

Abstract
This account chronicles our efforts at the development of a catalytic asymmetric Stetter reaction using chiral triazolium salts as small molecule organic catalysts. Advances in the mechanistically related azolium-catalyzed asymmetric benzoin reaction are discussed, particularly as they apply to catalyst design. A chronological treatise of reaction discovery, catalyst optimization and reactivity extension follows.
Keywords: asymmetric synthesis, triazolium salts, quaternary stereocenters, carbene catalysis, nucleophilic catalysis, organocatalysis, Stetter reaction, benzoin reaction
1. Introduction
N-heterocyclic carbenes (NHCs) have seen tremendous advances in the last two decades and their impact as ligands in transition metal catalyzed reactions is well established.1 NHCs have also begun to play a critical role in the growing field of small molecule catalysis and in recent years have been applied to a broad range of bond forming reactions.2 By far the most valuable application of NHCs as catalysts is found in their ability to render aldehydes nucleophilic, thereby reversing their normal mode of activity, referred to as umpolung reactivity.3
Traditional methods for the conversion of aldehydes into umpolung reagents involve the use of dithianes or protected cyanohydrins derivatives.4 These methods are stoichiometric and often require strong bases to generate the acyl anion equivalent. Recent advances in umpolung reactivity of carbonyls offer an unconventional, catalytic, and mild method to traditional carbonyl bond forming reactions. Designing asymmetric catalytic systems capable of rendering aldehydes nucleophilic has been a fascinating area of research for over 40 years, in part due to the in situ ability to form acyl anion equivalents under mild conditions.
Two classic reactions that take advantage of umpolung reactivity of aldehydes, the benzoin5 and Stetter6 reaction, have seen a rebirth of activity in the last decade. The key element of the benzoin and Stetter reactions is the catalytically generated acyl anion equivalent that can facilitate the formation of a new carbon-carbon bond (Figure 1).
Figure 1.
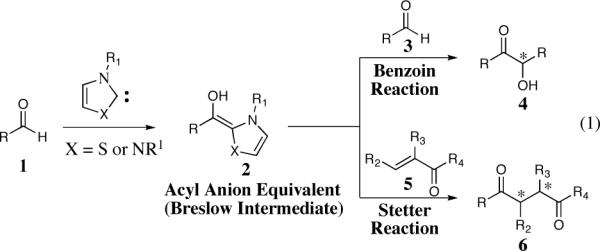
The benzoin and Stetter reaction
The benzoin reaction generates α-hydroxy ketones with a new stereogenic center when the in situ generated acyl anion equivalent 2 adds to another aldehyde.
The conjugate addition of the acyl anion equivalent 2 to α,β-unsaturated carbonyl 5, commonly known as the Stetter reaction, is a straightforward route to the synthesis of 1,4-dicarbonyl compounds. The application of prochiral Michael acceptors can result in the formation of two new contiguous stereocenters as illustrated by 6. 1,4-Dicarbonyl compounds are important synthons for the synthesis of natural products and the asymmetric Stetter reaction offers a practical route to the synthesis of this class of compounds.7
This account is a survey of recent advances in the catalytic asymmetric Stetter reaction that began over a decade ago. Stetter reactions that do not form enantioenriched products will not be included in this review. Important contributions from the asymmetric benzoin reaction will be highlighted to properly discuss the development of the asymmetric Stetter reaction.
2. The Proposed Mechanism of the benzoin and Stetter Reactions
To conduct a full discussion of catalyst effects and reaction development in the Stetter reaction, it is worthwhile to discuss the current mechanistic understanding of this transformation. As studies on the mechanism of the Stetter have yet to be reported, the following discussion will focus on the far-better studied benzoin mechanism. Lapworth proposed the currently accepted mechanism of the benzoin reaction in 1903 utilizing cyanide as the catalyst.8 In 1958 Breslow elucidated the mechanism of the benzoin reaction with thiazolium salts as precatalysts and showed the similarities between the active thiazolidine catalyst and cyanide.9 According to the proposed mechanism (Scheme 1), intermediate I results from nucleophilic attack of the carbene into the aldehyde. Proton transfer from the tetrahedral intermediate generates the acyl anion equivalent, commonly known as the Breslow intermediate II. Carbon-carbon bond formation results from nucleophilic attack of the acyl anion equivalent into a carbonyl or Michael acceptor, generating III. Proton transfer followed by catalyst expulsion generates the desired product and closes the catalytic cycle.
Scheme 1.
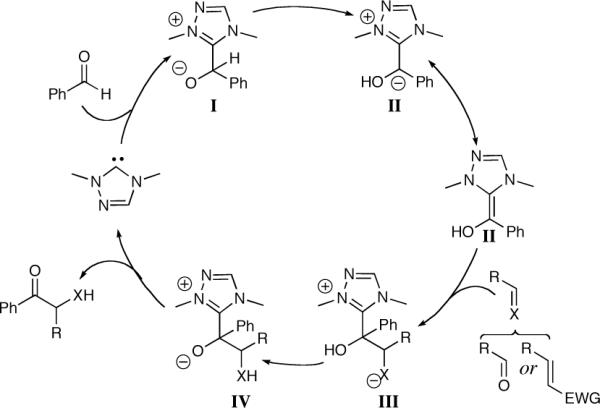
Proposed mechanism of the benzoin and Stetter reactions.
3. Benzoin Reaction
Nucleophilic acylation reactions have a long history dating back to 1832 when Wöhler and Liebig discovered that cyanide catalyzes the benzoin reaction.10 Over one hundred years later Ukai and co-workers reported that thiazolium salts in the presence of a base also catalyze the benzoin reaction,11 reminiscent of the action of thiamine pyrophosphate in biological systems.12 The successful implementation of thiazolium salts as precatalysts provides a scaffold for the rational design of chiral thiazolium catalysts and enabled the development of the asymmetric benzoin and Stetter reactions.
In 1966, Sheehan and Hunnemann were the first to validate the concept that enantioenriched thiazolium salts could relay chiral information to the newly formed α-hydroxy ketone products.13 Subsequently, Sheehan and Hara established a benchmark for the asymmetric benzoin reaction that lasted more than 20 years, achieving 52% ee with chiral thiazolium salt 9 albeit in 6% yield (Scheme 2).14 For the next two decades, numerous researchers worked on improving the efficiency and selectivity of the asymmetric benzoin reaction utilizing new chiral thiazolium catalyst scaffolds (examples of catalyst scaffolds are illustrated in Scheme 2 by 10–12). Despite the variation in the steric and electronic properties of the new thiazolium catalysts little to no improvements were observed.15
Scheme 2.

Catalysts for the asymmetric benzoin reaction
Inspired by the isolation and characterization of stable carbenes,16,17 most notably NHCs, Enders, Teles and coworkers accomplished a significant advance in the asymmetric benzoin reaction in 1996, employing a novel chiral triazolium salt 13.18 Utilizing only 1.25 mol% of catalyst 13 they were able to perform the asymmetric benzoin reaction in 75% ee and a 66% yield.
Prior to 1997, chiral catalysts applied to the asymmetric benzoin reaction possessed an element of free rotation adjacent to the reacting center. Leeper19 and subsequently Rawal20 were the first to report the rational design of chiral bicyclic thiazolium salts, which they hypothesized would decrease the degrees of freedom during the carbon-carbon bond forming event and lead to higher enantioselectivity. Although their initial fused bicyclic thiazolium salts (14–17) did not lead to vast improvements in the asymmetric benzoin reaction, they laid the foundation for future catalyst design. Leeper and co-workers later illustrated that chiral bicyclic triazolium salt 18 could bestow higher enantioselectivity for the asymmetric benzoin reaction, albeit in modest yield (45%).21 In 2002, Enders reported a chiral bicyclic triazolium salt 21 capable of catalyzing the benzoin reaction in 90% ee and good overall yield, which is the current state of the art in the asymmetric benzoin reaction.22
A major, inherent limitation of the benzoin reaction is the need for two equivalents of the same aldehyde. Hetero- or cross-benzoin reactions would greatly increase the power of this transformation, but the use of two different partners in this reaction typically leads to a thermodynamically controlled mixture of products. Recently several practical solutions have been developed to overcome some of these limitations. Müller and coworkers developed an enzyme catalyzed cross-benzoin reaction that takes advantage of donor-acceptor properties of a given aldehyde pair to obtain excellent yields and ee's of the crossed benzoin products.23 Enders24 and Suzuki25 have independently developed an efficient asymmetric intramolecular cross-benzoin reaction between aldehydes and ketones. Miller and Mennen have developed an efficient macrocyclization protocol that relies on the intramolecular crossed-benzoin reaction.26
In a related process, Johnson and co-workers introduced chiral metallophosphites as new umpolung catalysts capable of performing a regiospecific cross silyl benzoin reaction between acyl silanes and aldehydes (Scheme 3).27 The reaction is general for aryl acylsilanes and aryl or heterocyclic aldehydes with good to excellent yields (65–87%) and enantioselectivities (81–91% ee).
Scheme 3.
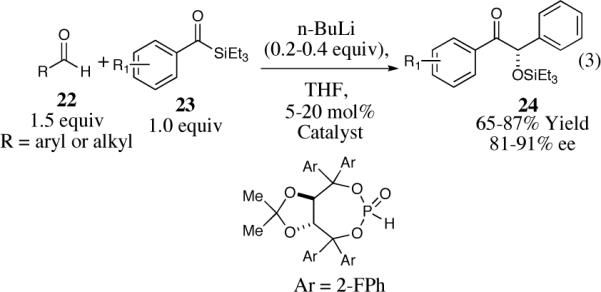
The enantioselective cross silyl benzoin reaction
4. The Synthesis of Chiral Bicyclic Triazolium Salts
Based on the chiral architecture of the catalysts employed in the asymmetric benzoin reaction, a number of general design features were identified to be advantageous for high reactivity and enantioselectivity: triazolium salts as first introduced by Enders and Teles were clearly the most attractive and a bicyclic restriction as advanced by Leeper and Rawal would lead to higher selectivities. In an effort to develop the utility of N-heterocyclic carbenes in asymmetric catalysis, we pursued a practical and efficient synthesis of a new family of triazolium salts. At the start of our work in 2000, we wanted to synthesize a family of chiral bicyclic triazolium salts that were easily modified and would come from readily available chiral precursors.28 Amino acid derivatives were chosen as the starting point due to their diverse steric profile and ready availability as either optically pure antipode. In addition, we envisioned tuning the triazolium salts by incorporating sterically and electronically different hydrazine moieties.29 This analysis led us to design two families of catalyst with tunable architectures: the morpholine-fused triazolium salts 25 and the pyrrolidine-fused triazolium salts 26, Scheme 4.
