

Abstract
The transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is a master regulator of metabolism in peripheral tissues, and it has been proposed that PGC-1α plays a similar role in the brain. Recent evidence suggests that PGC-1α is concentrated in GABAergic interneurons, so we investigated whether male and female PGC-1α −/− mice exhibit abnormalities in interneuron gene expression and/or function. We found a striking reduction in the expression of the Ca2+-binding protein parvalbumin (PV), but not other GABAergic markers, throughout the cerebrum in PGC-1α +/− and −/− mice. Furthermore, PGC-1α overexpression in cell culture was sufficient to robustly induce PV expression. Consistent with a reduction in PV rather than a loss of PV-expressing interneurons, spontaneous synaptic inhibition was not altered in PGC-1α −/− mice. However, evoked synaptic responses displayed less paired-pulse depression and dramatic facilitation in response to repetitive stimulation at the gamma frequency. PV transcript expression was also significantly reduced in retina and heart of PGC-1α −/− animals, suggesting that PGC-1α is required for proper expression of PV in multiple tissues. Together these findings indicate that PGC-1α is a novel regulator of interneuron gene expression and function and a potential therapeutic target for neurological disorders associated with interneuron dysfunction.
Introduction
Locally projecting interneurons that release the inhibitory neurotransmitter GABA are arguably the most diverse cell population in the brain (Markram et al., 2004; Danglot et al., 2006). While certain interneuron subsets are preferentially affected in psychiatric (Lewis and Gonzalez-Burgos, 2008) and developmental (Di Cristo, 2007) disorders, only a limited number of transcriptional cues by which specific interneuron populations mature and function during early postnatal brain development have been identified. We have previously found that the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is selectively localized to interneuron nuclei by the second week of postnatal life (Cowell et al., 2007). However, little is known about the function of PGC-1α in this neuronal population.
In non-neuronal tissues, PGC-1α has been coined the “master regulator of metabolism” based on its ability to induce gene programs controlling mitochondrial biogenesis and antioxidant production (Lin et al., 2005). PGC-1α is highly expressed in tissues with high energy demands, such as brown adipose tissue (BAT), heart, liver, skeletal muscle, and brain (Puigserver et al., 1998), and PGC-1α is robustly induced under physiological conditions of cold in BAT, fasting in liver, and exercise in skeletal muscle. Key to PGC-1α's differential effects in specific tissues is its ability to coactivate a wide array of transcription factors. As a transcriptional coactivator, PGC-1α facilitates the interactions of transcription factors and their activating proteins to form functional DNA-binding complexes. Thus, its downstream targets are dependent on coexpressed transcription factors.
Accumulating evidence suggests that PGC-1α plays a role in neuroprotection by activating genes involved in reactive oxygen species metabolism (St-Pierre et al., 2006; Cowell et al., 2009). Adult mice lacking PGC-1α exhibit severe neurodegeneration throughout the striatum, cortex, and hippocampus and manifest behavioral abnormalities reminiscent of neurological disorders (Lin et al., 2004; Leone et al., 2005). Reductions in PGC-1α have been observed in brain and muscle from patients with Huntington disease (HD) (Cui et al., 2006; Weydt et al., 2006; Chaturvedi et al., 2009), potentially due to pathogenic interactions of mutant huntingtin with transcriptional regulators of PGC-1α (Cui et al., 2006). In addition, PGC-1α may be involved in the pathologies of Alzheimer's disease (Qin et al., 2009) and Friedreich's ataxia (Marmolino and Acquaviva, 2009). However, given its developmental regulation, we hypothesized that analyses of GABAergic gene expression and function during the first month of life in PGC-1α −/− mice would reveal primary downstream targets for PGC-1α in the brain.
To test our hypothesis, we evaluated the expression of a battery of GABAergic markers using region-specific quantitative RT-PCR and immunohistochemistry. We also investigated alterations in GABAergic synaptic transmission with whole-cell electrophysiology. We found that PGC-1α is required for mRNA and protein expression of the Ca2+-binding protein parvalbumin (PV) throughout the cerebrum, and abnormalities in inhibitory signaling are consistent with loss of PV from perisomatic GABAergic terminals. These studies have revealed a unique role for PGC-1α in interneuron gene expression and function and suggest that disruptions in PGC-1α expression could have profound consequences for interneuron function.
Materials and Methods
Animals.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Two independent lines of PGC-1α knock-out mice (Lin et al., 2004; Leone et al., 2005) were maintained on a C57BL/6J genetic background and housed two to five in a cage at 26 ± 2°C room temperature with food and water ad libitum. PGC-1α +/+, +/−, and −/− mice were obtained from offspring of PGC-1α +/− breeding pairs. All experiments were conducted with male and female mice from the line described by Lin et al. (2004) on postnatal day 30 (P30) unless otherwise specified.
Immunohistochemistry.
Animals were anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (5 mg/kg) and perfused intracardially with PBS, pH 7.4, and 4% paraformaldehyde in PBS. Brains were removed, postfixed in 4% paraformaldehyde for 24–72 h, cryoprotected in graded sucrose (5–20%), embedded in a mixture of 20% sucrose and Tissue-Tek O.C.T. Compound (Sakura Finetek), and frozen at −80°C. Tissue blocks were sectioned at 20 μm, mounted onto charged slides (Fisher), and allowed to dry overnight before freezing at −80°C. Slides were thawed, washed in PBS, and blocked with 10% serum from the host of the secondary antibody in PBS for 1 h. Optimal staining of parvalbumin (Sigma) and GAD67 (Millipore Bioscience Research Reagents) required antigen retrieval by incubation in citrate buffer (10 mm citric acid, pH 6.0) at 37°C for 10 min and room temperature for 20 min and then washing in PBS. Sections were then incubated in the primary antibody overnight with 5% serum from the host of the secondary antibody in PBS with Triton X-100 (PBST; Sigma) at 4°C. Slides were washed in PBST and PBS and incubated for 1 h at room temperature with the corresponding secondary antibody with 5% serum for the host of the secondary antibody in PBST. Slides were washed in PBST, peroxidase quenched with 0.3% hydrogen peroxide in methanol, and incubated with horseradish peroxidase-labeled avidin-biotin complex (Vector Laboratories) for 30 min. After washes in PBS, antibody binding was visualized with diaminobenzamide (Vector Laboratories), and sections were dehydrated in graded ethanols and xylenes and mounted with nonaqueous mounting media. Selected images were captured using a SPOT camera and software system (Diagnostic Instruments) attached to a Leica DM5000B microscope (Leica) with 4×, 10×, 20×, and 60× objectives, and images were imported into Adobe Photoshop CS3 (Adobe) for adjustments to contrast and brightness.
Gene expression analyses.
