

Abstract
Objective
p40phox is an important regulatory subunit of NADPH oxidase, but its role in endothelial reactive oxygen species (ROS) production remains unknown.
Methods and Results
Using coronary microvascular endothelial cells isolated from wild-type and p47phox knockout mice, we found that knockout of p47phox increased the level of p40phox expression, whereas depletion of p40phox in wild-type cells increased p47phox expression. In both cases, the basal ROS production (without agonist stimulation) was well preserved. Double knockout of p40phox and p47phox dramatically reduced (≈65%) ROS production and cells started to die. The transcriptional regulation of p40phox and p47phox expressions involves HBP1. p40phox was prephosphorylated in resting cells. PMA stimulation induced p40phox swift dephosphorylation (within 1 minute) in parallel with the start of p47phox phosphorylation. p40phox was then rephosphorylated, and this was accompanied with an increase in ROS production. Depletion of p40phox resulted in ≈67% loss in agonist-induced ROS production despite the presence of p47phox. These were further supported by experiments on mouse aortas stimulated with angiotensin II.
Conclusion
p40phox is prephosphorylated in resting endothelial cells and can compensate p47phox in keeping basal ROS production. Dephosphorylation of p40phox is a prerequisite for agonist-induced p47phox phosphorylation, and p40phox through its dynamic dephosphorylation and rephosphorylation is involved in the regulation of agonist-induced ROS production.
Keywords: NADPH oxidase, endothelial cells, gene regulation, reactive oxygen species
It has been well established that endothelial cells (ECs) express constitutively a multi-subunit NADPH oxidase that generates a low level of O2.− under basal physiological conditions to modulate redox-sensitive intracellular signaling pathways and to maintain normal EC function.1,2 The activity of EC NADPH oxidase can be upregulated by agonists such as PMA (PKC activator), TNFα,3 and angiotensin II (AngII).4,5 Increased reactive oxygen species (ROS) production causes EC dysfunction, which is involved in the pathogenesis of inflammation and many cardiovascular disorders such as atherosclerosis, hypertension, and diabetes.2
NADPH oxidase contains a cytochrome b558 (consisting of 1 member of the Nox family and a p22phox subunit), and at least 4 regulatory subunits, eg, p40phox, p47phox, p67phox, and rac1. Previous studies have shown that p47phox plays a pivotal role in promoting agonist-induced EC NADPH oxidase activation, and knockout of p47phox severely compromises the endothelial ROS response to AngII and TNFα stimulation.4 However, the loss of p47phox did not reduce the basal (without agonist stimulation) ROS production. On the contrary, the basal NADPH-dependent ROS production was significantly higher in coronary microvascular ECs (CMECs) isolated from p47phox knockout (KO) mice compared to wild-type (WT) controls.4-6 The mechanism involved is not clear so far.
The p40phox shares high sequence homology with the p47phox.7,8 It contains similar SH3 domains that could interact with proline-rich domains in p67phox,7 become phosphorylated during NADPH oxidase activation,9-11 and is able to translocate to the plasma membrane in a similar manner to p47phox.12,13 Our hypothesis was that p40phox might compensate p47phox and maintain the basal NADPH oxidase activity in p47phox KO ECs. In this study, we investigated this hypothesis by looking at the relationship between the levels of p40phox and p47phox expression and ROS production in CMECs isolated from p47phox KO and WT mice. We also examined the differences in PMA- and AngII-induced phosphorylation of p40phox and p47phox in primary cultures of mouse CMECs as well as in mouse aortas.
Materials and Methods
Cell Culture
CMECs were isolated from 10-week-old wild-type (WT) and p47phox knockout (KO) mice on a 129sv background.5 All studies were performed in accordance with protocols approved by the Home Office under the Animals (Scientific Procedures) Act 1986 UK. Six hearts were used for each CMEC isolation.
Gene Transfection of CMECs
The full-length human p47phox and p40phox cDNAs (kindly provided by F. Wientjes, UCL) were subcloned with sense and antisense orientations into a mammalian cell expression vector PcDNA3.1 (Invitrogen) and confirmed by molecular sequencing. Transfection of CMEC was undertaken with Lipofectamine 2000 and Plus reagent.14 The success of transfection was examined by either real-time PCR or Western blotting.
ROS Production
Three independent complementary methods were used for ROS measurement. NADPH-dependent O2.− production by EC homogenates was assessed by lucigenin (5 μmol/L)-enhanced chemiluminescence.14 The ROS production by living cells was measured using (1) DCF fluorescence visualized and quantified under confocal microscopy; (2) dihydroethidium fluorescence measured by flow cytometer.