Scheme 4.
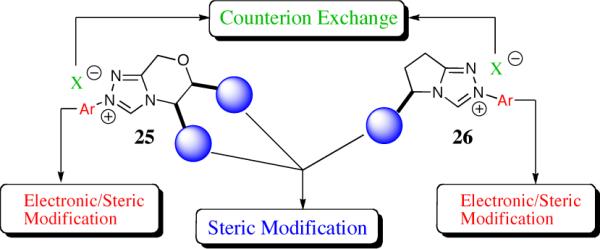
Designing new chiral bicyclic triazolium scaffolds
The amino-indanol chiral scaffold, fortuitously included due to its demonstrated success in other catalyzed reactions,30 was found to be the optimal scaffold to impart high enantioselectivity on the desired product in the morpholinone-derived triazolium salt series. Subtle changes in the electronic nature of the N-aryl substituent of the triazolium salts have a dramatic affect on the efficiency of the intramolecular Stetter reaction. The pyrrolidine derived triazolium salts are less sensitive to electronic variation on the N-aryl substituent. Phenylalanine was identified as the optimal chiral building block in the pyrrolidine derived series. In general, an increase in enantioselectivity and reactivity is observed for both chiral triazolium scaffolds, when an electron withdrawing substituent is placed on the N-aryl group.
4.1 Aminoindanol-Derived Bicyclic Scaffold
Commencing from readily available amino alcohols, optically pure morpholinone 28 was obtained in two steps (Scheme 5).31 A subsequent one-pot three-step sequence can be applied to obtain the three electronically different catalysts 33–35 in good overall yield as crystalline material.
Scheme 5.
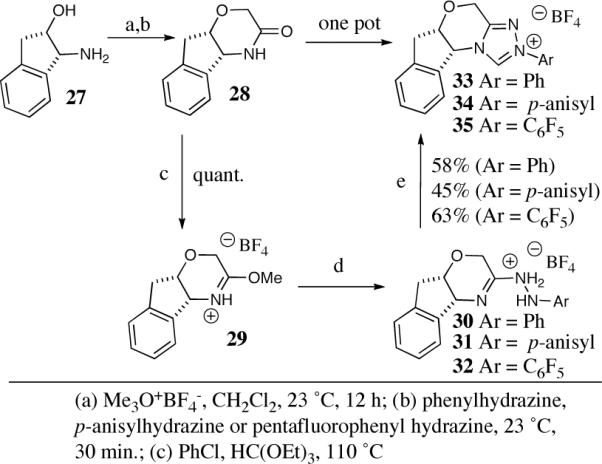
Synthesis of aminoindanol-derived triazolium salts.
4.2 Phenylalanine-Derived Bicyclic Scaffold
Chiral triazolium salts 40 and 41 can be prepared in a similar manner starting with commercially available phenylalanine (Scheme 6). Synthesis of the optically pure pyrrolidinone 39 proceeds in five chemical steps without column purification in good overall yield.32 Both chiral triazolium salts 40 and 41 can be obtained from this common intermediate in a one-pot three-step sequence. Salts 33–35 and 40–41 are air stable solids that retain their catalytic activity with no special precautions (to our knowledge they can be stored indefinitely on the bench in a desiccator).
Scheme 6.
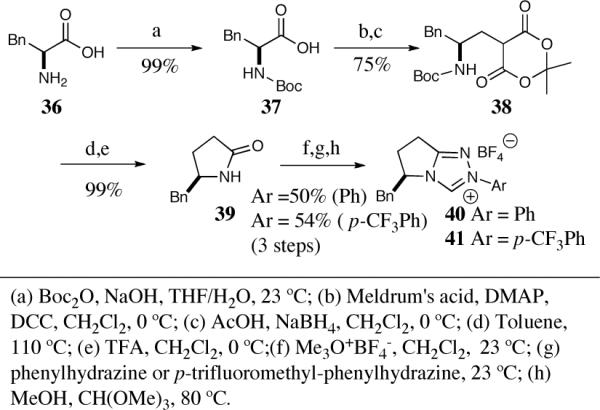
The synthesis of phenylalanine-derived triazolium salts.
5. The Intermolecular Stetter Reaction
In the early 1970's Stetter significantly extended the synthetic utility of the acyl anion equivalent by incorporating Michael acceptors as the electrophilic counterpart, affording 1,4-dicarbonyls. Stetter and co-workers demonstrated that a variety of aromatic and aliphatic aldehydes could participate in the intermolecular Stetter reaction. In addition, they were able to successfully employ a variety of different Michael acceptors. The Michael acceptors that contained a β-substituent often resulted in diminished reactivity and were generally limited to chalcones. Consequently, the asymmetric intermolecular Stetter reaction has received less attention than the benzoin reaction and has met with limited success. Enders and co-workers reported two examples of an intermolecular Stetter reaction utilizing a chiral thiazolium pre-catalyst, both as footnotes in reviews.33 The more selective case is illustrated in Scheme 7: use of thiazolium salt 44 in a biphasic solvent system results in formation of the desired 1,4-dicarbonyl 45 in 39% ee but only 4% overall yield.33
Scheme 7.
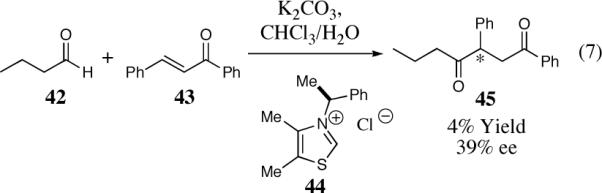
The first asymmetric intermolecular Stetter seaction.
Recently, Johnson and co-workers have developed an elegant solution to the enantioselective synthesis of 1,4-dicarbonyls by taking advantage of the umpolung reactivity of acyl silanes catalyzed by chiral metallophosphites.34 Acyl silanes are effective aldehyde surrogates capable of forming an acyl anion equivalent that can add to a variety of electrophiles in the presence of nucleophilic catalysts. A key feature in the utility of acyl silanes is the [1,2] Brook rearrangement that offers a unique approach to the in situ generation of the acyl anion equivalent in umpolung reactions.35 Taking advantage of this process Johnson was able to fashion the enantioselective synthesis of 1,4-dicarbonlyls in 89–97% ee and good chemical yield for α,β-unsaturated amides (Scheme 8). Simple recrystallization led to increased enantioselectivity in several cases.
Scheme 8.
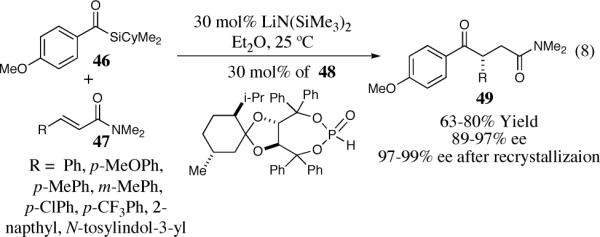
Asymmetric intermolecular Stetter reaction of acyl silanes.
Scheidt and co-workers have reported the application of silyl-protected thiazolium carbinols as stoichiometric carbonyl anions for the intermolecular acylation of nitroalkenes.36 The newly formed stereocenters can be controlled by the stoichiometric addition of a chiral thiourea with 74% ee obtained in the desired product.
6. Asymmetric Intramolecular Stetter Reaction
6.1 Recent Contributions to the Asymmetric Intramolecular Stetter Reaction
Surprisingly, the intramolecular version of the Stetter reaction was not investigated until Ciganek's report appeared in 1995 (Scheme 9).37 Unlike the intermolecular variant, the asymmetric intramolecular Stetter reaction has made significant progress in the last decade.
Scheme 9.
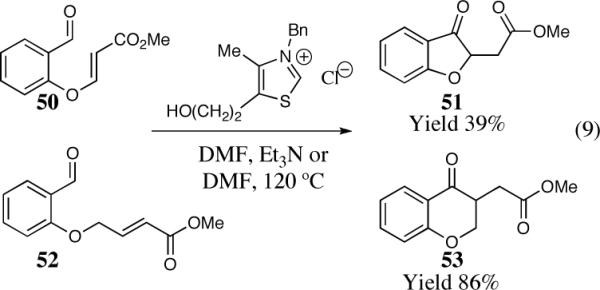
The first detailed study of the intramolecular Stetter reaction.
Enders and co-workers reported the first asymmetric intramolecular Stetter reaction in 1996 utilizing the increased reactivity of chiral triazolium carbene 13, resulting in the synthesis of chroman-4-ones in modest enantioselectivity and yields (Scheme 10).38
Scheme 10.
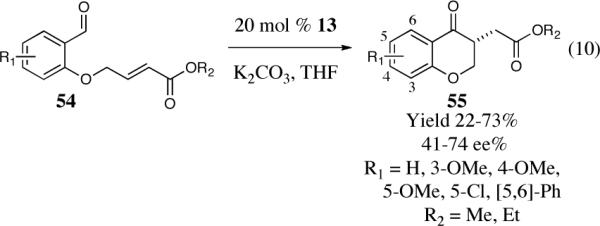
The first asymmetric intramolecular Stetter reaction
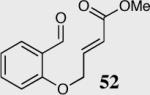
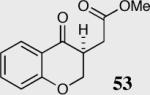
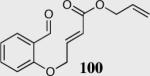
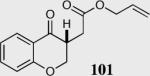
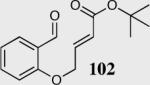
The cyclization of the salicylaldehyde-derived substrate 52 originally reported by Ciganek has become the benchmark test for comparing catalyst efficiency in the intramolecular Stetter reaction. Three additional examples of an asymmetric intramolecular Stetter reaction have appeared subsequent to our initial contributions to this field. Bach reported a unique chiral thiazolium salt 56 that is capable of imparting modest enantioselectivity in the synthesis of 53 (Scheme 11).39
Scheme 11.
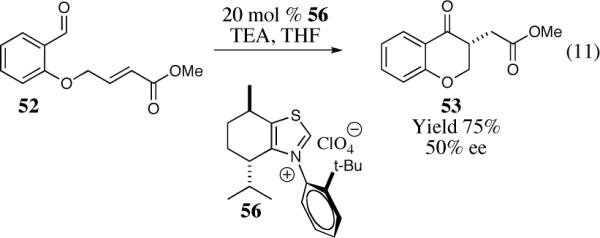
Bach's asymmetric intramolecular Stetter reaction.
Miller and co-workers have extended their minimal peptide catalyst research by installing a terminal thiazolium salt on the peptide backbone (Scheme 12).40 Miller observed substantial variation in the efficiency and selectivity of the reaction using simple derivatives of a thiazolylalanine (Taz) derived catalyst. The highest selectivity in the Taz series was found by employing catalyst 57, resulting in chromanone 60 (R1=H) in 80% ee and 40% yield.