Mice were anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (5 mg/kg) before they were killed by decapitation. Brains were rapidly removed and dissected by gross anatomical markers. Eyes, hearts, and mixed-type skeletal muscle from quadriceps femoris were also removed. Tissue pieces were collected in centrifuge tubes, flash frozen on dry ice, and stored at −80°C. At least 12 h before homogenization, RNAlater-ICE (Ambion) cooled to −80°C was added to tissue pieces on dry ice, and samples were stored at −20°C. RNA was isolated by the Trizol method following the manufacturer's instructions (Invitrogen). Briefly, tissue was homogenized with Tissue-Tearor (Biospec) in Trizol reagent. RNA was isolated using chloroform-isopropanol, and concentrations were quantified at A260. Equivalent amounts of RNA (1 μg) were treated with DNase I (Promega) at 37°C for 30 min, and DNase was inactivated at 65°C for 15 min. RNA was reverse transcribed using the High-Capacity cDNA Archive Kit (Applied Biosystems). Taqman PCR was performed with JumpStart Taq Readymix (Sigma) and Applied Biosystems primers for PGC-1α (catalog no. Mm00447183_m1), GAD67 (Mm00725661_s1), GAD65 (Mm00484623_m1), parvalbumin (Mm00443100_m1), calbindin (Mm00486645_m1), calretinin (Mm00801461_m1), somatostatin (Mm00436671_m1), cholecystokinin (Mm00446170_m1), KV3.1 (Mm00657708_m1), manganese superoxide dismutase (MnSOD) (Mm01344233_g1), glucose transporter 4 (Glut4) (Mm00436615_m1), mitochondrial transcription factor A (Tfam) (Mm00447485_m1), mitofusin 2 (Mfn2) (Mm00500120_m1), and β-actin.
Human-specific primers for SH-SY5Y experiments (see below) included primers for parvalbumin (Hs00161045_m1) and 18S rRNA (Hs99999901_s1). Reactions were performed with an initial ramp time of 2 min at 50°C and 10 min at 95°C and 40 subsequent cycles of 15 s at 95°C and 1 min at 60°C. Relative concentrations of the genes of interest were calculated in comparison to a standard curve made with 1:5, 1:10, and 1:20 dilutions of cDNA from a +/+ littermate control. Values were normalized to β-actin values for the same sample and then expressed as fold of the averaged values for +/+ mice ± SEM.
Cell culture experiments.
SH-SY5Y neuroblastoma cells were transfected using an adenovirus encoding green fluorescent protein (GFP) alone or GFP and PGC-1α in tandem (see Cowell et al., 2009). Cells were collected 48 h later in Trizol reagent (n = 4/group), and RNA was isolated as above. Representative data are shown from one of three replicate experiments.
Whole-cell recordings.
Mice (P25–P36) were killed by decapitation under isoflurane anesthesia. Brains were placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 110 choline chloride, 26 d-glucose, 2.5 MgCl2, 2.5 KCl, 1.25 Na2PO4, 0.5 CaCl2, 1.3 Na-ascorbate, 3 Na-pyruvate, and 25 NaHCO3, bubbled with 95% O2/5% CO2, and 350-μm-thick hippocampal slices were prepared using a vibratome (Vibratome 3000EP). Slices were incubated at 37°C for ∼30 min in recording solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 25 d-glucose bubbled with 95% O2/5% CO2, then transferred to room temperature in the same solution. Slices were superfused continuously with oxygenated recording ACSF at room temperature. Whole-cell patch-clamp recordings were acquired from visually identified mature granule cells of the dentate gyrus using Dodt optics on an upright microscope (Olympus BX51). Cells were voltage clamped at −70 mV, using an internal solution containing the following (in mm): 129 CsCl, 2 MgCl2, 10 EGTA, 10 HEPES, 3 Na-ATP, 0.3 GTP, and 2 QX-314, pH 7.2. Pipette tip resistance was 3–4 MΩ, and series resistances were <15 MΩ. Voltage-clamp recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 kHz. Input resistance and series resistance were monitored by applying a 5 mV voltage step, and data were discarded when series resistance changed by >20%. Spontaneous and evoked IPSCs were pharmacologically isolated with NBQX (10 μm) and d-APV (50 μm). Analysis of spontaneous IPSCs (sIPSCs) was performed using the event detection package with a variable amplitude template in AxoGraphX (Axograph Scientific). All sIPSCs that fit the template and passed visual inspection were included in the analysis. A patch pipette filled with recording solution was used to focally stimulate at the border of the granule cell layer and inner molecular layer. To calculate the paired-pulse ratio, the amplitude of the second IPSC was measured relative to the baseline set immediately before the second stimulus. The decay of IPSCs was fit with two exponentials and the weighted decay τ was calculated from the equation A1τ1 + A2τ2, where A is the relative amplitude of each component and τ is its time constant. NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide), APV (dl-2-amino-5-phosphonovaleric acid), and picrotoxin (PTX) were diluted from stock solutions to final concentrations in ACSF. All drugs were from Tocris Bioscience.
Field potential recordings.
Animals were killed under isoflurane anesthesia using a rodent guillotine. The brain was immersed in ice-cold cutting saline [CS (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, and 0.6 ascorbate]. Transverse hippocampal slices (400 μm) were prepared with a Vibratome (The Vibratome Company) and equilibrated in a mixture of 50% CS and 50% ACSF (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 glucose) at room temperature. Slices were further equilibrated in 100% ACSF for 45 min at room temperature, followed by a final incubation in 100% ACSF at 32°C for 1 h. Oxygenated ACSF (95%/5% O2/CO2) was perfused into an interface recording chamber (Fine Science Tools) at a rate of 1 ml/min. Extracellular stimuli were administered on the border of areas CA3 and CA1 along the Schaffer collaterals using Teflon-coated, bipolar platinum electrodes. fEPSPs were recorded in stratum radiatum with an ACSF-filled glass recording electrode (1–3 MΩ). Experimental stimuli were set to an intensity that evoked a fEPSP that had a slope of 50% of the maximum fEPSP slope. Long-term potentiation (LTP) was induced with two 100 Hz tetani (1 s) with an interval of 20 s between tetani. Synaptic efficacy was monitored 0.5 h before and 3 h following induction of LTP by recording fEPSPs every 20 s (traces were averaged for every 2 min interval). All electrophysiological recordings and analysis was performed blind to the genotype.
Statistical analysis.
For gene expression studies, one-way ANOVAs were used to analyze each gene of interest with genotype (mouse experiments) or multiplicity of infection (cell culture experiments) as the independent variable and gene expression as the dependent variable. Post hoc analyses were performed using two-tailed t tests. For electrophysiological recordings, statistical differences were determined with one-tailed t tests or, if the data failed tests for normality, Mann–Whitney U tests. For all tests, α = 0.05.
Results
PGC-1α is required for PV protein and transcript expression throughout the cerebrum
To initially determine whether specific subtypes of GABAergic neurons were affected in PGC-1α-deficient animals, we evaluated the intensity and distribution of GAD67 and several subtype-specific GABAergic markers in +/+, +/−, and −/− littermates (Lin et al., 2004) throughout the lifespan. Drastic reductions in immunoreactivity for the interneuron-specific Ca2+-binding protein PV were noted in +/− and −/− compared to +/+ animals (Fig. 1), while no major changes were apparent in any other protein tested (data not shown). Lack of PV protein was apparent as early as P14 (data not shown), when cortical protein expression was first observed in +/+ mice, and was more pronounced by P30, when protein expression reaches adult levels. Decreased staining was still evident in adult animals (9 months of age; data not shown).