In Vitro 32P Labeling
Cells were labeled with 32P-orthophosphate (100 μCi/mL) at 37°C overnight in phosphate-free medium.3 Cells were stimulated with PMA (100 ng/mL) and harvested for immunoprecipitation and p40phox or p47phox phosphorylation by autoradiography.
Statistics
Data were presented as means±SD from at least 3 experimental results taken from 3 independent cultures for each condition. Comparisons were made by unpaired t test, with Bonferonni correction for multiple testing. P<0.05 was considered statistically significant.
Results
Increased p40phox and p67phox Expression in p47phox KO CMECs
The mRNA expressions of Nox1, Nox2, Nox4, p40phox, p67phox, and NoxO1 were examined by quantitative real-time PCR in WT and p47phox KO CMECs (90% confluence) (Figure 1). Three Nox isoforms, Nox1, Nox2, and Nox4, were all detected in CMECs, and their mRNA expression levels were Nox1<Nox2<Nox4. Comparing to WT levels, p47phox KO cells had significantly higher mRNA levels of Nox2 (1.29±0.1-fold), p40phox (2.3±0.3-fold), p67phox (1.5±0.2-fold), and NoxO1 (1.24±0.1-fold), and the largest increase was in p40phox (Figure 1A). We then examined protein levels of the 3 major regulatory subunit, p40phox, p47phox, and p67phox, and found that p47phox KO cells had significantly higher levels of p40phox (1.7±0.5-fold) and p67phox (1.4±0.7-fold) compared to WT controls (Figure 1B). These changes were accompanied by significant increases in basal NADPH-dependent O2.− production by p47phox KO CMEC homogenates (45±4%) detected by lucigenin-chemiluminescence (Figure 1C, left), and in living adherent cells (15±3%) detected by DCF fluorescence in (Figure 1C, right and supplement Figure IIA, available online at http://atvb.ahajournals.org; all P<0.05).
Figure 1.
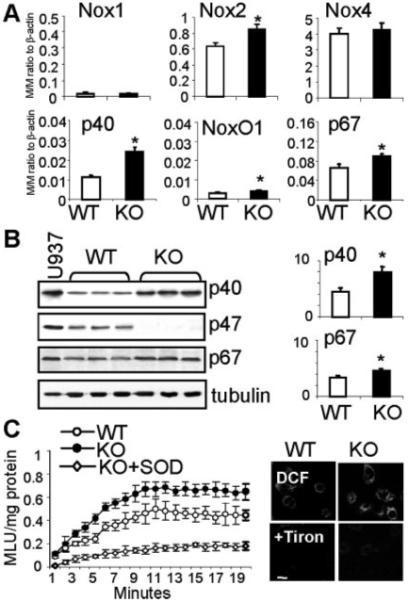
Expression and activity of NADPH oxidase subunits in WT and p47phox KO CMEC. WT: wild-type; KO: p47phox knockout. A) Quantitative real-time PCR. The mRNA levels of Nox1, Nox2, Nox4, p40phox, NoxO1 and p67phox were normalized to the levels of β-actin detected in the same samples and expressed as molar/molar ratio to β-actin. B) Western blotting. For quantification, specific protein bands were normalized to α-tubulin detected in the same samples and expressed as arbitrary units. C) ROS production. Right panel: NADPH-dependent lucigenin-chemiluminescence. MLU: mean light units. Left panel: DCF fluorescence detection of ROS production in adherent CMEC (for quantification see supplement Figure IIA). n=3 separate CMEC isolations. *p<0.05 for KO values versus WT values.