Scheme 12.
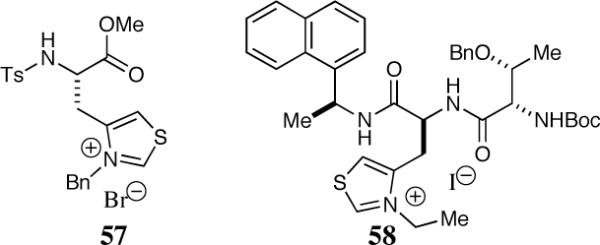
Chiral peptide derived thiazolium salts
Embedding the thiazolium salt in the small peptide (58) increased the efficiency of the reaction to 67% isolated yield and the scope of the reaction was studied utilizing these optimal conditions (Scheme 13).
Scheme 13.
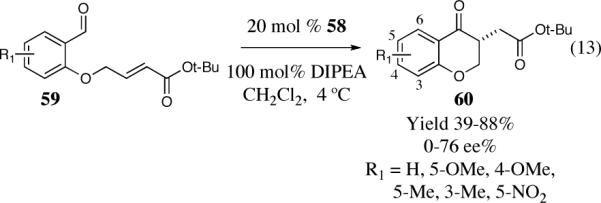
The optimal peptide derived catalyst for the asymmetric Stetter reaction
Tomioka and co-workers have developed C2-symmetric imidazolidene carbenes and successfully applied them to the intramolecular Stetter reaction of aliphatic aldehydes (Scheme 14).41 Changes to the steric and electronic nature of the Michael acceptor caused the efficiency of the reaction to vary slightly. The optimal conditions were found using the more bulky t-butyl ester, which generated the product in 59% yield and 80% ee.
Scheme 14.
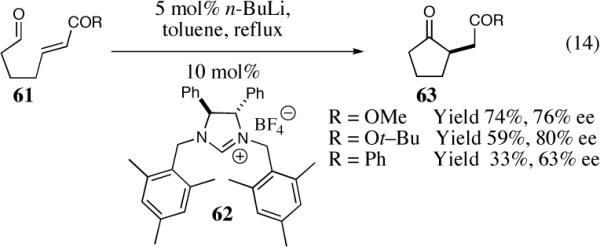
The asymmetric intramolucular Stetter reaction utilizing C2-symmetric imidazolidene carbene.
6.2 Comparison of the Asymmetric Intramolecular Stetter Reaction with Two Different Triazolium Carbene Scaffolds
As part of our effort to design and develop chiral nucleophilic carbenes for use in organic synthesis, we saw an opportunity to use these salts as precatalysts in the asymmetric intramolecular Stetter reaction. We initially studied the asymmetric intramolecular Stetter reaction of salicylaldehyde-derived substrate 52 with the newly synthesized chiral bicyclic triazolium salts.42 Our first few experiments using reaction conditions typically applied to the asymmetric benzoin reaction, notably protic solvents, led to the isolation of nearly racemic products (0–4% ee). Unphased by our initial results and driven by our youthful spirit we continued to evaluate our new catalyst under different reaction conditions. Needless to say, we were extremely excited when we obtained a 90% yield and 83% ee utilizing catalyst 40 in the presence of triethylamine and THF at room temperature (Scheme 15).
Scheme 15.
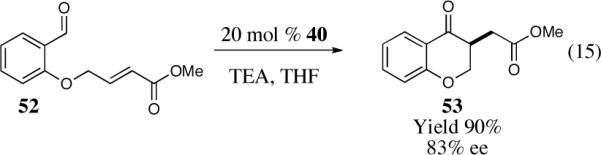
Initial results in the asymmetric intramolecular Stetter reaction.
Encouraged, we began to screen different bases, solvents and temperatures to increase the selectivity of the reaction. We ultimately found that non-polar solvents and strong bases with non-coordinating counterions afford optimal results. Initially we reported xylenes to be the optimal solvent but subsequent studies revealed anhydrous toluene to be similar in terms of reactivity and selectivity and beneficial from a practical perspective.
Fortuitously, both catalyst scaffolds behave similarly with respect to changes in base, solvent and temperature and therefore we were able to optimize the reaction conditions in tandem. This unpredicted circumstance has led to the development of two different catalysts that are each capable of performing the asymmetric intramolecular Stetter reaction in high enantioselectivity with excellent reactivity. Furthermore, the two different catalyst motifs have thus far offered complimentary reactivity for substrates that result in unsatisfactory reactivity or selectivity. On average, the amino-indanol chiral scaffold affords the desired product in higher enantioselectivity, entries 1–10 (Table 1). Conversely, the all carbon phenylalanine-derived scaffold is typically more reactive. Entries 7–10 reveal how the nature of the catalyst can increase the efficiency of the reaction without loss of enantioselectivity. Table 1 illustrates how the steric and electronic nature of the triazolinylidene carbenes can be tuned to afford high enantioselectivity and reactivity depending on the nature of the substrate.
Table 1.
Comparison of two different catalyst scaffolds. 
| Entry | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
|
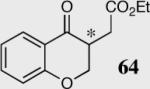
|
33 | 80 | 90 (R) | |
| 1 | 34 | 94 | 94 (R) | ||
| 2 | 40 | 94 | 90 (S) | ||
| 41 | 95 | 92 (S) | |||
| 3 |
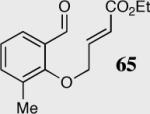
|
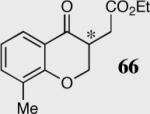
|
34 | 90 | 84 (R) |
| 4 | 40 | 94 | 80 (S) | ||
| 5 |
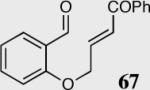
|
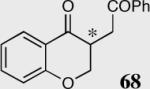
|
34 | 50 | 81 (R) |
| 6 | 40 | 85 | 68 (S) | ||
| 7 |
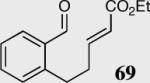
|
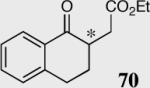
|
34 | 35 | 94 (S) |
| 8 | 40 | 90 | 92 (R) | ||
| 9 |
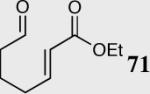
|
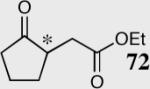
|
34 | 80 | 81 (S) |
| 10 | 40 | 81 | 95 (R) |
Based on this promising lead, we set out to test the scope of the intramolecular Stetter reaction by systematically investigating the different aspects that might influence the reaction. We began by changing the nature and length of the tethered Michael acceptor and then turned our attention to the effects of different substituents on the aromatic aldehyde backbone. Subsequently we investigated a range of tethered Michael acceptors that were capable of participating in the intramolecular Stetter reaction of aromatic aldehydes.
6.3 The Scope of the Intramolecular Stetter Reaction with Different Tethers
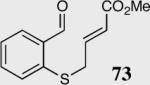
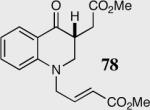
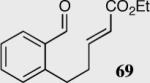
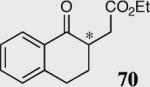
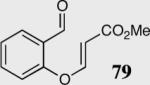
The length and nature of the tethered Michael acceptor of a variety of aromatic aldehydes was investigated to examine their effects on enantioselectivity and reactivity of the intramolecular Stetter reaction (Table 2). The reaction affords high yield and enantioselectivity for oxygen, sulfur, and nitrogen tethers (entries 1–4). Removal of the heteroatom in the linker results in only 35% yield utilizing catalyst 34 (entry 5). The all carbon tether requires the use of the more reactive catalyst 40, affording the desired product in 92% ee and 90% yield (entry 6). Substrate 7b, bearing an excised methylene relative to the other substrates, leads to racemic product in good yield, most likely a function of increased acidity of the newly formed stereocenter (entry 7).43 On the other hand, increasing the tether length by one methylene unit relative to the parent substrate results in complete suppression of the reaction and recovery of the unreacted starting material (entry 8).
Table 2.
Intramolecular Stetter reaction with different types of tether linkers. 
| Entry | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
| 1 |
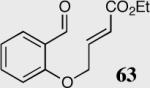
|
|
34 | 94 | 94 |
| 2 |
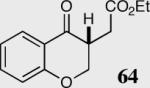
|
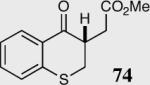
|
34 | 63 | 96 |
| 3 |
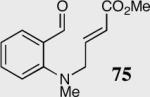
|
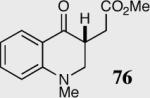
|
34 | 64 | 82 |
| 4 |
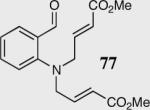
|
|
34 | 72 | 84 |
| 5 |
|
|
34 | 35 | 94(a) |
| 6 | 40 | 90 | 92b | ||
| 7 |
|
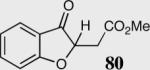
|
34 | 90 | <5 |
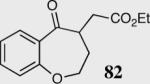
| 8 |
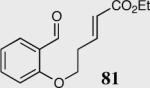
|
|
34 | 0 | NA |
S-enantiomer.
R-enantiomer.
6.4 Electronic Effects of the Aromatic Backbone of the Aldehyde on the Intramolecular Stetter Reaction
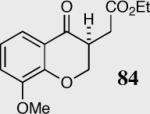
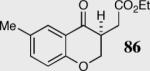
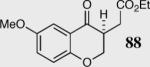
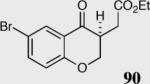
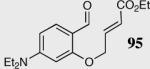
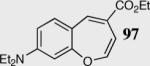
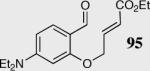
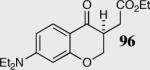
In an effort to understand how the nature of the aromatic aldehyde influences the reaction, we examined the role of electron-donating and -withdrawing groups on reactivity and selectivity (Table 3). Within this series, substitution of hydrogen at the 3 position of the aromatic ring by a larger group results in decreased selectivity (entries 1–2). The best results were obtained on substrates lacking steric bulk near the reactive site (entries 3–11). Electron-donating groups (EDG) afford higher enantioselectivity utilizing catalyst 40 relative to electron-withdrawing groups (EWG) in the same position on the aromatic ring. We observe that electron withdrawing substituents lead to product epimerization under the reaction conditions.44 Diminished enantioselectivities can be avoided by the implementation of the less basic catalyst 41. Increasing the electron-donating ability at the 4-position from methoxy to diethylamino results in an increase of an undesired aldol-elimination side reaction from 5% to 30% (97 is the side reaction product obtained for the diethylamino case). Once again, application of catalyst 41 increases the yield of the desired product without loss enantioselectivity and completely suppresses the formation of the undesired side reaction (entries 11 and 12).