Figure 1.
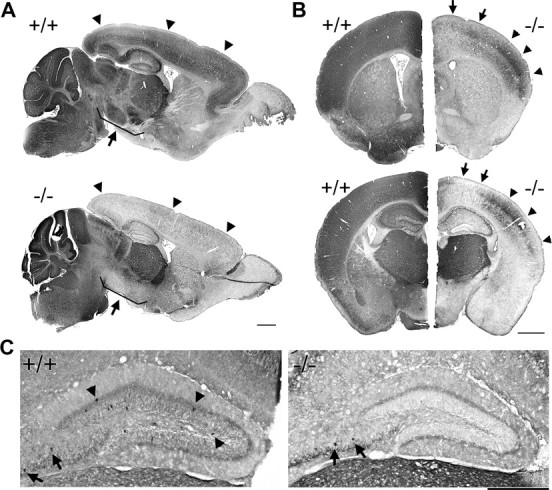
Loss of PV immunoreactivity throughout the cerebrum in PGC-1α −/− mice. Immunohistochemistry with an antibody specific to PV was conducted on 20 μm sagittal and coronal brain sections from PGC-1α +/+ and −/− littermates. A, Low-magnification photos in the sagittal plane demonstrate a striking absence of PV immunoreactivity in −/− animals compared to +/+ littermates throughout the cortex (arrowheads), hippocampus, and basal ganglia (nuclei shown by arrows). B, Low-magnification photos in the coronal plane reveal that loss of PV immunoreactivity was not uniform across the cerebrum. Motor cortex (arrows) was more affected than parietal cortex (arrowheads). This differential loss was more pronounced in posterior (bottom panels) than anterior (top panels) brain regions. C, Representative higher-magnification photos show that the dentate gyrus of −/− animals was devoid of PV-labeled cell bodies (arrowheads). Some preservation of labeled cell bodies was observed in other areas of hippocampus such as the CA4 region (arrows). n = 3/group. Scale bars, 1 mm.
Interestingly, deficiencies in PV immunoreactivity were most pronounced in brain regions originating from the telencephalon, including the hippocampus, cortex, and basal ganglia, while PV expression in the thalamus, cerebellum, and hindbrain appeared to be unaffected (Fig. 1A). Even subregions within the cerebrum did not exhibit homogenous loss of PV immunoreactivity. For instance, while PV staining intensity was greatly reduced in all areas of cortex, posterior regions appeared to be more affected than anterior regions, and while most cortical regions exhibited virtually no PV, some PV immunoreactivity was still present in parietal cortex (Fig. 1B). At higher magnification, it was clear that PV immunoreactivity was reduced in both cell bodies and processes (Fig. 1C).
General immunostaining observations were confirmed at the transcript level with region-specific q-RT-PCR in PGC-1α +/+, +/−, and −/− littermates at P30. Regions for transcript analyses were chosen based on immunohistochemistry observations, and q-RT-PCR was conducted on total RNA isolated from discrete anatomical regions. As expected, PGC-1α transcript was reduced ∼50% of +/+ levels in +/− animals and undetectable in −/− animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We found significant differences in PV transcript expression among genotypes in the hippocampus (F(2,17) = 24.95, p = 8.77 × 10−6), cortex (F(2,17) = 21.31, p = 2.34 × 10−5), and striatum (F(2,17) = 5.28, p = 0.02) but not the cerebellum (Fig. 2A). Significant decreases in PV expression were found in +/− and −/− compared with +/+ animals in the hippocampus, cortex, and striatum (two-tailed t tests, p < 0.05). PGC-1α −/− animals also had significantly decreased PV expression compared with +/− animals in the hippocampus and cortex (two-tailed t tests, p < 0.05), indicating a dose-dependent relationship between PGC-1α and PV transcript expression in these areas. In fact, simple linear regressions revealed that PGC-1α transcript expression significantly predicted PV transcript expression in the hippocampus (b = 0.74, t(19) = 5.83, p = 1.58 × 10−5), cortex (b = 0.84, t(19) = 7.53, p = 5.76 × 10−7), striatum (b = 0.50, t(19) = 2.92, p = 0.009), and cerebellum (b = 0.21, t(17) = 2.21, p = 0.04).
Figure 2.

PGC-1α is necessary and sufficient for neural expression of PV. A, Region-specific q-RT-PCR was conducted on homogenates of hippocampus, cortex, striatum, and cerebellum of PGC-1α +/+, +/−, and −/− mice. Significant decreases in PV transcript expression were found in PGC-1α +/− and −/− compared to +/+ mice in the hippocampus, cortex, and striatum but not the cerebellum. In the hippocampus and cortex, PV expression in −/− animals was significantly decreased compared to +/− animals, indicating a dose-dependent relationship between PGC-1α and PV. B, The dose-dependent decreases in PGC-1α and PV transcript expression were replicated in the hippocampus of a separate line of PGC-1α-deficient animals (Leone et al., 2005). Values for mRNA abundance were normalized to actin and presented as fold +/+ ± SEM. Group sizes (n) are indicated on the bars. C, PV expression was robustly induced in neuroblastoma cells transfected with an adenovirus (AdV) for PGC-1α (multiplicity of infection 10:1 and 20:1, n = 4/group) as compared to cells transfected with GFP alone. Values for mRNA abundance were normalized to 18S and presented as percentage maximal expression ± SEM. One-way ANOVA was used, followed by two-tailed t tests. *p < 0.05, **p < 0.01, ***p < 0.005.
Because these findings demonstrated a tight correlation between PGC-1α and PV expression within these regions, we examined PGC-1α and PV transcript expression in the hippocampus of a second independent line of PGC-1α-deficient animals (Leone et al., 2005). We found significant differences among genotypes in both PGC-1α (F(2,22) = 75.52, p = 1.40 × 10−10) and PV (F(2,22) = 9.23, p = 0.001) transcript expression. Again, expression of PGC-1α and PV was significantly different among all genotypes (two-tailed t tests, p < 0.05), indicating a dose-dependent relationship between PGC-1α and PV transcript expression in this region (Fig. 2B).
Overexpression of PGC-1α in neuroblastoma cells robustly induces PV expression
We transfected SH-SY5Y neuroblastoma cells with either an adenovirus for green fluorescent protein or PGC-1α (10:1 or 20:1 multiplicities of infection) and found a significant effect of PGC-1α overexpression on PV transcript expression (F(2,9) = 18.78, p = 0.0006) (Fig. 2C). These results indicate that PGC-1α is sufficient to induce PV in cultured cells and suggest that PGC-1α may be a component of the transcriptional machinery required for PV expression.