Changes in p47phox Expression Inversely Affected the Expression of p40phox
We then reexpressed p47phox into p47phox KO CMECs. The success of gene transfection was confirmed by real-time PCR (data not shown) and the appearance of p47phox bands on Western blot (Figure 2A). Equal loading of samples was confirmed by reprobing of the p47phox membrane for α-tubulin. Reexpression of p47phox into p47phox KO cells decreased (40±3%) the levels of p40phox expression (Figure 2A) and was accompanied by a reduction in basal O2.− production (Figure 2D; all P<0.05). This observation was further confirmed by depletion of p47phox in WT CMECs using a full length antisense p47phox cDNA. This technique had proven to be very successful in our previous studies6,14 and avoided possible cross-silencing of genes that have the high sequence similarity with p47phox and p40phox. Transient depletion of p47phox, as confirmed by the Western blot, was accompanied by a reciprocal increase (60±6%) in the p40phox expression (Figure 2B) and a substantial increase in basal ROS production (Figure 2D; all P<0.05), which mimicked what we observed in p47phox KO cells. However, overexpression of p47phox into WT cells had no significant effect on the p40phox expression (Figure 2C) and ROS production (data not shown). The p67phox expression was unchanged (Figure 2A through 2C). The enzymatic source of ROS production in WT CMECs after depletion of p47phox was examined using enzyme inhibitors (supplemental Figure IIB). The ROS production was virtually abolished in the presence of a flavoprotein inhibitor, diphenyleneiodonium (DPI), or a cell-permeable O2− scavenger, tiron, and was significantly inhibited (≈70%) by superoxide dismutase (SOD) or an NADPH oxidase inhibitor (apocynin). However, the inhibitors of xanthine oxidase (oxypurinol), NOS (L-NAME, N-ω-nitro-l-arginine methyl ester), or mitochondrial complex I enzymes (rotenone) had no significant effect. Similar results were obtained for p47phox KO CMECs (data not shown).
Figure 2.
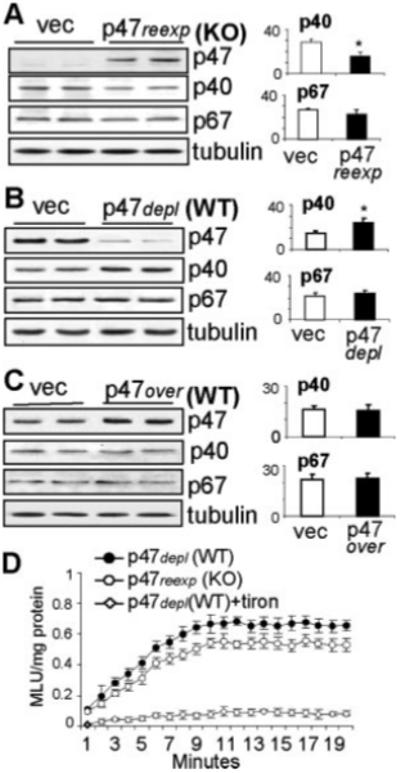
Genetic manipulation of p47phox expression in WT and p47phox KO CMEC. WT: wild-type; KO: p47phox knockout; vec: Vector controls. A-C) Western blotting. For quantification, specific protein bands were normalized to α-tubulin detected in the same samples and expressed as arbitrary units. D) NADPH-dependent lucigenin-chemiluminescence. n=3 separate transfections of independent CMEC isolations. *P<0.05 for indicated values versus vector control values.
The Effects of Transient Overexpression or Depletion of p40phox in WT Cells
Overexpression of p40phox in WT CMECs, as shown by Western blot, had no significant effect on p47phox expression (Figure 3A), but significantly increased (30±2%) the basal ROS production detected by lucigenin-chemiluminescence in cell homogenates and by dihydroethidium (DHE) flow cytometry (right shift) in living cells (Figure 3B). In contrast, transient depletion of p40phox expression in WT CMECs caused a reciprocal significant increase (49±4%) in p47phox protein expression (Figure 3A), which maintained the basal ROS production without significant change compared to the vector controls (Figure 3B). The protein levels of p67phox were not affected (Figure 3A). Compared to vector transfected controls, increased basal ROS production attributable to overexpression of p40phox in WT cells was accompanied by a 22±11% increase in cell number and 46±5% increase in proliferation (MTS assay) (Figure 3C, P<0.05). However, depletion of p40phox in WT cells did not cause significant change in basal ROS production and in cell proliferation, which might be attributable to the presence of p47phox.
Figure 3.
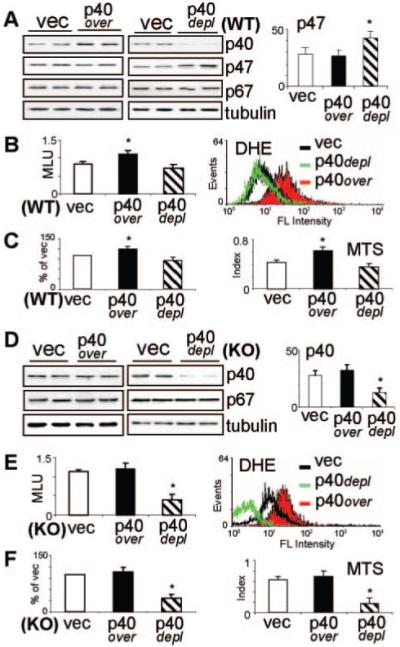
Genetic manipulation of p40phox in CMEC. WT: wild-type (A-C); KO: p47phox knockout (D-F); vec: Vector controls. p40over: overexpression of p40phox; p40depl: depletion of p40phox. A) and D): Western blotting. For quantification, specific protein bands were normalized to α-tubulin detected in the same samples and expressed as arbitrary units. B) and E): ROS production detected in cell homogenates by NADPH-dependent lucigenin-chemiluminescence (left panel) and in living cells by DHE flow cytometry (right panel). C) and F) Cell viability and proliferation detected by counting living cell numbers and expressed as % of vector controls (left panel) and MTS cell proliferation assay expressed as proliferation index (right panel). n=3 separate transfections of independent CMEC isolations. *P<0.05 for indicated values versus vector control values.
