Clinical and Translational Science grant number 5UL1RR024143-03 and The Rockefeller University. Jean-Laurent Casanova was an International Scholar of the Howard Hughes Medical Institute.
References
- 1.Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342(14):1027–38. doi: 10.1056/NEJM200004063421407. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss RA. Epidemiology and pathogenesis of Kaposi’s sarcoma-associated herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356(1408):517–34. doi: 10.1098/rstb.2000.0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 4.Gao SJ, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, et al. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. N Engl J Med. 1996;335(4):233–41. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- 5.Mendez JC, Procop GW, Espy MJ, Smith TF, McGregor CG, Paya CV. Relationship of HHV8 replication and Kaposi’s sarcoma after solid organ transplantation. Transplantation. 1999;67(8):1200–1. doi: 10.1097/00007890-199904270-00022. [DOI] [PubMed] [Google Scholar]
- 6.Ziegler JL, Kaongole-Mbidde E. Kaposi’s sarcoma in childhood: an analysis of 100 cases from Uganda and relationship to HIV infection. Int J Cancer. 1996;65(2):200–3. doi: 10.1002/(SICI)1097-0215(19960117)65:2<200::AID-IJC12>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Tamariz-Martel R, Maldonado MS, Carrillo R, Crespo D, Perez-Caballero C, Munoz A. Kaposi’s sarcoma after allogeneic bone marrow transplantation in a child. Haematologica. 2000;85(8):884–5. [PubMed] [Google Scholar]
- 8.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infacy. Science. 2007;317(5838):617–619. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 9.Picard C, Casanova JL, Abel L. Mendelian traits that confer predisposition or resistance to specific infections in humans. Curr Opin Immunol. 2006;18(4):383–90. doi: 10.1016/j.coi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Camcioglu Y, Picard C, Lacoste V, Dupuis S, Akcakaya N, Cokura H, et al. HHV-8-associated Kaposi sarcoma in a child with IFNgammaR1 deficiency. J Pediatr. 2004;144(4):519–23. doi: 10.1016/j.jpeds.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Picard C, Mellouli F, Duprez R, Chedeville G, Neven B, Fraitag S, et al. Kaposi’s sarcoma in a child with Wiskott-Aldrich syndrome. Eur J Pediatr. 2006;165(7):453–7. doi: 10.1007/s00431-006-0107-2. [DOI] [PubMed] [Google Scholar]
- 12.Akman ES, Ertem U, Tankal V, Pamir A, Tuncer AM, Uluoglu O. Aggressive Kaposi’s sarcoma in children: a case report. Turk J Pediatr. 1989;31(4):297–303. [PubMed] [Google Scholar]
- 13.Bisceglia M, Amini M, Bosman C. Primary Kaposi’s sarcoma of the lymph node in children. Cancer. 1988;61(8):1715–8. doi: 10.1002/1097-0142(19880415)61:8<1715::aid-cncr2820610833>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 14.Dutz W, Stout AP. Kaposi’s sarcoma in infants and children. Cancer. 1960;13:684–94. doi: 10.1002/1097-0142(196007/08)13:4<684::aid-cncr2820130408>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Erdem T, Atasoy M, Akdeniz N, Parlak M, Ozdemir S. A juvenile case of classic Kaposi’s sarcoma. Acta Derm Venereol. 1999;79(6):492–3. doi: 10.1080/000155599750010102. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari A, Casanova M, Bisogno G, Cecchetto G, Meazza C, Gandola L, et al. Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol. 2002;39(2):109–14. doi: 10.1002/mpo.10078. [DOI] [PubMed] [Google Scholar]
- 17.Hussein MR. Cutaneous and lymphadenopathic Kaposi’s sarcoma: a case report and review of literature. J Cutan Pathol. 2008;35(6):575–8. doi: 10.1111/j.1600-0560.2007.00844.x. [DOI] [PubMed] [Google Scholar]
- 18.Landau HJ, Poiesz BJ, Dube S, Bogart JA, Weiner LB, Souid AK. Classic Kaposi’s sarcoma associated with human herpesvirus 8 infection in a 13-year-old male: a case report. Clin Cancer Res. 2001;7(8):2263–8. [PubMed] [Google Scholar]
- 19.Zurrida S, Agresti R, Cefalo G. Juvenile classic Kaposi’s sarcoma: a report of two cases, one with family history. Pediatr Hematol Oncol. 1994;11(4):409–16. doi: 10.3109/08880019409140540. [DOI] [PubMed] [Google Scholar]