Other GABAergic markers in the hippocampus are unaffected by loss of PGC-1α
To determine whether the reduction in PV transcript and protein expression was attributable to a loss of interneurons in PGC-1α-deficient animals, we investigated the expression of several other GABAergic markers. The hippocampus was specifically chosen as a model region because of the drastic deficiency of PV in −/− and +/− animals in this region. Immunoreactivity for the pan-GABAergic marker GAD67 was not altered in intensity or distribution in any area of the hippocampus. Even in the dentate gyrus, an area devoid of PV staining, labeling for GAD67 appeared to be comparable among genotypes (Fig. 3A). Using region-specific q-RT-PCR, we then measured transcript expression of GAD67 and GAD65 in this region and found no significant differences among genotypes (Fig. 3B). Of note, transcript expression of KV3.1, a potassium channel concentrated specifically in PV-positive neurons, was also unaltered in +/− and −/− animals. To investigate whether loss of PV was compensated by increases in other types of Ca2+-binding proteins or neuropeptides expressed by interneurons in the hippocampus, we measured transcript expression of calbindin, calretinin, cholecystokinin, and somatostatin and found no significant differences among genotypes (Fig. 3C).
Figure 3.
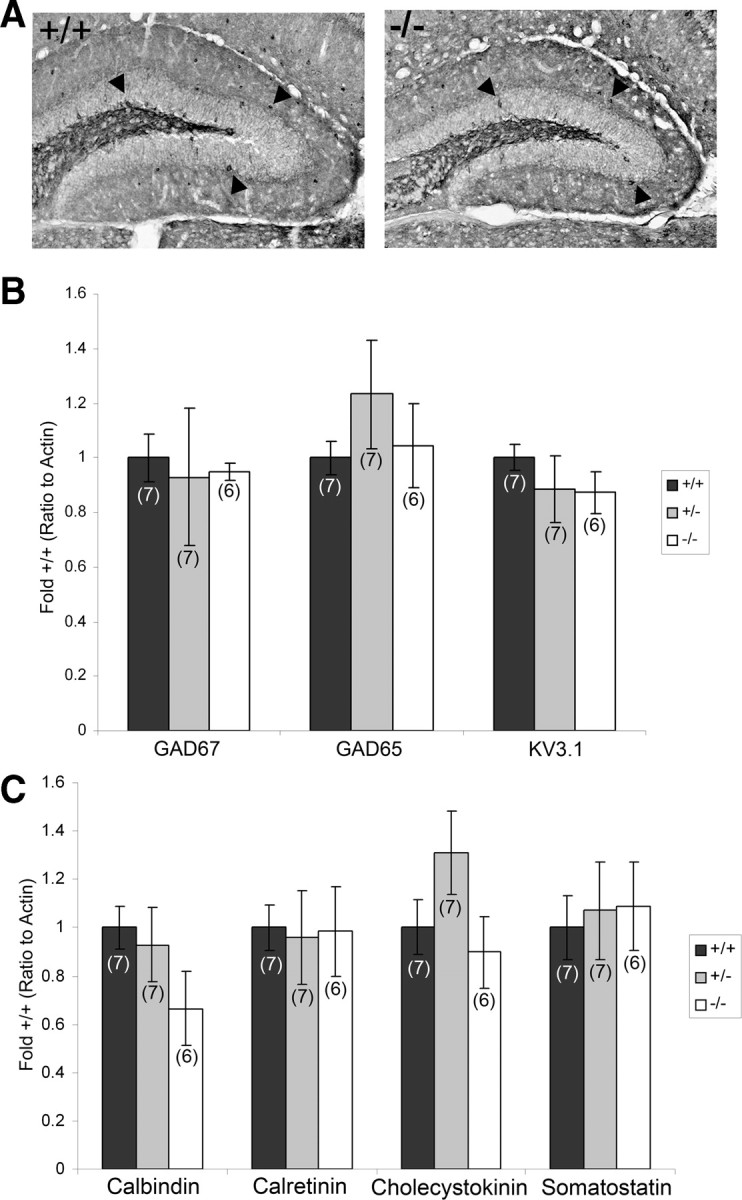
Other GABAergic markers are unaffected by loss of PGC-1α in the hippocampus. A, Immunohistochemistry with an antibody specific to GAD67 was conducted on 20 μm sagittal and coronal brain sections from PGC-1α +/+ and −/− littermates. Representative high-magnification photos of the dentate gyrus in the coronal plane demonstrate that immunoreactivity for GAD67 (arrowheads) was not observably different in intensity or distribution between +/+ and −/− animals. n = 3/group. B, Region-specific q-RT-PCR was conducted on homogenates of gross anatomical dissections of hippocampus of PGC-1α +/+, +/−, and −/− mice. Transcript expression of GAD67, GAD65, and the PV-interneuron-specific potassium transporter KV3.1 was not significantly different among genotypes. C, Expression of other interneuron-specific transcripts was not significantly altered among genotypes. Values for mRNA abundance were normalized to actin and presented as fold +/+ ± SEM. Group sizes (n) are indicated on the bars. One-way ANOVAs were used, and no significant differences were found.
Metabolic gene targets are not affected in the hippocampus of PGC-1α −/− animals
We next investigated whether well established metabolic gene targets of PGC-1α were altered in the brain of PGC-1α-deficient animals. We used region-specific q-RT-PCR to analyze transcript expression of the previously identified neural targets of PGC-1α (Cowell et al., 2009) MnSOD, Mfn2, Tfam, and Glut4 in the hippocampus and cerebellum of PGC-1α −/−, +/−, and +/+ littermates. In stark contrast to the sharp decline of PV expression, PGC-1α loss did not affect the expression of these metabolic genes in the hippocampus at this age (Fig. 4A). As PGC-1α is expressed in low levels in the hippocampus, we also measured these targets in the cerebellum, the brain region with the highest expression of PGC-1α (Cowell et al., 2007). We found significant differences in expression of MnSOD (F(2,15) = 5.57, p = 0.02) and Mfn2 (F(2,15) = 4.47, p = 0.03) in this region. For both of these genes, the significant differences were found only between −/− and +/+ animals (two-tailed t tests, p < 0.05) (Fig. 4B).
Figure 4.

Metabolic gene targets are not affected in the hippocampus of PGC-1α −/− animals. PGC-1α metabolic gene targets MnSOD, Mfn2, Tfam, and Glut4 were analyzed in homogenates of gross anatomical dissections of the hippocampus and cerebellum by region-specific q-RT-PCR in PGC-1α +/+, +/−, and −/− littermates. A, No significant differences were found for these targets in the hippocampus. B, Transcript expression of MnSOD and Mfn2 was significantly decreased in −/− compared to +/+ mice in the cerebellum. Values for mRNA abundance were normalized to actin and presented as fold +/+ ± SEM. Group sizes (n) are indicated on the bars. One-way ANOVA was used, followed by two-tailed t tests. *p < 0.05, **p < 0.01.