The Effects of Transient Overexpression or Depletion of p40phox in p47phoxKO CMECs
In p47phox KO cells that had already a higher level of p40phox (see Figure 1), transfection of p40phox cDNA failed to further increase the p40phox expression (Figure 3D), and there was no significant change in basal O2.− production (Figure 3E) or in cell proliferation (Figure 3F). In contrast, depletion of p40phox expression in p47phox KO cells (double knockout), as shown by the Western blot (Figure 3D), resulted in a dramatic 64±9% reduction (P<0.05) in basal O2− production by cell homogenates and in living cells (Figure 3E). Cells stopped proliferating and died, with the cell number dropping to 35±11%, and the proliferation index (MTS assay) reduced to 27±7% of the vector controls (Figure 3F).
The Kinetics of PMA-Induced p40phox Dephosphorylation and Rephosphorylation and ROS Production
The p40phox was already prephosphorylated in resting cells (time 0). PMA stimulation induced a swift dephosphorylation of p40phox within the fist minute, followed by a progressive rephosphorylation up to 30 minutes of PMA stimulation (Figure 4A). In contrast, p47phox had very little prephosphorylation in resting cells. PMA induced p47phox phosphorylation, which started in the first minute, peaked at ≈15 minutes, and then remained constant (Figure 4A). We then examined the ROS production in the same cells. In WT cells, PMA significantly increased the O2.− production with a time course curve similar to the curve of p47phox phosphorylation (Figure 4B). However, in p47phox KO cells, PMA initially reduced the ROS production in parallel to the kinetic of p40phox dephosphorylation. Although the ROS production in p47phox KO cells recovered slowly afterward, it was only 52±7% of the WT level at 30 minutes of PMA stimulation. Depletion of p40phox in WT cells so severely damaged PMA-induced ROS production such that it was only 47±8% of the WT level at 30 minutes of PMA stimulation (Figure 4B). The relationship between p40phox and p47phox phosphorylation in response to PMA (30 minutes) stimulation was further examined by immunoprecipitation of p40phox and p47phox followed by immunoblotting using phos-serine specific antibodies. We found that a full phosphorylation response of p40phox and p47phox to agonist stimulation requires the presence of both subunits. Knockout of p47phox significantly reduced (≈42%) PMA (30 minutes)-induced p40phox serine-phosphorylation, and depletion of p40phox also severely compromised (≈50%) PMA-induced p47phox serine-phosphorylation (Figure 4C).
Figure 4.