Table 3.
Electronic effects of the aromatic aldehyde on the intramolecular Stetter reaction. 
| Entry | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
| 1 |
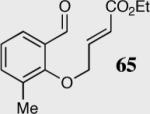
|
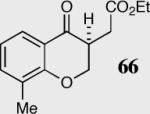
|
40 | 94 | 80 |
| 2 |
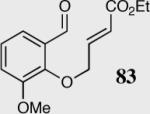
|
|
40 | 95 | 80 |
| 3 |
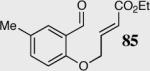
|
|
40 | 94 | 90 |
| 4 |
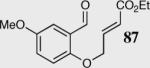
|
|
40 | 94 | 90 |
| 5 |

|
|
40 | 95 | 89 |
| 6 | 41 | 94 | 92 | ||
| 7 |
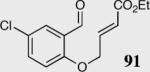
|
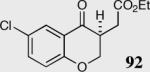
|
40 | 94 | 88 |
| 8 | 41 | 94 | 91 | ||
| 9 |
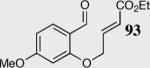
|
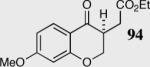
|
40 | 45 | 90 |
| 10 | 41 | 94 | 91 | ||
| 11 |
|
|
40 | 30 | -- |
| 12 |
|
|
41 | 55 | 95 |
| 13 |
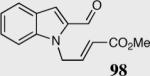
|
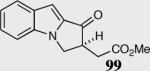
|
40 | 80 | 65 |
6.5 The Effects of the Michael Acceptor on the Asymmetric Intramolecular Stetter Reaction
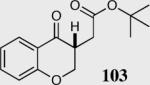
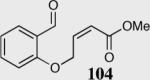
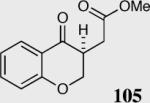
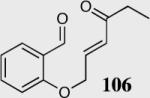
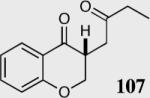
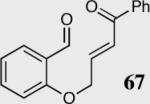
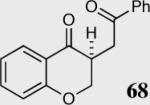
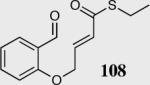
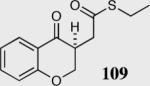
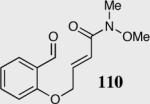
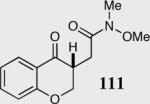
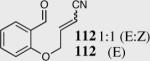
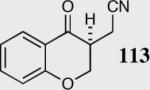
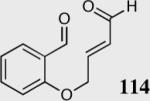
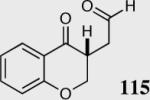
We initially investigated the effects of the Michael acceptor on the intramolecular Stetter reaction using catalyst 33.45 However catalyst 33 was unable to induce cyclization of α,β-unsaturated aldehydes, amides or Michael acceptors that possess Z alkenes. With access to a larger family of chiral catalysts we re-examined the effects of the Michael acceptor (Table 4). The reaction is remarkably broad with respect to the α,β-unsaturated Michael acceptor employed, with the successful cyclization of α,β-unsaturated aldehyde, amide, nitrile, esters, thioester, and ketones utilizing catalyst 35. Increasing the steric bulk of the α,β-unsaturated esters from methyl to tert-butyl results in a slight increase in selectivity (entries 1–3). To our gratification cyclization of the (Z)-α,β-unsaturated methyl ester 104 can now be achieved albeit in low enantioselectivity (entry 4). The cyclization of α,β-unsaturated ketones affords the desired product in excellent overall yield; however, there was a decrease in selectivity from the α,β-unsaturated ethyl ketone 106 to the phenyl ketone 67 (entries 5–6). To our delight, the α,β-unsaturated thioesters 108 and amides 110 are well tolerated in the intramolecular Stetter reaction (entries 7–8). The incorporation of the thioester and the Weinreb amide offers an excellent handle for further manipulations. α,β-Unsaturated nitrile 112 was subjected to the reaction as an inseparable 1:1 mixture of E/Z olefin isomers and to our surprise afforded the desired product in good yield and 80% enantioselectivity. Astonishingly, the enantioselectivity of the product was approximately identical to the reaction commencing from (E)-α,β-unsaturated nitrile 112 (entry 10). This unexpected result clearly illustrates that the Z olefin isomer is tolerated but the resulting enantioselectivity depends on the nature and size of the Michael acceptor. The α,β-unsaturated aldehyde affords the desired product in 50% isolated yield, although the product is isolated in only 30% ee.
Table 4.
Effects of the Micheal acceptor on the intramolecular Stetter reaction. 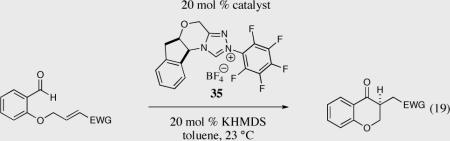
| Entry | Substrate | Product | Yield % | ee% |
|---|---|---|---|---|
| 1 |
|
|
94 | 95 |
| 2b |
|
|
94 | 93 |
| 3b |
|
|
94 | 97 |
| 4 |
|
|
80 | 22 |
| 5b |
|
|
94 | 92 |
| 6 |
|
|
94 | 78 |
| 7 |
|
|
85 | 70 |
| 8b |
|
|
94 | 92 |
| 9 |
|
|
88 | 80 |
| 10* | 80 | 78 | ||
| 11b |
|
|
50 | 30 |
20 mol % of catalsyt 33 was used.
Opposite antipode of the chiral triazolium salt 35 was used.
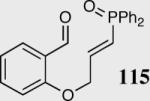
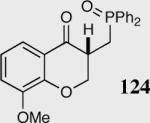
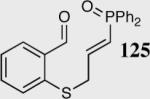
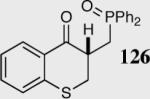
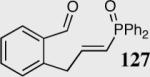
Encouraged by the ability of the acyl anion equivalent to add to a variety of Michael acceptors and owing to the synthetic utility of organophosphorus compounds, we decided to evaluate the ability of vinylphosphine oxides and vinylphosphonates as the elecrophilic component in the intramolecular Stetter reaction. While many nucleophiles add readily to the β-position of vinylphosphine oxides and vinylphosphonates, few examples have been shown to do so asymmetrically.46 To our gratification, employing a vinylphosphine oxide or vinylphosphonate as the electrophilic acceptor affords the Stetter product in typically high yields and enantioselectivities (Table 5).47
Table 5.
Vinylphosphine oxides and vinylphosphonate acceptors. 
| Enty | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
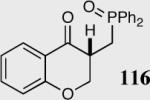
| 1 |
|
|
35 | 90 | 86 |
| 2 | 40a | 74 | |||
| 3 |
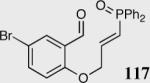
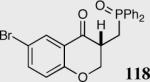
|
|
35 | 88 | 96 |
| 4 |
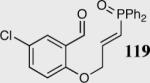
|
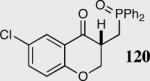
|
35 | 90 | 94 |
| 5 | 40a | 75 | |||
| 6 |
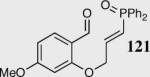
|
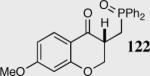
|
35 | 86 | 93 |
| 7 |
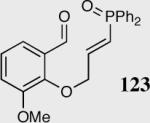
|
|
35 | 75 | 87 |
| 8 | 40a | 91 | |||
| 9 |
|
|
35 | 45 | 90 |
| 9 |
|
|
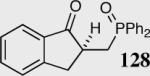
35 | 99 | 90 |
| 10 |
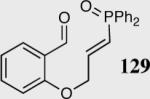
|
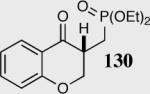
|
35 | 65 | 80 |
Reactions were performed using an analog of catalyst 40 where the N-Ph group was substitued with N-C6F5.
6.6 Asymmetric Intramolecular Stetter Reaction of Aliphatic Aldehydes
After successfully performing a highly enantioselective intramolecular Stetter reaction on a broad range of aromatic aldehydes we turned our attention to the potentially more difficult aliphatic aldehydes. Most aliphatic aldehydes bear acidic hydrogens α to the aldehyde and initially we were concerned that this could be a problem under our basic reaction conditions. We were delighted to learn that a variety of aliphatic aldehydes with different tethered Michael acceptors were also tolerated in the intramolecular Stetter reaction as illustrated in Table 6. Cyclopentanones could be generated in excellent yield and selectivity (entries 1, 2, 7, 8 and 10). Nitrogen is tolerated in the tether, affording the desired product in excellent enantiomeric excess, offering a new approach towards optically enriched pyrolidinones (entry 3). The intramolecular Stetter reaction resulting in cyclohexanone products was substantially more challenging and the cyclization of these substrates resulted in recovered starting material (entries 4 and 11). We were forced to use more activated Michael acceptors for the successful synthesis of cyclohexanones (entries 5–6). Exchanging the acceptor to a vinylphosphonate or vinylphosphine oxide also leads to successful intramolecular Stetter reactions to give the desired products in high yield and enantioselectivity (entries 7–10).
Table 6.