Basal GABA release is unaffected by PGC-1α deficiency
In the brain, fast and precise release of GABA from PV-positive interneurons is important for generating fast oscillatory activity in hippocampus and cortex (Bartos et al., 2007). To examine functional consequences of PV loss, we first evaluated basal GABA release in PGC-1α +/+ and −/− mice by measuring sIPSCs in dentate granule cells (Fig. 5A). There was no difference in sIPSC frequency or amplitude between genotypes. The frequency of sIPSCs was 8.4 ± 1.4 Hz in +/+ mice (n = 16) compared to 7.6 ± 1.0 Hz in −/− mice (n = 13), and the average amplitude of sIPSCs was 69 ± 8 pA in +/+ mice compared to 56 ± 5 pA in −/− mice. The similarity in the frequency and amplitude of sIPSCs between genotypes confirmed that deletion of PGC-1α does not reduce the total number of GABAergic synapses or the overall level of spontaneous GABA release. The half-width of sIPSCs was unaltered (7.9 ± 0.2 ms in +/+ mice and 7.8 ± 0.3 ms in −/− mice), but the 20–80% rise time of sIPSCs tended to be slower in −/− mice (1.5 ± 0.09 ms compared to 1.3 ± 0.08, p = 0.05), possibly suggesting that more distal synapses make a larger contribution to sIPSCs in the absence of PGC-1α.
Figure 5.

Ablation of PGC-1α alters short-term plasticity of evoked IPSCs but not basal GABA release. A, Representative traces of sIPSCs (blocked by the GABAA receptor antagonist PTX) in +/+ and −/− animals. Quantification of spontaneous IPSCs revealed that the IPSC frequency and amplitude were not different between genotypes. B, Paired-pulse depression was reduced in PGC-1α −/− mice. Example IPSCs are the average of 15 traces with stimulus artifacts blanked for clarity (left). Reduced paired-pulse depression was seen at the 20 and 30 ms interstimulus intervals (right, n = 12 per interval). Mann–Whitney U tests. *p < 0.05.
PGC-1α −/− mice exhibit less paired-pulse depression
Since PV controls short-term plasticity of GABA release by shaping the time course of the presynaptic Ca2+ transient (Caillard et al., 2000; Collin et al., 2005), we next tested the paired-pulse ratio of IPSCs evoked by focal stimulation of perisomatic inhibitory terminals in the granule cell layer. Consistent with reduced paired-pulse depression of IPSCs in PV −/− mice (Caillard et al., 2000), loss of PV in the dentate gyrus was associated with significantly less paired-pulse depression at short interstimulus intervals (Fig. 5B). There was no difference in the average amplitude of evoked IPSCs (813 ± 75 pA in +/+ mice, n = 12, and 729 ± 49 pA in −/− mice, n = 12), and the 20–80% rise times of evoked IPSCs were also not different between genotypes (1.45 ± 0.24 ms in +/+ mice and 1.87 ± 0.29 ms in −/− mice). Interestingly, the weighted decay τ of evoked IPSCs was significantly slower in −/− mice (37 ± 3 ms compared to 30 ± 1.5 ms in +/+ mice, p = 0.04) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), an effect that could result from delayed release events that appeared as inflections on the decay phase of the IPSC. Supporting this possibility, delayed synaptic events were apparent in individual traces from most cells in −/− mice, and the average number of delayed release events per cell was greater in −/− mice compared to +/+ mice (p = 0.007) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Delayed release events in −/− mice were not due to differences in the rate of sIPSCs between genotypes but rather could reflect a greater contribution of non-PV-expressing terminals that display asynchronous GABA release (Hefft and Jonas, 2005).
Repetitive stimulation at the gamma frequency enhances GABA release
PV-expressing fast spiking basket cells play a key role in generating gamma-frequency oscillations (Bartos et al., 2007). We thus tested whether GABA release in response to gamma-frequency stimulation is altered in PGC-1α −/− mice. Stimulation trains (500 ms duration) at 40 Hz delivered to axons surrounding granule cell soma caused depression of GABA release throughout the train in +/+ mice, measured by the normalized amplitude of the last IPSC in the train (0.53 ± 0.05, n = 12). In −/− mice, however, there was a robust increase in GABA release during the train (normalized last amplitude, 1.37 ± 0.26, n = 12, p = 0.002) (Fig. 6A), as well as a significant increase in the total charge during the train (942 ± 186 A · s in −/− mice compared to 328 ± 28 A · s in +/+ mice, p = 0.001) (Fig. 6B). Consistent with PV contributing to the synchronicity of GABA release (Hefft and Jonas, 2005), asynchronous release was evident in individual traces during 40 Hz train stimulation in −/− mice, whereas release was highly synchronous in +/+ mice (Fig. 6C). We quantified asynchronous release after the train (i.e., delayed release) by normalizing the amplitude of the last IPSC in the averaged response and then measuring the area from the last stimulus to the end of the recorded episode. The area after the train was significantly greater in −/− mice (166 ± 7 pA · ms compared to 87 ± 8 pA · ms, p = 0.001, data not shown), suggesting that there is enhanced delayed release. Altogether these results demonstrate that the short-term plasticity and the synchrony of GABAergic signaling are impaired in PGC-1α −/− mice in a manner consistent with the loss of PV from perisomatic inhibitory terminals.
Figure 6.

Repetitive stimulation at the gamma frequency enhances GABA release. Stimulation trains (500 ms duration) at 40 Hz delivered to axons surrounding the somas of mature granule cells resulted in robust increases in GABA release in PGC-1α −/− mice. A, The amplitude of the last IPSC in the train (gray dotted lines) was significantly larger in −/− compared to +/+ animals. Reponses were normalized to the peak amplitude of the first IPSC (black dotted line). B, Total charge during the train (gray area) was also significantly increased in −/− compared to +/+ mice. Traces shown in A and B are the average normalized response from all 12 cells in +/+ and −/− mice. C, Asynchronous release was apparent in individual IPSCs during and after the 40 Hz train in recordings from −/− mice. Seven individual episodes from one cell in a +/+ and −/− mouse are overlaid.
PGC-1α −/− mice exhibit reduced long-term potentiation
We expected that enhanced GABA release following train stimulation could impair the induction of long-term plasticity at excitatory synapses. Consistent with this idea, the magnitude of LTP induced by 100 Hz train stimulation at CA1 Schaffer collateral synapses was reduced in PGC-1α −/− mice (p = 0.005) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Importantly, no changes were observed in the input/output function or the paired-pulse ratio of evoked excitatory field responses (supplemental Fig. 3B,C, available at www.jneurosci.org as supplemental material), suggesting that excitatory synaptic transmission was normal. These results further suggest that there is a selective dysregulation of inhibitory synaptic transmission in PGC-1α-deficient mice.