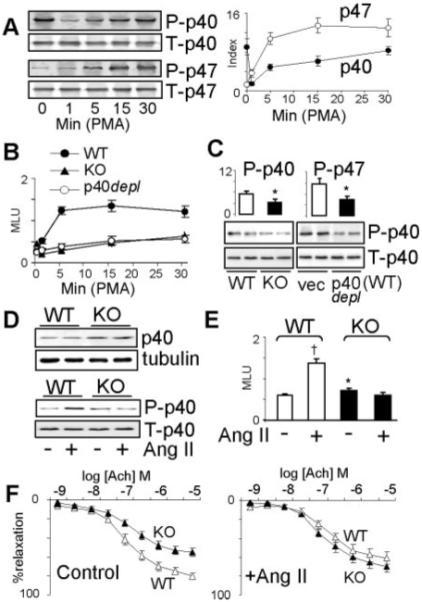
Agonist-induced p40phox and p47phox phosphorylation and ROS production.: CMEC (A-C); Aortic rings (D-F). WT: wild-type; KO: p47phox knockout; vec: Vector controls. p40depl: depletion of p40phox. P-p40: phosphorylated p40phox; T-p40: Total p40phox. For the detection of phosphorylation, p40phox or p47phox were immunoprecipitated down and the loading control of IP products was calculated to the equal levels of total p40phox or p47phox in different samples. The phosphorylations of p40phox or p47phox were detected by 32P autoradiography (A) or immunoblot using phos-serine specific monoclonal antibody (B and D lower panel). Total p40phox or p47phox were detected by immunoblot. For quantification, the levels of phosphorylation bands were normalized to the bands of the total protein detected in the same sample. A) Time course of PMA stimulation. B) Time course of PMA-induced NADPH-dependent ROS production detected by lucigenin-chemiluminescence. C) Differences in serine phosphorylation after 30 min. of PMA stimulation. n=3 separate experiments (A-C). D) Upper panel: Immunoblot for the difference in p40phox expression between aortas isolated from WT and p47phox KO mice. Lower panel: Differences in Ang II-induced p40phox serine phosphorylation between WT and p47phox KO aortas. E) Differences in NADPH-dependent ROS production between WT and p47phox KO aortas stimulated with or without Ang II. The results were expressed as mean light units (MLU) per mg protein. F) Endothelial-dependent vessel relaxation to acetylcholine (Ach) with or without Ang II stimulation. n=6 mice (D-F). *P<0.05 for indicated values versus WT values or vector values. †P<0.05 for indicated values versus values without AngII in WT group.
Effects of Acute Ang II Treatment on Aortas Isolated From WT and p47phox KO Mice
Compared to WT controls, aortas from p47phox KO mice had higher levels of p40phox expression (Figure 4D, upper panels) and ROS production (Figure 4E) and reduced endothelium-dependent vessel relaxation to acetylcholine (Ach; Figure 4F, left). Acute Ang II (200 nmol/L, 30 minutes) treatment of WT vessels increased p40phox serine phosphorylation (Figure 4D, lower) and the ROS production (Figure 4E), and these were accompanied by a reduced endothelium-dependent vessel relaxation (Figure 4F, right). However, these Ang II effects were absent in p47phox KO vessels.
The Roles of Transcription Factor HBP1 in the Regulation of p40phox and p47phox Expression
Compared to WT cells, the levels of HBP1 (HMG box-containing protein 1) were significantly higher (≈2.3 folds) in p47phox KO cells (Figure 5A, left), and were significantly lower in p40phox depleted WT CMECs (Figure 5A, right). The role of HBP1 in the transcriptional regulation of p40phox and p47phox expression was further examined by transient in vitro knockdown of HBP1 using shRNA15 in human microvascular ECs (HMEC1). Knockdown of HBP1, as shown by the Western blot, resulted in a significant increase in p47phox expression and this was accompanied with a significant reduction in p40phox expression (Figure 5B)
Figure 5.

The role of HBP1 in the regulation of p40phox and p47phox expression and p40phox regulation of EC ROS production. WT: wild-type; KO: p47phox knockout; vec: Vector controls. p40depl: depletion of p40phox. A) HBP1 expression in CMEC. For quantification, HBP1 bands were normalized to α-tubulin detected in the same samples and expressed as arbitrary units. B) The effect of knockdown HBP1 on the levels of p40phox and p47phox expression in HMEC1. *P<0.05 for indicated values versus WT value or vector values of the same protein. n=3 independent experiments. C) Schematic diagram of the proposed concept of p40phox regulation of NADPH oxidase activity through its dynamic phosphorylation. Pre-phosphorylated p40phox contributes to the basal ROS production and inhibits p47phox phosphorylation. PMA stimulation causes p40phox dephosphorylation, which allows p47phox phosphorylation to happen. Once the p47phox is phosphorylated, the rephosphorylation of p40phox synergizes p47phox effects and results in the full activation of NADPH oxidase.
Discussion
The Relationship Between the Levels of p40phox and p47phox Expressions in ECs
NADPH oxidase contains at least 4 major regulatory subunits: p40phox, p47phox, p67phox, and rac. The canonical view of the NADPH oxidase activation is that it requires the association of all essential regulatory subunits, ie, p47phox, p67phox, and Rac1 with cytochrome b558.16 However, this view has been changed by reports that in a cell-free system, NADPH oxidase could be activated in the total absence of p47phox if high concentrations of p67phox and rac were present, albeit at a lower maximal rate than in the presence of p47phox.17,18 Previously, we have also shown that p47phox KO CMECs had a slightly but significantly higher basal NADPH-dependent O2− production, but the underlying mechanisms were not clear at that time.6 Here we report that the levels of p40phox and p67phox are indeed elevated at both mRNA and protein levels in p47phox KO CMECs.