The intramolecular Stetter reaction of aliphatic aldehydes. 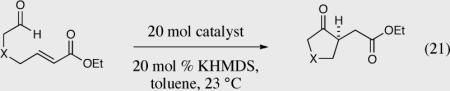
| Entry | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
| 1 |
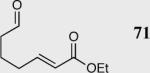
|
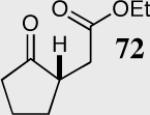
|
40 | 81 | 95 |
| 2 |
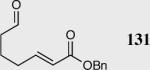
|
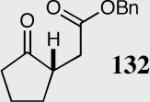
|
40 | 85 | 90 |
| 3 |
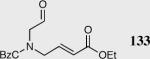
|
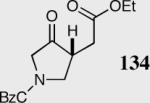
|
40 | 80 | >99 |
| 4 |
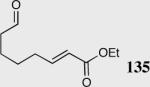
|
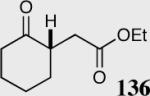
|
40 | 0 | -- |
| 5 |
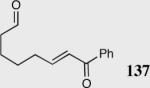
|
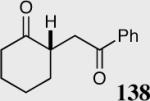
|
40 | 60 | 42 |
| 6 |
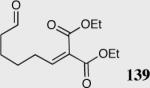
|
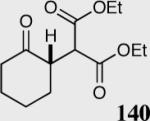
|
40 | 97 | 82 |
| 7 |
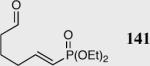
|
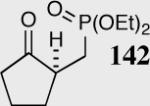
|
35 | 66 | 74 |
| 8 |
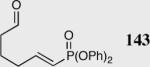
|
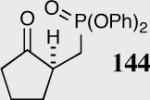
|
35 | 80 | 90 |
| 9 |
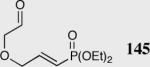
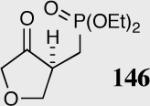
|
|
35 | 94 | 88 |
| 10 |
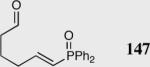
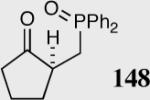
|
|
35 | 96 | 90 |
| 11 |
|
|
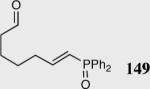
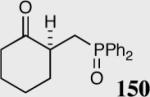
35 | 0 | -- |
7. The Effect of Pre-Existing Stereocenters in the Intramolecular Stetter Reaction
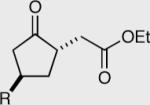
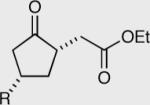
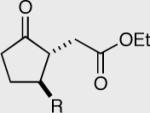
Investigation of the aliphatic substrates led us to probe the effects that a pre-existing stereocenter would have on the asymmetric intramolecular Stetter reaction.48 With this in mind we evaluated the impact of substitution at each position between the aldehyde and the tethered Michael acceptor (substrates are racemates). Substitution at the 3 or 4 position has no effect on the overall course of the reaction. The products are isolated in a 1:1 trans:cis ratio each in high enantioselectivity. On the other hand substitution at the 2-position (α to the aldehyde) appears to override the catalyst selectivity preference, thus resulting in low enantioselectivity. At the onset of this project we were optimistic that we might be able to perform some type of kinetic resolution utilizing substrates that contained pre-existing stereocenters, but as can be seen by these results, the current reaction conditions are not amenable to this hypothesis.
8. The Synthesis of Quaternary Stereocenters via Asymmetric Intramolecular Stetter Reaction
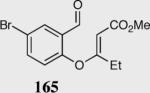
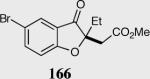
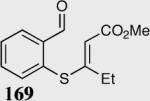
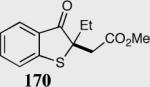
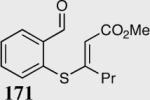
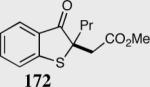
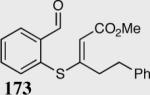
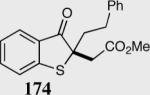
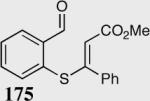
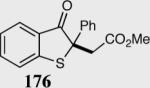
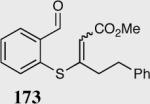
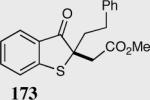
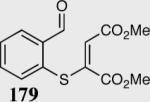
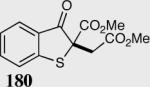
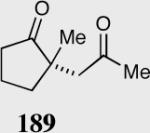
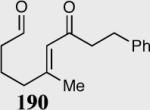
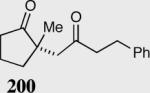
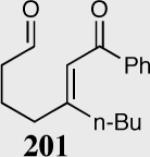
The asymmetric formation of quaternary stereocenters is a formidable challenge in organic chemistry.49 Our successful cyclization of various β-substituted Michael acceptors encouraged us to investigate the more challenging construction of quaternary stereocenters (Table 8).50 The formation of quaternary stereocenters utilizing an intramolecular Stetter reaction was first illustrated by Trost and co-workers during the synthesis of hirsutic acid; however, three equivalents of achiral thiazolium salt were required to perform the reaction.51 Vinylogous carbonate 163 was chosen as a test substrate and previously optimized reaction conditions afforded the desired tertiary ether in 99% ee and 45% yield. As illustrated above, we have been able to tune the electronics of the catalyst to increase the efficiency of the reaction and therefore a series of aminoindanol-derived catalysts were screened with pentafluorophenyl catalyst 35 identified as the most effective. With these optimized reaction conditions, the scope of the reaction was tested; benzofuranones, benzothiophenone and indanone products were all formed in excellent chemical yield and enantioselectivity. The corresponding six-membered ring suffers from lower reactivity, although the generation of a quaternary stereocenter is still achievable in 99% ee (entry 8). Treatment of thioether 177 leads to recovered starting material under the standard reaction conditions; however, the desired product could be obtained in > 99% ee by applying the more reactive triazolium catalyst 40, albeit in only 11% yield (entry 9).
Table 8.
Formation of quaternary stereocenters from aromatic aldehydes via the asymmetric Stetter reaction. 
| Entry | Substrate | Product | Yield % | ee% |
|---|---|---|---|---|
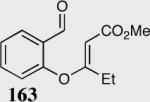
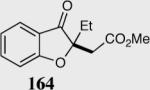
| 1 |
|
|
96 | 97 |
| 2 |
|
|
92 | 89 |
| 3 |
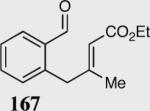
|
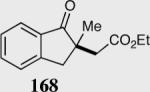
|
95 | 99 |
| 4 |
|
|
95 | 92 |
| 95a | 92a | |||
| 5 |
|
|
54 | 87 |
| 83a | 98a | |||
| 6 |
|
|
33 | 88 |
| 91a | 99a | |||
| 7 |
|
|
11 | 82 |
| 15a | 82a | |||
| 8b |
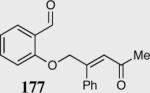
|
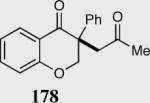
|
55 | 99 |
| 9 |
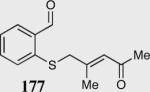
|
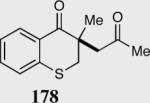
|
0 | --- |
| 11c | >99c |
KOt-Bu was used as the base.
Catalyst added in two portions.
Catalyst 40 was used.
A further study of the synthesis of enantioenriched benzothiophenones was initiated.52 Easy access to both geometric alkene isomers of the starting material allowed for further investigation of the requirements for obtaining good reactivity and enantioselectivity. Previously, we had observed higher reactivity and selectivity for (E)-α,β-unsaturated Michael acceptors and in some cases a dramatic decrease in enantioselectivity was observed. Therefore it was not surprising that the E alkene isomers lead to higher reactivity and enantioselectivity; however, it was nonetheless gratifying to learn that the Z alkene isomer could still lead to excellent reactivity and good enantioselectivity for the synthesis of various benzothiophenones (Table 9). Overall, the intramolecular Stetter reaction tolerates aromatic aldehydes with varied β,β-substitution of the Michael acceptor and heteroatom tethers.
Table 9.
Further investigation of the asymmetric Stetter reaction 
| Entry | Substrate | Product | Yield % | ee% |
|---|---|---|---|---|
| 1 |
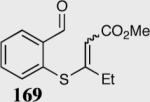
|
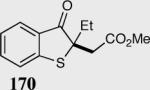
|
(E)90 | 97 |
| (Z)89 | 86 | |||
| 2 |
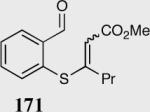
|
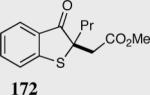
|
(E)83 | 98 |
| (Z)85 | 89 | |||
| 3 |
|
|
(E)91 | 99 |
| (Z)92 | 84 | |||
| 4a |
|
|
(Z)85 | 90 |
The opposite antipode of catalyst 34 was used.
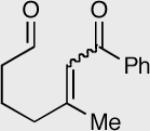
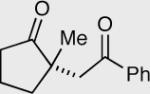
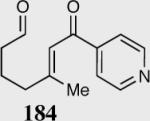
Similar to our previous results, the incorporation of aliphatic aldehydes that contain an acidic proton α to the aldehyde also provided competent substrates for the formation of quaternary stereocenters. Exposure of α,β-unsaturated phenyl ketone 181 to the reaction conditions affords an 85% yield and 96% ee. The corresponding isomer 183 was examined under identical reaction conditions and found to proceed more sluggishly giving lower yield and enantioselectivity. Recall that diminished selectivity, albeit to a lesser extent, results when Z-alkene isomers are employed (Table 9).
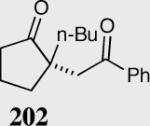
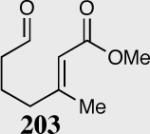
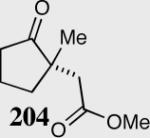
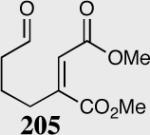
A variety of aliphatic substrates containing a trans relationship between the pendant aldehyde and Michael acceptor were investigated. Aliphatic aldehydes with tethered aromatic or aliphatic α,β-unsaturated ketones with modest steric bulk at the β-position are well tolerated (Table 10). Attempts to cyclize aliphatic aldehydes that contained a less activating Michael acceptor such as 203 failed, and starting material was recovered. Fortunately, tuning the electronics of the alkene and catalyst allows for the successful cyclization of β,β-disubstituted unsaturated esters (entries 9–11).
Table 10.
Formation of quaternary steocenters from aliphatic aldehydes tethered to different Michael acceptors. 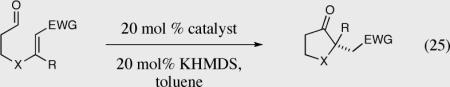
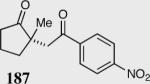
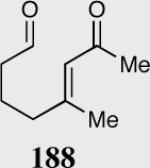
| Entry | Substrate | Product | Catalyst | Yield % | ee% |
|---|---|---|---|---|---|
|
|
||||
| 1 | 181 E-isomer | 182 R = 4-Py | 35 | 85 | 96 |
| 2 | 183 Z-isomer | 183 R = p-NO2Ph | 35 | 50 | 56 |
| 3 |
|
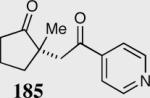
|
35 | 85 | 96 |
| 4 |
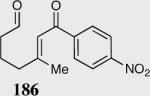
|
|
35 | 90 | 84 |
| 5 |
|
|
35 | 81 | 95 |
| 6 |
|
|
35 | 63 | 99 |
| 7 |
|
|
35 | 71 | 98 |
| 8 |
|
|
35 | 0 | -- |
| 9 |
|
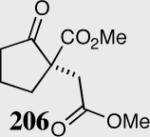
|
35 | 78 | 65 |
| 40 | 86 | 90a | |||
| 10 |
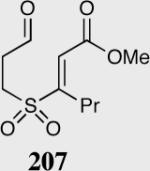
|
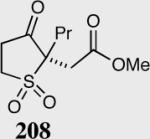
|
40 | 98 | 80a |
| 11 |
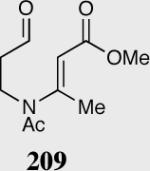
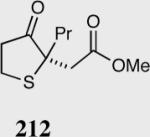
|
|
35 | 65 | 95 |
| 12 |
|
|
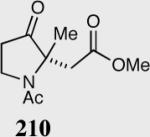
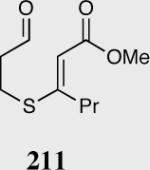
35 | 0 | -- |
| 40 | 0 | -- |
Opposite antipode was obtained.