PGC-1α regulates PV in a tissue-specific manner
As PV is not exclusively expressed in the brain, we questioned whether PV expression in peripheral tissues was also affected by loss of PGC-1α. PGC-1α is abundantly expressed in heart and skeletal muscle (Puigserver et al., 1998), and these tissues express PV as well (Heizmann et al., 1982; Vongvatcharanon et al., 2008). We also wanted to investigate the expression of PV in the retina; although the expression of PGC-1α has not yet been examined in the retina, PV is found in AII amacrine cells (Wässle et al., 1993). Using q-RT-PCR, we measured transcript expression of PGC-1α and PV in retina, heart, and mixed-type skeletal muscle of PGC-1α +/+, +/−, and −/− littermates at P30. Significant differences among genotypes were found for expression of PGC-1α in retina (F(2,17) = 33.94, p = 1.16 × 10−6), heart (F(2,23) = 107.31, p = 2.17 × 10−12), and skeletal muscle (F(2,17) = 32.88, p = 1.44 × 10−6) (Fig. 7A). In retina and heart, PGC-1α transcript expression differed among all genotypes (two-tailed t tests, p < 0.05); however, in skeletal muscle, expression of PGC-1α did not differ between +/+ and +/− animals, suggesting some compensation in the remaining allele in +/− mice. Expression of PV was significantly different among genotypes in the retina (F(2,17) = 5.51, p = 0.01) and heart (F(2,23) = 5.73, p = 0.01) but not skeletal muscle, consistent with the fact that PGC-1α is primarily expressed in slow-twitch muscle fibers (Lin et al., 2002), while PV is expressed in fast-twitch muscle fibers (Celio and Heizmann, 1982). In the retina, decreases in PV expression were found in +/− and −/− compared to +/+ mice; whereas, in the heart, decreases were in −/− compared to +/+ and +/− mice (Fig. 7B).
Figure 7.
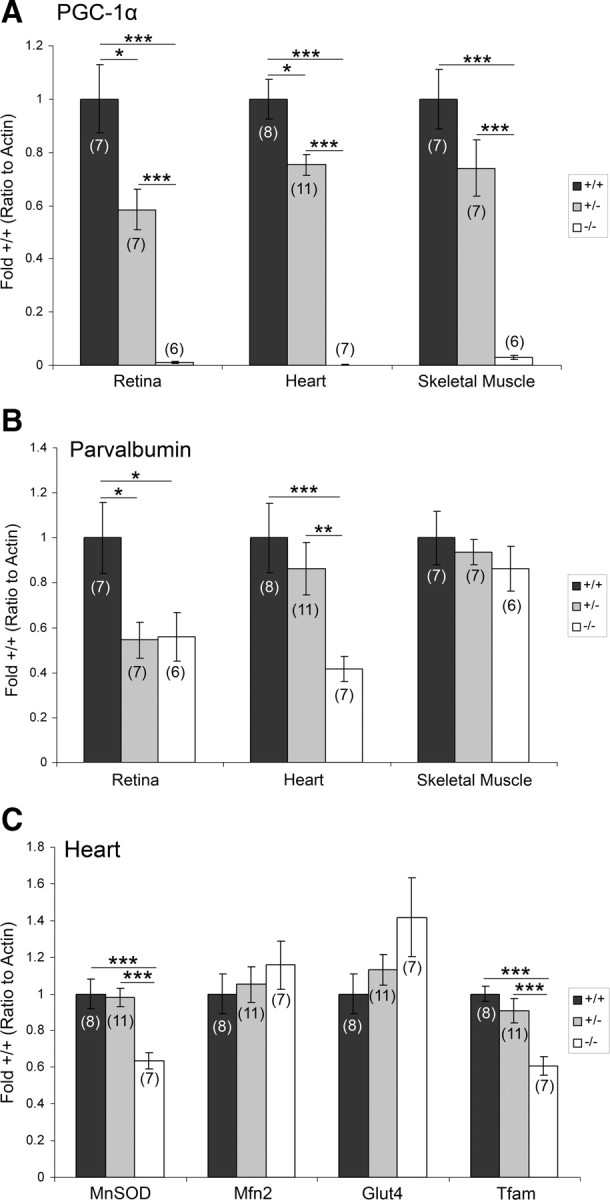
PGC-1α regulates PV in a tissue-specific manner. Transcript expression was analyzed by q-RT-PCR in homogenates of retina, heart, and skeletal muscle taken from PGC-1α +/+, +/−, and −/− mice. A, Transcript expression of PGC-1α significantly differed among all genotypes in the retina and heart, but only −/− mice differed from +/+ and +/− mice in skeletal muscle. B, Decreased PV transcript expression was found in −/− mice compared with +/+ and +/− mice in the retina and heart. C, Cardiac expression of MnSOD and Tfam was decreased in −/− mice compared to +/+ and +/− mice. Values for mRNA abundance were normalized to actin and presented as fold +/+ ± SEM. Group sizes (n) are indicated on the bars. One-way ANOVA was used, followed by two-tailed t tests. *p < 0.05, **p < 0.01, ***p < 0.005.
To investigate whether PV was decreased to a similar extent as traditional PGC-1α gene targets, expression of MnSOD, Mfn2, Glut4, and Tfam was also analyzed in the heart (Fig. 7C). Significant differences among genotypes were found in transcript expression for MnSOD (F(2,23) = 12.06, p = 0.0003) and Tfam (F(2,23) = 11.95, p = 0.0003). Decreases in −/− compared to +/+ and +/− mice accounted for these differences for both transcripts. Of note, PV expression (41 ± 6% of +/+) in the heart of −/− mice was decreased to an extent similar to MnSOD (63 ± 4% of +/+) and Tfam (61 ± 5% of +/+).
Discussion
Previous work has demonstrated that PGC-1α localizes to the nuclei of GABAergic interneurons by the second week of postnatal life during the maturation of the rodent brain (Cowell et al., 2007). To elucidate the roles of PGC-1α in this neuronal population, we measured the expression of a variety of GABAergic protein and gene targets in mice lacking PGC-1α. We found a striking and selective reduction in expression of the Ca2+-binding protein PV in PGC-1α +/− and −/− mice within the first month of life. Expression of other interneuron-specific transcripts, such as GAD67, GAD65, calbindin, calretinin, cholecystokinin, and somatostatin, was not significantly affected at this age, leading us to the hypothesis that the PV-expressing interneuron population was still present but lacking PV. This hypothesis was supported by our electrophysiological investigations in the dentate gyrus of the hippocampus, which demonstrate that basal GABA release was not affected (GABAergic terminals still intact) but that GABA release was enhanced with gamma-frequency stimulation, as seen in a PV −/− animals (see below).
Interestingly, loss of PV expression was limited to the hippocampus, cortex, and basal ganglia. PV interneurons, as well as those containing somatostatin, in the hippocampus, cortex, and striatum are thought to rise from progenitors in the medial ganglionic eminence during embryonic development and tangentially migrate to these regions (Marin et al., 2000; Danglot et al., 2006; Wonders and Anderson, 2006). However, due to the postnatal induction of PGC-1α expression (Cowell et al., 2007), it is unlikely that PGC-1α mediates fate determination of interneurons. Rather, our studies of GABAergic abnormalities in the hippocampus suggest that PGC-1α regulates the expression of PV within PV interneurons rather than PV-interneuron differentiation or migration. First, loss of PV protein and transcript expression is not accompanied by decreases in the pan-GABAergic marker GAD67 or changes in spontaneous inhibitory signaling, which would be expected if an entire population of interneurons were absent. Second, we do not observe changes in expression of the PV-interneuron-specific potassium channel KV3.1 (Chow et al., 1999; Rudy and McBain, 2001), and we would predict that KV3.1 expression would mirror changes PV expression if the interneurons themselves were missing. Third, certain electrophysiological changes in inhibitory signaling in PGC-1α −/− mice mimic the changes observed in PV −/− animals, in which PV interneurons are present but lack PV.