It is known that p40phox has high sequence homology with p47phox.7 Based on molecular similarity, it seemed possible for p40phox to replace p47phox and to maintain the basal O2− production in p47phox KO ECs. This hypothesis was confirmed by gene transfection experiments showing that in vitro depletion of p47phox in WT CMECs increased p40phox expression and the basal ROS production, whereas reexpression of p47phox into p47phox KO CMECs reduced p40phox expression, which decreased the basal ROS production back to the WT level. The physiological significance of our findings is not only to ascribe a molecular consequence to the loss of p47phox, but also to define an important compensatory mechanism involved in the regulation of NADPH oxidase activity in ECs.
The Role of p40phox in EC Basal ROS Production and Cell Proliferation
p40phox was originally discovered in the complex copurified and coimmunoprecipitated down with p47phox and p67phox.19 Despite extensive studies describing the interaction of p40phox with p67phox and p47phox in cell-free assays,13,16,19,20 the role of p40phox in living cells has not been well defined. Paradoxically there are virtually as many reports on an inhibitory role20,21 as on an activating role9,10 of p40phox on NADPH oxidase activation. Our current study, using a series of experiments of in vitro overexpression or depletion of p40phox, provides evidences that p40phox is critically involved in the regulation of EC basal ROS production required for EC growth. Overexpression of p40phox in WT CMECs increased basal levels of ROS production, which was accompanied by an increase in cell proliferation. However, depletion of p40phox in WT ECs had no significant effect on both basal levels of ROS production and cell proliferation, presumably because of the compensatory effect from increased p47phox expression. Double knockout of both subunits ie, depletion of p40phox in p47phox KO ECs caused a massive drop in basal ROS production, which was accompanied by cell proliferation arrest and cell death. Different from Nox2, Nox4 does not exhibit a binding site for p47phox, and the known regulatory subunits are not required for its activity.22 Although Nox4 has been found to be involved in mediating insulin-induced cell differentiation in preadipocytes,23 its levels were not affected by the p47phox knockout. The mRNA of a newly discovered homologue of p47phox, NoxO1,24 was also detected in CMECs and was slightly increased in p47phox KO ECs. However, its expression levels were very low compared to the levels of p40phox, and it was not able to compensate the loss of p47phox. Put together, our results strongly suggest that as long as 1 subunit, p40phox or p47phox, is present in ECs, that is enough to maintain the basal ROS production from NADPH oxidase and to keep ECs alive and proliferating. However, the loss of both p40phox and p47phox severely compromises the basal ROS production and EC survival.
Dynamic Phosphorylation and Dephosphorylation of p40phox in ECs
An important discovery from the current study is the prephosphorylation of p40phox in resting ECs, and the dynamic dephosphorylation and rephosphorylation of p40phox during PMA stimulation. Interestingly, one recent study reported that p40phox was also in a basal phosphorylated state in resting neutrophils and undergoes further phosphorylation on multiple sites by PKC stimulation. They found in a cell-free assay that both phosphorylated and nonphosphorylated p40phox were able to interact with p47phox and p67phox, but only phosphorylated p40phox inhibited NADPH oxidase activation if added before full activation of the enzyme.21 Based on this previous report and our experimental results, we suggest that prephosphorylated p40phox interacts with unphosphorylated p47phox in resting ECs to keep the NADPH oxidase activity at low but optimized basal levels.
PMA is often used to bypass cell surface receptors and directly causes PKC-dependent phosphorylation of p47phox. Interestingly, p40phox has also been shown to be phosphorylated by PKC at its serines.11 In the present study, we have extended this knowledge by showing a rapid dephosphorylation followed by a progressive rephosphorylation of p40phox when ECs were stimulated with PMA. The biphasic phosphorylation of p40phox has never been reported, and this may contribute to the complexity of its roles in NADPH oxidase activation and the discrepancy between previous studies.9,10,20,21 Although the mechanisms of p40phox in EC Nox2 regulation requires further investigation, our hypothesis (Figure 5C) is that prephosphorylated p40phox inhibits p47phox phosphorylation. Agonist stimulation induces a rapid p40phox dephosphorylation which allows p47phox phosphorylation to occur and to bind to p22phox. However, once the p47phox is phosphorylated, rephosphorylation of p40phox (maybe at different sites) synergizes the action of phosphorylated p47phox to further promote EC ROS production. The optimal NADPH oxidase response to agonist stimulation requires the presence of both p47phox and p40phox, and either depletion of p40phox or knockout of p47phox severely compromises agonist-induced O2.− production. The pathophysiological significance of p40phox in the regulation of vascular ROS production and endothelial function was further demonstrated by experiments on WT and p47phox KO aortic vessels stimulated with or without Ang II. In p47phox KO vessels, acute Ang II stimulation failed to induce p40phox phosphorylation, and there was no increase in O2.− production and the endothelium-dependent vessel relaxation to Ach was well preserved as compared to WT controls. Superoxide anions are dismutated to H2O2. Recently, endothelial H2O2 production and H2O2-mediated hyperpolarization response to Ach have been found to be closely coupled to nitric oxide synthases (NOSs) system.25 The potential relationship between p40phox and NOSs system requires further investigation.