9. The Synthesis of Contiguous Stereocenters via Asymmetric Intramolecular Stetter Reaction
Our research on the enantio- and diastereoselective intramolecular Stetter reaction began during investigations involving the deuteroaldehyde 213 (Scheme16).53 Following the Stetter reaction of 213 by 2H NMR, we noted the formation of two diastereomers with a 3:1 selectivity, along with a new deuterium signal in an area that did not correspond to any protons in either starting material or product. We ultimately determined that the conjugate acid of the base used to generate the carbene was undergoing partial deuteration. In the absence of this conjugate acid (HMDS, hexamethyldisilazane), the diastereoselectivity of deuteration increased to 9:1 by 2H NMR.
Scheme 16.
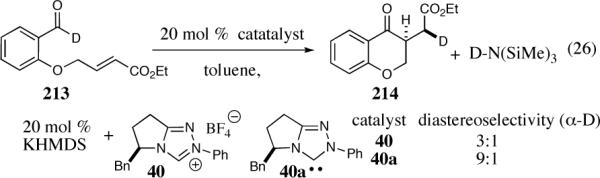
Initial discovery of the enantio- and diastereoselective intramolecular Stetter reaction.
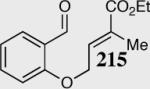
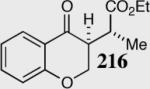
Having realized the viability of a diastereoselective deuteron transfer, we turned our attention to prochiral trisubstituted Michael acceptors. The cyclization of α-methyl substituted α,β-unsaturated ethyl ester 214 was chosen as a model substrate for our investigation (Scheme 17). To our gratification, optimal reaction conditions were conducted in the absence of HMDS (a catalyst that we term the free carbene) and catalyst optimization revealed an increase in enantio- and diastereoselectivity utilizing the slightly more electron-deficient carbene 41. The nature of the catalyst and the presence of the conjugate acid HMDS were both shown to diminish the diastereoselectivity of this reaction, presumably by a base induced mechanism.
Scheme 17.
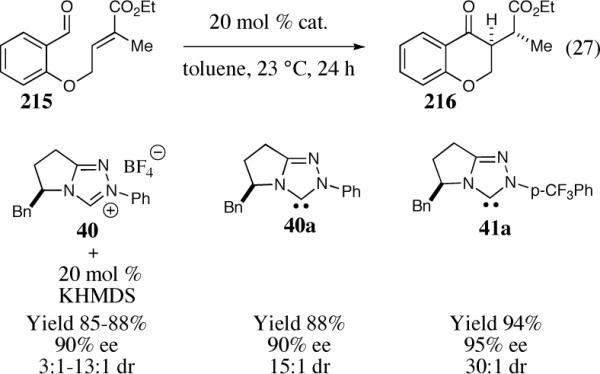
Optimization of the enantio- and diastereoselective intramolecular Stetter reaction.
With optimized reaction conditions in hand a series of prochiral trisubstituted Michael acceptors were prepared to test the scope of the enantio- and diastereoselective Stetter reaction (Table 11).
Table 11.
A highly enantio- and diastereoselective intramolecular Stetter reaction. 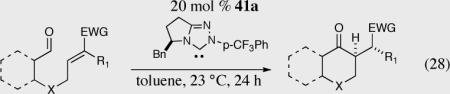
| Entry | Substrate | Product | Yield (%) | ee (%)a | dr (%)a |
|---|---|---|---|---|---|
| 1 |
|
|
94 | 95 | 30:1 |
| 2 |
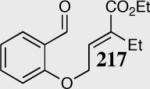
|
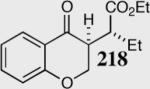
|
95 | 92 | 35:1 |
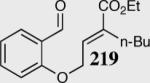
| 3b |
|
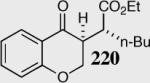
|
53 | 94 | 12:1 |
| 4 |
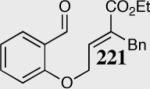
|
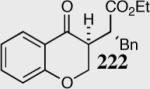
|
80 | 84 | 20:lc |
| 5 |
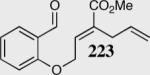
|
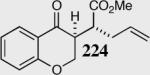
|
95 | 83 | 13:1 |
| 6 |
|
|
95 | 94 | 10:1 |
| 7 |
|
|
80 | 95 | 18:1 |
| 8 |
|
|
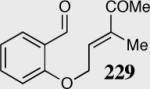
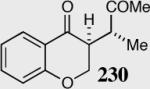
85 | 55 | 10:1 |
| 9 |
|
|
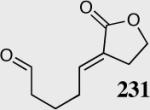
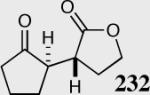
94 | 99 | 50:1 |
| 10 |
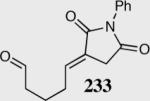
|
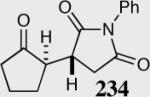
|
80 | 88 | 15:1 |
Enantiomeric excess of the major diasteromer and diastereomeric ratio determined by HPLC or GC analysis using a chiral stationary phase.
Catalyst added in two portions, see supporting information.
Diastereomeric ratio determined by 1H NMR.
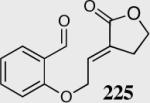
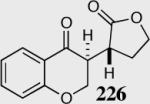
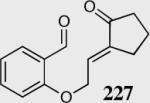
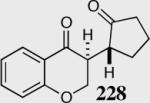
The reaction displays excellent generality with respect to nature and size of the α-substituted Michael acceptors. Moderate steric bulk can be tolerated at the α-position of various α-disubstituted α,β-unsaturated esters (entries 1–5). Alkylidene lactone and cyclopentanone each afford the desired product in high enantio- and diastereoselectivity (entries 6–7). Aliphatic aldehydes are also viable substrates, affording the desired product in >80% yield with good enantio- and diastereoselectivity (entries 9–10). The formation of the bicyclic and tricyclic products in entries 6,7, 9 and 10 is particularly exciting. The construction of adjacent rings that are joined by a single carbon-carbon bond with each end being a stereogenic center remains difficult using other approaches.
The relative stereochemistry of the newly formed contiguous stereocenters was assigned as syn based on single crystal analysis for 216 and 226. This stereochemistry was concluded to arise from a diastereoselective proton transfer event from the sterically more hindered face of the enolate, subsequent to carbon-carbon bond formation (Scheme 19).54
Scheme 19.

Possible intermediates for the enantio- and diastereoselective intramolecular Stetter reaction
Support for the intramolecular proton transfer was gained by examining isomeric Michael acceptors. According to the model 240 and 244 the relative diastereoselectivity should be dependent on the olefin geometry of the Michael acceptor assuming proton transfer is faster than bond rotation. Substrates 235 and 237 were subjected to the reaction conditions and found to provide complementary diastereoselectivity. The (E)-isomer proved highly enantio- and diastereoselective, affording 236 in 42:1 diastereoselectivity (Scheme 18), while the (Z)-isomer preferentially affords 238 in 1:6 diastereoselectivity, albeit low enantioselectivity (Scheme 18).
Scheme 18.
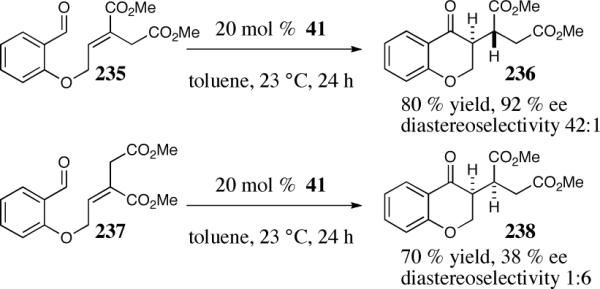
Control of the relative diastereoselectivity depending on the alkene geometry.
The control of the relative diastereoselectivity suggests enolate protonation must occur prior to bond rotation (Scheme 19).55 Intramolecular protonation of enolate 240 or 244 would result in the observed diastereoselectivity. Rapid bond rotation relative to proton transfer would access a common enolate intermediate that would result in the same relative diastereoselectivity.
10. The Asymmetric Synthesis of Hydrobenzofuranones via the Intramolecular Stetter Reaction
In an ongoing effort to expand the generality of the asymmetric Stetter reaction and to construct complex products from simple starting materials we investigated the asymmetric Stetter reaction of cyclohexadienones.56 The requisite substrates are readily accessible in two steps from hypervalent iodine-mediated oxidation of para-substituted phenols and trapping with glycols followed by oxidation of the primary alcohol. Treatment of a range of substrates to optimized reaction conditions affords product in high yields and enantioselectivities with two contiguous stereocenters. This reaction once again illustrated a subtle impact on enantioselectivity and reactivity arising from electronic variations in the catalyst with triazolium salt 34 proving to be the optimal catalyst. Alkyl and aryl substitution at the 4-position leads to good to excellent enantioselectivity (82–94% ee) and proceeds with greater than 95:5 diastereoselectivity (by 1H NMR).
Expedient access to highly substituted phenol-derived cyclohexadienones allowed us to synthesize hexahydrobenzofuranones possessing three contiguous stereocenters. To our gratification, the reaction is remarkably fast (5 minutes) and generates a single diastereomer in up to >99% ee for a variety of different substitutions at the 2,4 and 6 position. In addition, 3,4,5-trimethylphenol-derived substrate allowed for the synthesis of 273 containing one quaternary stereocenter adjacent to a tertiary ether in excellent enantioselectivity, diastereoselectivity and chemical yield.
By far the most surprising aspect of this reaction, gleaned from a solvent study, was the effect of alcoholic solvents on the reaction, which invariably afford the opposite enantiomer using the same series of catalyst (Table 13).57 We speculate this inversion in selectivity occurs due to hydrogen bonding that changes the chiral environment, ultimately affecting the stereochemical outcome of the reaction.
Table 13.