The dose-dependent decrease in PV transcript expression in the hippocampus and cortex in PGC-1α +/− and −/− animals suggests that PGC-1α may directly regulate PV in these regions. In accordance with this view, overexpression of PGC-1α in a neuroblastoma cell line robustly induced PV transcript expression in a dose-responsive manner. As PGC-1α cannot directly bind DNA, we speculate that the regional specificity of PV deficiency is due to differential expression of transcription factors activated by PGC-1α.
PV is one of three Ca2+-binding proteins that, together with calbindin and calretinin, are expressed in largely non-overlapping populations of GABAergic neurons. Unlike calbindin and calretinin, PV has slow Ca2+-binding kinetics that accelerate the initial fast decay phase of Ca2+ transients (Chard et al., 1993; Lee et al., 2000). Paired-pulse modulation in PV −/− animals reveals that loss of PV from presynaptic terminals switches synapse phenotype from depressing to facilitating, an effect attributed to PV speeding the fast component of the presynaptic Ca2+ transient to ensure a small-amplitude residual component that minimizes facilitation and asynchronous release (Caillard et al., 2000; Collin et al., 2005). We show that loss of PGC-1α reduces paired-pulse depression of IPSCs only at short interstimulus intervals that are likely affected by the residual component of the presynaptic Ca2+ transient. The inhibitory effect of PV on facilitation may be magnified during repetitive stimulation at short interstimulus intervals, resulting in more dramatic facilitation during gamma-frequency stimulation, similar to the marked enhancement of postsynaptic inhibition upon repetitive stimulation in PV −/− mice (Vreugdenhil et al., 2003). In future studies, it would interesting to test whether other abnormalities in PV −/− mice, such as increased power of gamma oscillations, are also present in PGC-1α −/− animals.
Slow Ca2+ binding by PV is also predicted to slow the residual component of the Ca2+ transient that develops during repetitive high-frequency stimulation (Collin et al., 2005). This slow component drives delayed transmitter release and, accordingly, the duration of delayed GABA release at cerebellar synapses is reduced in PV−/− mice (Collin et al., 2005). In contrast, we observed an increase in asynchronous and delayed GABA release in PGC-1α −/− mice. This discrepancy could stem from differences in Ca2+ handling at the specific synapses tested. Significantly delayed GABA release occurs at PV-positive inhibitory terminals in the cerebellum (Collin et al., 2005). However, in the dentate gyrus, delayed release occurs only at non-PV-positive basket cell terminals, whereas PV-positive basket terminals display highly synchronous release (Hefft and Jonas, 2005). Thus, loss of PV in the dentate gyrus would be expected to enhance asynchronous release, mimicking release mediated by non-PV-expressing perisomatic terminals. Overall, the loss of PV is sufficient to underlie the alterations in short-term plasticity and timing of GABA release that we observed in PGC-1α −/− mice. However, we cannot rule out the possibility that changes in metabolic activity within PV-expressing interneurons (see below) also makes a contribution.
Unlike PV −/− animals, adult PGC-1α −/− mice (3–4 months of age) have global reductions in key metabolic and antioxidant genes, such as MnSOD (Lin et al., 2004; St-Pierre et al., 2006). Unexpectedly, none of the traditional PGC-1α target genes were altered in +/− or −/− animals in the hippocampus at P30. As we and others have found that overexpression of PGC-1α in vitro increases the expression of key mitochondrial proteins in neural cell lines (Cowell et al., 2009; Wareski et al., 2009), we suspect that PGC-1α serves to regulate metabolic function within interneurons. However, due to the fact that interneurons comprise such a small percentage of the total cells in a homogenate, the cell-specific decreases in ubiquitous transcripts are undetectable. Single-cell transcriptional studies are required to determine whether metabolic targets are reduced specifically in interneurons in PGC-1α −/− animals.
In light of the reductions in MnSOD and other PGC-1α metabolic targets in older PGC-1α-deficient animals, it is possible that the roles for PGC-1α become more important in cell types besides interneurons as animals age. With age, PGC-1α-null mice develop spongiform vacuolizations throughout the striatum, hippocampus, and cortex (Lin et al., 2004; Leone et al., 2005). Interestingly, these are the same regions that we observe blunted expression of PV, but it is unlikely that loss of PV alone is sufficient to give rise to lesions. Indeed, vacuolizations in PGC-1α-deficient animals are thought to be predominantly localized to white matter tracts; accordingly, these animals also exhibit reduced expression of myelin-associated oligodendrocyte basic protein (Lin et al., 2004). Experiments involving the cell-specific deletion of PGC-1α in different cell types, such as interneurons and glia, are critical to identify the relative contributions of each cell population to the transcriptional, anatomical, and behavioral phenotype in PGC-1α −/− mice.
A link between PV and PGC-1α is not that surprising considering the role of PGC-1α in the normal function of mitochondria, which act as temporary Ca2+ stores and are thus involved with the shaping of intracellular Ca2+ transients (Gunter et al., 2004). In muscle, chemically induced Ca2+ influx increases PGC-1α expression and PGC-1α-mediated mitochondrial biogenesis (Wu et al., 2002; Wright et al., 2007), and accumulating evidence suggests that neural expression of PGC-1α is also governed by an activity-dependent Ca2+ pathway. Depolarization of neurons by NMDA receptor activation or potassium chloride increases PGC-1α expression (Meng et al., 2007), which can be prevented by NMDA antagonists or Ca2+ channel blockade, respectively (Luo et al., 2009; Liang et al., 2010). Conversely, blockade of neuronal activity by tetrodotoxin or sensory deprivation decreases PGC-1α expression in visual cortex (Liang and Wong-Riley, 2006). While it is currently unknown whether Ca2+-mediated induction of PGC-1α results in mitochondrial biogenesis in neurons, an inverse relationship appears to exist between PV and mitochondria in neurons such that overexpression of PV decreases mitochondrial density (Maetzler et al., 2004) and loss of PV increases mitochondrial density (Chen et al., 2006). Thus PGC-1α, PV, and mitochondria may coordinately regulate Ca2+ homeostasis in a cell-specific manner in the brain.