The HBP1 is a member of the HMG box family transcriptional factors26 and has been found to play a role in transcriptional repression of p47phox gene.27 The promoter of p47phox gene contains 6 adjacent tandem high-affinity HBP1 binding sites, which are required for the transcriptional repression.27 HBP1 has also been reported to enhance promoter activity of genes with lower affinity or a single HBP1 binding site.28,29 Interestingly, the promoter region of p40phox has also a HBP1 binding site, which makes p40phox a possible candidate for HBP1 to activate.27 We propose that HBP1 may play a dual roles (ie, to repress p47phox and in the mean time to promote p40phox) and to regulate ROS production by NADPH oxidase. This hypothesis is supported by our results that knockdown of HBP1 significantly increased the p47phox expression and reduced p40phox expression. There is also a feedback mechanism from p47phox to HBP1 because knockout of p47phox remarkably increased HBP1 expression. Although HBP1 may represent a transcriptional mechanism involved in the regulation of ROS production by NADPH oxidase in ECs, more detailed investigations are required to fully address this question.
In summary, we have reported, for the first time, a critical role of p40phox in the regulation of basal and agonist-induced ROS production by NADPH oxidase in ECs. The p40phox is prephosphorylated in resting ECs and plays an inhibitory role to keep the basal ROS production, and is able to substitute for p47phox in p47phox KO ECs. p40phox undergoes rapid dephosphorylation and then rephosphorylation when ECs are stimulated with PMA, and the biphasic phosphorylation of p40phox is involved in the regulation of NADPH oxidase activity. Depletion of p40phox resulted in a loss in agonist-induced EC ROS production despite the presence of p47phox. Inhibition of p40phox phosphorylation may have a potential therapeutic application in treating EC dysfunction-related diseases.
Supplementary Material
Acknowledgments
Sources of Funding
This work is supported by the Wellcome Trust (grant 07863/Z/05/Z) and the Biotechnology and Biological Sciences Research Council, UK (grant BB/D009510/1).
Footnotes
Disclosures
None.
References
- 1.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 2.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J-M, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25:2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J-M, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II: Role of the p47phox subunit. J Biol Chem. 2003;278:12094–12100. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- 5.Li J-M, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2− production, vascular tone, and mitogen-activated protein kinase activation. Circulation. 2004;109:1307–1313. doi: 10.1161/01.CIR.0000118463.23388.B9. [DOI] [PubMed] [Google Scholar]
- 6.Li J-M, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, Shah AM. Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α. Circ Res. 2002;90:143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- 7.Wientjes FB, Hsuan JJ, Totty NF, Segal AW. p40phox, a third cytosolic component of the activation complex of the NADPH oxidase to contain src homology 3 domains. Biochem J. 1993;296:557–561. doi: 10.1042/bj2960557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matute JD, Arias AA, Dinauer MC, Patiño PJ. p40phox: The last NADPH oxidase subunit. Blood Cells Mol Dis. 2005;35:291–302. doi: 10.1016/j.bcmd.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Bouin A-P, Grandvaux N, Vignais PV, Fuchs A. p40phox is phosphorylated on threonine 154 and serine 315 during activation of the phagocyte NADPH oxidase: Implication of a protein Kinase C-type kinase in the phosphorylation process. J Biol Chem. 1998;273:30097–30103. doi: 10.1074/jbc.273.46.30097. [DOI] [PubMed] [Google Scholar]
- 10.Someya A, Nunoi H, Hasebe T, Nagaoka I. Phosphorylation of p40-phox during activation of neutrophil NADPH oxidase. J Leukoc Biol. 1999;66:851–857. doi: 10.1002/jlb.66.5.851. [DOI] [PubMed] [Google Scholar]