Screen of solvents. 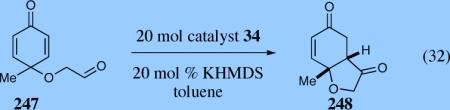
| Entry | Solvent | Yield % | ee% |
|---|---|---|---|
| 1 | toluene | 90 | 88 |
| 2 | xylenes | 81 | 75 |
| 3 | benzene | 88 | 89 |
| 4 | Et2O | 67 | 87 |
| 5 | DMF | 71 | 73 |
| 6 | THF | 77 | 89 |
| 7 | CH2Cl2 | 16 | 67 |
| 8 | MeOH | 29 | −11 |
| 9 | EtOH | 63 | −42 |
| 10 | CF3CH2OH | 0 | NA |
| 11 | i-PrOH | 64 | −63 |
| 12 | i-BuOH | 30 | −57 |
11. Applications to Total Synthesis
Although the bulk of our early work has been focused on the development of the asymmetric Stetter reaction, an increasing amount of effort in recent years has been dedicated to applying this transformation to complex molecule total synthesis. The success of the racemic or achiral Stetter reaction in total synthesis58 has spurred our own efforts to apply our family of catalysts to these types of problems.
With this in mind, we turned our focus on a family of natural products possessing a spirofuranone-lactam core, exemplified by FD-838 (Scheme 20). Subjection of maleimide 276 to the asymmetric Stetter reaction using catalyst 32 provides the spirofuranone succinimide as a single enantiomer in good yield. A short sequence of steps provided a functionalized advanced intermediate.59
Scheme 20.
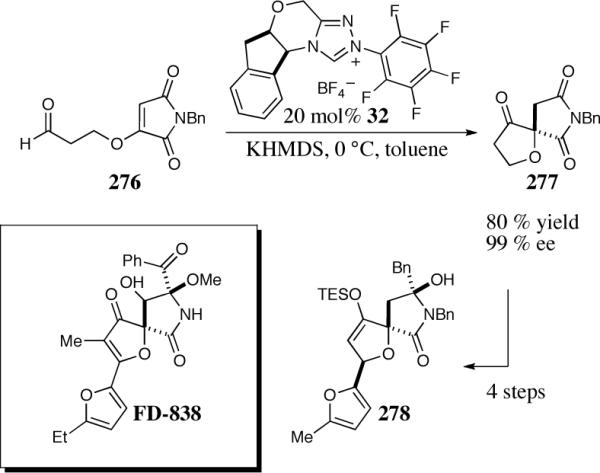
Model study approach to FD-838.
12. Summary and Outlook
The application of chiral heteroazolium carbenes for the asymmetric Stetter reaction has seen tremendous advances in the last decade, especially in the asymmetric intramolecular variant. A continued understanding of the effects that govern reactivity and selectivity of the asymmetric Stetter reaction will undoubtedly aide in further advances. In addition, the development of new and more efficient catalysts that are capable of overcoming the current limitations of the asymmetric Stetter reaction are necessary, particularly in the asymmetric intermolecular Stetter reaction. With the latter point in mind, we have recently discovered that electron-deficient pyrrolidine catalyst 279 mediates the intramolecular Stetter reaction between glyoxamides and alkylidine malonates in excellent yields and selectivities, eq 33.60 Further studies in this area hold significant promise at ultimately delivering a broadly useful and general intermolecular asymmetric Stetter reaction.

Table 7.
The effexts of pre-existing stereocenters in the intramolecular Stetter reaction (substrates are racemates). 
|
|
trans:cis | |
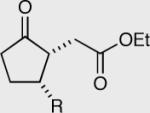
| R = Me l51 98% ee | R = Me l53 94% ee | 50:50 | Yield = 97% |
| R = Ph 152 98% ee | R = Ph 154 96% ee | 50:50 | Yield = 96% |
|
|
||
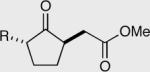
| R = Me 155 90% ee | R = Me 157 95% ee | 50:50 | Yield = 90% |
| R = iPr 156 84% ee | R = Ph 158 96% ee | 50:50 | Yield = 95% |
| aR = iPr 156 94% ee | aR = Ph 158 98% ee | a50:50 | Yield = 95% |
|
|
||
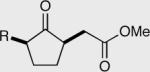
| R = Bn 159 <5% ee | R = Bn 161 <5% ee | 15:85 | Yield = 95% |
| aR = Cy 160 20% ee | aR = Cy l62 6% ee | a3.5:96.5 | Yield = 74% |
20 mol% catalyst 40 was used.
Table 12.
Asymmetric synthesis of hydrobenzofuranones via the intramolecular Stetter reaction 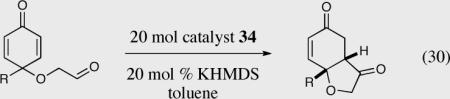
| Entry | R | Substrate | Product | Yield % | ee% |
|---|---|---|---|---|---|
| 1 | Me | 247 | 248 | 90 | 92 |
| 2 | Et | 248 | 249 | 86 | 94 |
| 3 | i-Pr | 250 | 251 | 87 | 94 |
| 4 | t-Bu | 252 | 253 | 86 | 94 |
| 5 | Ph | 254 | 255 | 87 | 88 |
| 6 | 4-BrPh | 256 | 257 | 78 | 85 |
| 7 | CH2OAc | 258 | 259 | 86 | 83 |
| 8 | CH2CH2OMe | 260 | 261 | 86 | 82 |
| 9 | CH2CH2CO2Me | 262 | 263 | 94 | 87 |
Table 12.
Synthesis of three contiguous stereocenters via the asymmetric Stetter reaction. 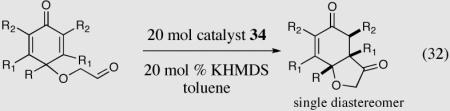
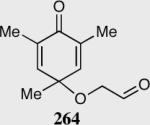
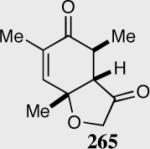
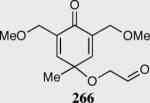
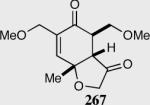
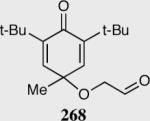
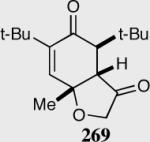
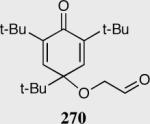
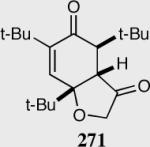
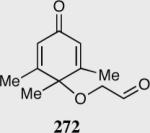
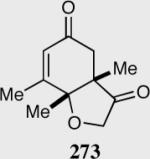
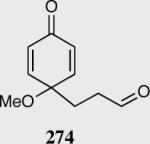
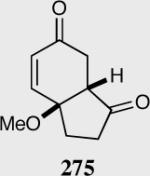
Reaction proceeds in > 95:5 dr.
The free carbene was used.
Acknowledgment
Advances described in this account would not have been possible without the intellectual and experimental contributions of our coworkers, whose names are listed in the relevant references. We are particularly grateful to Mark Kerr who, along with the two of us, started this project. We gratefully acknowledge the NSF (CAREER) and NIGMS (GM72586) for generous support of our programs. J. R. thanks the National Institutes of Health (minority supplement and Ruth L. Kirschstein Minority Pre-doctoral fellowship) and Colorado PEAKS AGEP (graduate fellowship). T. R. thanks Merck, Johnson and Johnson, GlaxoSmithKline, Eli Lilly, Amgen and Boehringer-Ingelheim for unrestricted support. T. R. thanks the Herman Frasch Foundation, the A. P. Sloan Foundation, and the Monfort Family Foundation. We gratefully acknowledge various individuals at Merck Research Laboratories, most recently Donald Gauthier, for generous gifts of aminoindanol.
References
- 1.For reviews, see: Regitz M. Angew. Chem., Int. Ed. Engl. 1996;35:725.. Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem. Rev. 2000;100:39. doi: 10.1021/cr940472u.. Herrmann WA. Angew. Chem. Int. Ed. 2002;41:1291. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y..
- 2.For reviews, see: Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j.. Johnson JS. Angew. Chem. Int. Ed. 2004;43:1326. doi: 10.1002/anie.200301702.. Pohl M, Lingen B, Müller M. Chem. Eur. J. 2002;8:5288. doi: 10.1002/1521-3765(20021202)8:23<5288::AID-CHEM5288>3.0.CO;2-F.. Nair V, Bindu S, Sreekumar V. Angew. Chem. Int. Ed. 2004;43:5130. doi: 10.1002/anie.200301714.. Zeitler K. Angew. Chem. Int. Ed. 2004;43:7506.. Christmann M. Angew. Chem. Int. Ed. 2005;44:2632. doi: 10.1002/anie.200500761.. Webber P, Krische MJ. Chemtracts: Org. Chem. 2007;19:262..
- 3.Seebach D. Angew. Chem., Int. Ed. Engl. 1979;18:239. [Google Scholar]
- 4.For reviews, see: Albright JD. Tetrahedron. 1983;39:3207.. Aitken RA, Thomas AW. Adv. Heterocyclic Chem. 2001;79:89..
- 5.For examples of the benzoin reaction that are not referenced later in the text, see; Hachisu Y, Bode JW, Suzuki K. J. Am. Chem. Soc. 2003;125:8432. doi: 10.1021/ja035308f.. Enders D, Niemeier O, Balensiefer T. Angew. Chem. Int. Ed. 2006;45:1463. doi: 10.1002/anie.200503885.. For examples of the benzoin reaction with acyl silanes see; Linghu X, Johnson JS. Angew. Chem. Int Ed. 2003;42:2534. doi: 10.1002/anie.200250554.. Linghu X, Potnick JR, Johnson JS. J. Am. Chem. Soc. 2004;126:3070. doi: 10.1021/ja0496468.
- 6.Stetter H, Kuhlmann H. In: Organic Reactions. Paquette LA, editor. Vol. 40. Wiley; New York: 1991. p. 407. [Google Scholar]
- 7.(a) Tomioka K, Koga K. Noncatalytic Additions to alpha,beta-Unsaturated Carbonyl Compounds. In: Morrison JD, editor. Asymmetric Synthesis. Vol. 2. AP; New York: 1983. p. 201. [Google Scholar]; (b) Yoshikoshi A, Miyashita M. Acc. Chem. Res. 1985;18:284. [Google Scholar]; (c) Rosini G, Balleni R. Synthesis. 1988:833. [Google Scholar]
- 8.Lapworth A. J. Am. Chem. Soc. 1903;83:995. [Google Scholar]
- 9.For the mechanism of the thiamine-catalyzed benzoin reaction, see: Breslow R. J. Am. Chem. Soc. 1958;80:3719.. Breslow R, Kim R. Tetrahedron Lett. 1994;35:699.. White M, Leeper F. J. Org. Chem. 2001;66:5124. doi: 10.1021/jo010244h..