Of clinical relevance, decreased expression of PGC-1α has been reported in striatum and muscle from patients with HD (Cui et al., 2006; Weydt et al., 2006; Chaturvedi et al., 2009). While there are conflicting reports regarding the relative number of PV-positive interneurons in postmortem tissue from HD patients (Cicchetti et al., 2000), animal models clearly implicate the involvement of PV interneurons in HD pathology. Intranuclear inclusions of mutant huntingtin (mtHtt) have been localized to 86% of PV-positive interneurons in the striatum in the R6/2 mouse model (Kosinski et al., 1999; Meade et al., 2002), and PV mRNA and protein expression is also reduced in these animals (Luthi-Carter et al., 2002; Giampà et al., 2009). Others have demonstrated that mtHtt expression in GABAergic interneurons is required for HD-related pathology in the cortex (Gu et al., 2005), and detailed electrophysiological characterization of PV-positive neurons in the cortex of the BACHD model has shown that abnormalities in signaling in PV-positive neurons in the cortex are observed around the onset of mild motor symptoms before pyramidal neurons show changes in signaling or viability (Spampanato et al., 2008). Considering the concentration of PGC-1α in interneurons and our demonstration of the requirement of PGC-1α for proper PV expression, we speculate that the alterations of PGC-1α expression in HD reflect an underlying interneuron pathology. Future studies are required to define the roles of PV-positive interneurons in the etiology and progression of HD.
Footnotes
This work was funded by National Institutes of Health (NIH) Grants MH077955-01 (R.M.C.) and NS064025 (L.O.W.), the University of Alabama Health Service Foundation's General Endowment Fund Scholar Award (R.M.C.), the Civitan International Research Center Emerging Scholar's Award (E.K.L.), and NIH Neuroscience Blueprint Core Grant NS57098 to the University of Alabama at Birmingham. We thank Pratik Talati for assistance with cell culture experiments.
References
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A. 2000;97:13372–13377. doi: 10.1073/pnas.230362997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR, Heizmann CW. Calcium-binding protein parvalbumin is associated with fast contracting muscle fibres. Nature. 1982;297:504–506. doi: 10.1038/297504a0. [DOI] [PubMed] [Google Scholar]
- Chard PS, Bleakman D, Christakos S, Fullmer CS, Miller RJ. Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. J Physiol. 1993;472:341–357. doi: 10.1113/jphysiol.1993.sp019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Racay P, Bichet S, Celio MR, Eggli P, Schwaller B. Deficiency in parvalbumin, but not in calbindin D-28k upregulates mitochondrial volume and decreases smooth endoplasmic reticulum surface selectively in a peripheral, subplasmalemmal region in the soma of Purkinje cells. Neuroscience. 2006;142:97–105. doi: 10.1016/j.neuroscience.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Chow A, Erisir A, Farb C, Nadal MS, Ozaita A, Lau D, Welker E, Rudy B. K+ channel expression distinguishes subpopulations of parvalbumin- and somatostatin-containing neocortical interneurons. J Neurosci. 1999;19:9332–9345. doi: 10.1523/JNEUROSCI.19-21-09332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Brain Res Rev. 2000;34:80–101. doi: 10.1016/s0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, Marty A, Llano I. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502:1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Talati P, Blake KR, Meador-Woodruff JH, Russell JW. Identification of novel targets for PGC-1alpha and histone deacetylase inhibitors in neuroblastoma cells. Biochem Biophys Res Commun. 2009;379:578–582. doi: 10.1016/j.bbrc.2008.12.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus. 2006;16:1032–1060. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- Di Cristo G. Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin Genet. 2007;72:1–8. doi: 10.1111/j.1399-0004.2007.00822.x. [DOI] [PubMed] [Google Scholar]
- Giampà C, Middei S, Patassini S, Borreca A, Marullo F, Laurenti D, Bernardi G, Ammassari-Teule M, Fusco FR. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Heizmann CW, Berchtold MW, Rowlerson AM. Correlation of parvalbumin concentration with relaxation speed in mammalian muscles. Proc Natl Acad Sci U S A. 1982;79:7243–7247. doi: 10.1073/pnas.79.23.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski CM, Cha JH, Young AB, Mangiarini L, Bates G, Schiefer J, Schwarz M. Intranuclear inclusions in subtypes of striatal neurons in Huntington's disease transgenic mice. Neuroreport. 1999;10:3891–3896. doi: 10.1097/00001756-199912160-00031. [DOI] [PubMed] [Google Scholar]
- Lee SH, Schwaller B, Neher E. Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol. 2000;525:419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, González-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- Liang HL, Wong-Riley MT. Activity-dependent regulation of nuclear respiratory factor-1, nuclear respiratory factor-2, and peroxisome proliferator-activated receptor gamma coactivator-1 in neurons. Neuroreport. 2006;17:401–405. doi: 10.1097/01.wnr.0000204980.98876.11. [DOI] [PubMed] [Google Scholar]
- Liang HL, Dhar SS, Wong-Riley MT. p38 mitogen-activated protein kinase and calcium channels mediate signaling in depolarization-induced activation of peroxisome proliferator-activated receptor gamma coactivator-1alpha in neurons. J Neurosci Res. 2010;88:640–649. doi: 10.1002/jnr.22222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhu W, Jia J, Zhang C, Xu Y. NMDA receptor dependent PGC-1alpha up-regulation protects the cortical neuron against oxygen-glucose deprivation/reperfusion injury. J Mol Neurosci. 2009;39:262–268. doi: 10.1007/s12031-009-9196-5. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Maetzler W, Nitsch C, Bendfeldt K, Racay P, Vollenweider F, Schwaller B. Ectopic parvalbumin expression in mouse forebrain neurons increases excitotoxic injury provoked by ibotenic acid injection into the striatum. Exp Neurol. 2004;186:78–88. doi: 10.1016/j.expneurol.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Marmolino D, Acquaviva F. Friedreich's ataxia: from the (GAA)n repeat mediated silencing to new promising molecules for therapy. Cerebellum. 2009;8:245–259. doi: 10.1007/s12311-008-0084-2. [DOI] [PubMed] [Google Scholar]
- Meade CA, Deng YP, Fusco FR, Del Mar N, Hersch S, Goldowitz D, Reiner A. Cellular localization and development of neuronal intranuclear inclusions in striatal and cortical neurons in R6/2 transgenic mice. J Comp Neurol. 2002;449:241–269. doi: 10.1002/cne.10295. [DOI] [PubMed] [Google Scholar]
- Meng H, Liang HL, Wong-Riley M. Quantitative immuno-electron microscopic analysis of depolarization-induced expression of PGC-1alpha in cultured rat visual cortical neurons. Brain Res. 2007;1175:10–16. doi: 10.1016/j.brainres.2007.07.063. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Gu X, Yang XW, Mody I. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience. 2008;157:606–620. doi: 10.1016/j.neuroscience.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Vongvatcharanon S, Vongvatcharanon U, Boonyoung P. Immunohistochemical localization of parvalbumin calcium-binding protein in the heart tissues of various species. Acta Histochem. 2008;110:26–33. doi: 10.1016/j.acthis.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol. 2003;89:1414–1422. doi: 10.1152/jn.00576.2002. [DOI] [PubMed] [Google Scholar]
- Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem. 2009;284:21379–21385. doi: 10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wässle H, Grünert U, Röhrenbeck J. Immunocytochemical staining of AII-amacrine cells in the rat retina with antibodies against parvalbumin. J Comp Neurol. 1993;332:407–420. doi: 10.1002/cne.903320403. [DOI] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:18793–18799. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]