- 11.Grandvaux N, Elsen S, Vignais PV. Oxidant-dependent phosphorylation of p40phox in B lymphocytes. Biochem Biophys Res Commun. 2001;287:1009–1016. doi: 10.1006/bbrc.2001.5665. [DOI] [PubMed] [Google Scholar]
- 12.Dusi S, Donini M, Rossi F. Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox. Biochem J. 1996;314:409–412. doi: 10.1042/bj3140409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stahelin RV, Burian A, Bruzik KS, Murray D, Cho W. Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J Biol Chem. 2003;278:14469–14479. doi: 10.1074/jbc.M212579200. [DOI] [PubMed] [Google Scholar]
- 14.Li J-M, Fan LM, George VT, Brooks G. Nox2 regulates endothelial cell cycle arrest and apoptosis via p21cip1 and p53. Free Radic Biol Med. 2007;43:976–986. doi: 10.1016/j.freeradbiomed.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Kim J, Ruthazer R, McDevitt MA, Wazer DE, Paulson KE, Yee AS. The HBP1 transcriptional repressor participates in RAS-induced premature senescence. Mol Cell Biol. 2006;26:8252–8266. doi: 10.1128/MCB.00604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wientjes FB, Reeves EP, Soskic V, Furthmayr H, Segal AW. The NADPH oxidase components p47phox and p40phox bind to moesin through their PX domain. Biochem Biophys Res Commun. 2001;289:382–388. doi: 10.1006/bbrc.2001.5982. [DOI] [PubMed] [Google Scholar]
- 17.Freeman JL, Lambeth JD. NADPH oxidase activity is independent of p47phox in vitro. J Biol Chem. 1996;271:22578–22582. doi: 10.1074/jbc.271.37.22578. [DOI] [PubMed] [Google Scholar]
- 18.Koshkin V, Lotan O, Pick E. The cytosolic component p47phox is not a sine qua non participant in the activation of NADPH oxidase but is required for optimal superoxide production. J Biol Chem. 1996;271:30326–30329. doi: 10.1074/jbc.271.48.30326. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs A, Dagher M-C, Vignais PV. Mapping the domains of interaction of p40phox with both p47phox and p67phox of the neutrophil oxidase complex using the two-hybrid system. J Biol Chem. 1995;270:5695–5697. doi: 10.1074/jbc.270.11.5695. [DOI] [PubMed] [Google Scholar]
- 20.Sathyamoorthy M, de Mendez I, Adams AG, Leto TL. p40phox down-regulates NADPH oxidase activity through interactions with its SH3 domain. J Biol Chem. 1997;272:9141–9146. doi: 10.1074/jbc.272.14.9141. [DOI] [PubMed] [Google Scholar]
- 21.Lopes LR, Dagher M-C, Gutierrez A, Young B, Bouin A-P, Fuchs A, Babior BM. Phosphorylated p40phox as a negative regulator of NADPH oxidase. Biochemistry. 2004;43:3723–3730. doi: 10.1021/bi035636s. [DOI] [PubMed] [Google Scholar]
- 22.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 23.Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29:239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 24.Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 25.Takaki A, Morikawa K, Tsutsui M, Murayama Y, Tekes E, Yamagishi H, Ohashi J, Yada T, Yanagihara N, Shimokawa H. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. doi: 10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yee AS, Paulson EK, McDevitt MA, Rieger-Christ K, Summerhayes I, Berasi SP, Kim J, Huang C-Y, Zhang X. The HBP1 transcriptional repressor and the p38 MAP kinase: Unlikely partners in G1 regulation and tumor suppression. Gene. 2004;336:1–13. doi: 10.1016/j.gene.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Berasi SP, Xiu M, Yee AS, Paulson KE. HBP1 repression of the p47phox gene: Cell cycle regulation via the NADPH oxidase. Mol Cell Biol. 2004;24:3011–3024. doi: 10.1128/MCB.24.7.3011-3024.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavender P, Vandel L, Bannister AJ, Kouzarides T. The HMG-box transcription factor HBP1 is targeted by the pocket proteins and E1A. Oncogene. 1997;14:2721–2728. doi: 10.1038/sj.onc.1201243. [DOI] [PubMed] [Google Scholar]
- 29.Lin KM, Zhao W-G, Bhatnagar J, Zhao W-D, Lu J-P, Simko S, Schueneman A, Austin GE. Cloning and expression of human HBP1, a high mobility group protein that enhances myeloperoxidase (MPO) promoter activity. Leukemia. 2001;15:601–612. doi: 10.1038/sj.leu.2402071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.