- 10.Wöhler F, Liebig J. Ann. Pharm. 1832;3:249. [Google Scholar]
- 11.Ugai T, Dokawa T, Tsubokawa SJ. Pharm. Soc. Jpn. 1943;63:296. [Google Scholar]
- 12.Pohl M, Lingen B, Muller M. Chem. Eur. J. 2002;8:5289. doi: 10.1002/1521-3765(20021202)8:23<5288::AID-CHEM5288>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 13.Sheehan JC, Hunnemann DH. J. Am. Chem. Soc. 1966;88:3666. [Google Scholar]
- 14.Sheehan JC, Hara T. J. Org. Chem. 1974;39:1196. [Google Scholar]
- 15.(a) Tagaki W, Tamura Y, Yano Y. Bull. Chem. Soc. Jpn. 1980;53:478. [Google Scholar]; (b) Martí J, Castells J, López–Calahorra F. Tetrahedron Lett. 1994;35:699. [Google Scholar]; (c) Yamashita K, Sasaki S–I, Osaki T, Nango M, Tsuda K. Tetrahedron Lett. 1995;36:4816. [Google Scholar]
- 16.Igau A, Grutzmacher H, Baceiredo A, Trinquier G, Betrand G. Angew. Chem. Int. Ed. 1989;28:621. [Google Scholar]
- 17.Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. [Google Scholar]
- 18.Enders D, Breuer K, Teles JH. Helv. Chim. Acta. 1996;79:1217. [Google Scholar]
- 19.(a) Knight RL, Leeper FJ. Tetrahedron Lett. 1997;38:3611. [Google Scholar]; (b) Gerhard AU, Leeper FJ. Tetrahedron Lett. 1997;38:3615. [Google Scholar]
- 20.Dvorak CA, Rawal VH. Tetrahedron Lett. 1998;39:2925. [Google Scholar]
- 21.Knight RL, Leeper FJ. J. Chem. Soc. Perkin Trans1. 1998:1891. [Google Scholar]
- 22.Enders D, Kallfass U. Angew. Chem. Int. Ed. 2002;41:1743. doi: 10.1002/1521-3773(20020517)41:10<1743::aid-anie1743>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 23.Dünkelmann P, Kolter-Jung D, Nitsche A, Demir AS, Siegert P, Lingen B, Baumann M, Pohl M, Müller M. J. Am. Chem. Soc. 2002;124:12084. doi: 10.1021/ja0271476. [DOI] [PubMed] [Google Scholar]
- 24.(a) Enders D, Niemeier O, Balensiefer T. Angew. Chem. Int. Ed. 2006;45:1463. doi: 10.1002/anie.200503885. [DOI] [PubMed] [Google Scholar]; (b) Enders D, Niemeier O, Raabe G. Synlett. 2006;15:2431. [Google Scholar]
- 25.Takikawa H, Hachisu Y, Bode JW, Suzuki K. Angew. Chem. Int. Ed. 2006;45:3492. doi: 10.1002/anie.200600268. [DOI] [PubMed] [Google Scholar]
- 26.Mennen SM, Miller SJ. J. Org. Chem. 2007;72:5260. doi: 10.1021/jo070676d. [DOI] [PubMed] [Google Scholar]
- 27.Linghu X, Potnick JR, Johnson JS. J. Am. Chem. Soc. 2004;126:3070. doi: 10.1021/ja0496468. [DOI] [PubMed] [Google Scholar]
- 28.Kerr MS, Read de Alaniz J, Rovis T. J. Org. Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rovis T. Chem. Lett. 2008:2. [Google Scholar]
- 30.Ghosh AK, Fidanze S, Senanayake CH. Synthesis. 1998:937. doi: 10.1055/s-1998-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norman BH, Kroin JS. J. Org. Chem. 1996;61:4990. [Google Scholar]
- 32.(a) Meyers AI, Tavares FX. J. Org. Chem. 1996;61:8207. doi: 10.1021/jo9613491. [DOI] [PubMed] [Google Scholar]; (b) Smrcina M, Majer P, Majerová E, Guerassina T, Eissenstat MA. Tetrahedron. 1997;53:12867. [Google Scholar]
- 33.(a) Enders D, Breuer K. Addition of acyl carbanion equivalents to carbonyl groups and enones. In: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I–III. Vol. 3. Springer-Verlag; Berlin, Germany: 1999. p. 1093. [Google Scholar]; (b) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]
- 34.Nahm MR, Potnick JR, White PS, Johnson JS. J. Am. Chem. Soc. 2006;128:2751. doi: 10.1021/ja056018x. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.For related examples of acyl silanes in the Stetter reaction see: Mattson AE, Bharadwaj AR, Scheidt KA. J. Am. Chem. Soc. 2004;126:2314. doi: 10.1021/ja0318380.. Bharadwaj AR, Scheidt KA. Org. Lett. 2004;6:2465. doi: 10.1021/ol049044t..
- 36.Mattson AE, Zuhl AM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2006;128:4932. doi: 10.1021/ja056565i. [DOI] [PubMed] [Google Scholar]
- 37.Ciganek E. Synthesis. 1995:1311.. For a seminal example of an intramolecular Stetter reaction, see; Trost BM, Shuey CD, DiNinno F, McElvain CD. J. Am. Chem. Soc. 1979;101:1284..
- 38.Enders D, Breuer K, Runsink J. Helv. Chim. Acta. 1996;79:1899. [Google Scholar]
- 39.Pesch J, Harms K, Bach T. Eur. J. Org. Chem. 2004:2025. [Google Scholar]
- 40.Mennen SM, Blank JT, Tran MB, Imbriglio JE, Miller SJ. Chem. Commun. 2005:195. doi: 10.1039/b414574g. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto Y, Tomioka K. Tetrahedron Lett. 2006;47:5843. [Google Scholar]
- 42.(a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (b) Read de Alaniz J, Kerr MS, Moore JL, Rovis T. J. Org. Chem. 2008;73:2033. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Investigation of the enantioselectivity as a function of conversion revealed that 80 is formed in 80% enantiomeric excess at 10% conversion, with rapid erosion to 50% ee at 30% conversion.
- 44.Similar observations have been noted by Miller; see reference 40.
- 45.Kerr MS, Rovis T. Synlett. 2003:1934. [Google Scholar]
- 46.(a) Minowa N, Hirayama M, Fukatsu S. Tetrahedron Lett. 1984;25:1147. [Google Scholar]; (b) Ruiz M, Vicente O, Shapiro G, Weber HP. Tetrahedron Lett. 1994;35:4551. [Google Scholar]; (c) Hayashi T, Senda T, Takaya Y, Ogasawara M. J. Am. Chem. Soc. 1999;121:11591. [Google Scholar]; (d) Fernández M, Diaz A, Guillin JJ, Blanco O, Ruiz M, Vicente O. J. Org. Chem. 2006;71:6958. doi: 10.1021/jo061072x. [DOI] [PubMed] [Google Scholar]; (e) Kondoh A, Yorimitsu H, Oshima K. J. Am. Chem. Soc. 2007;129:6996. doi: 10.1021/ja071622o. [DOI] [PubMed] [Google Scholar]; (f) Nishida G, Noguchi K, Hirano M, Tanaka K. Angew. Chem. Int. Ed. 2008;47:3410. doi: 10.1002/anie.200800144. [DOI] [PubMed] [Google Scholar]; (g) Sulzer-Mossé S, Tissot M, Alexakis A. Org. Lett. 2007;9:3749. doi: 10.1021/ol7015498. [DOI] [PubMed] [Google Scholar]; (h) Capuzzi M, Perdicchia D, Jørgensen KA. Chem. Eur. J. 2008;14:128. doi: 10.1002/chem.200701317. [DOI] [PubMed] [Google Scholar]
- 47.Cullen SC, Rovis T. Org. Lett. 2008;10:3141. doi: 10.1021/ol801047k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368. [Google Scholar]
- 49.(a) Corey EJ, Guzman–Perez A. Angew. Chem. Int. Ed. 1998;37:389. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Christoffers J, Baro A. Angew. Chem. Int. Ed. 2003;42:1688. doi: 10.1002/anie.200201614. [DOI] [PubMed] [Google Scholar]; (c) Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;126:8876. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]
- 51.Trost BM, Shuey CD, Dinnino F, McElvain SS. J. Am. Chem. Soc. 1979;101:1284. [Google Scholar]
- 52.Moore JL, Kerr MS, Rovis T. Tetrahedron. 2006;62:11477. [Google Scholar]
- 53.Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]
- 54.For examples of intramolecular proton transfer see; Zimmerman HE. Acc. Chem. Res. 1987;20:263.. Zimmerman HE, Wang P. Org. Lett. 2002;15:2593. doi: 10.1021/ol026251p. and references therein. Berrada S, Metzner P. Tetrahedron Lett. 1987;28:409..
- 55.It is also possible that the α-hydroxy-α-azolium anion adds to the Michael acceptor in concerted fashion, analogous to the reverse Cope elimination mechanism seen with hydroxylamine additions. See: Niu D, Zhao K. J. Am. Chem. Soc. 1999;121:2456.. Sibi MP, Liu M. Org. Lett. 2000;21:3393. doi: 10.1021/ol006500e.. Sibi MP, Prabagaran N, Ghorpade SG, Jasperse CP. J. Am. Chem. Soc. 2003;125:11796. doi: 10.1021/ja0372309..
- 56.Liu Q, Rovis T. J. Am. Chem. Soc. 2006;127:2552. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Q, Rovis T. Org. Proc. Res. Dev. 2007;11:598. doi: 10.1021/op600278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Stetter H, Kuhlmann H. Synthesis. 1975:379. [Google Scholar]; (b) reference 51.; (c) Baumann KL, Butler DE, Deering CF, Mennen KE, Millar A, Nanninga TN, Palmer CW, Roth BD. Tetrahedron Lett. 1992;33:2283. [Google Scholar]; (d) Galopin CC. Tetrahedron Lett. 2001;42:5589. [Google Scholar]; (e) Harrington PE, Tius MA. J. Am. Chem. Soc. 2001;123:8509. doi: 10.1021/ja011242h. [DOI] [PubMed] [Google Scholar]; (f) Anjaiah S, Chandrasekhar S, Gree R. Adv. Synth. Cat. 2004;346:1329. [Google Scholar]; (g) Nicolaou KC, Tang YF, Wang JH. Chem. Commun. 2007:1922. doi: 10.1039/b704589a. [DOI] [PubMed] [Google Scholar]
- 59.Orellana A, Rovis T. Chem. Commun. 2008:730. doi: 10.1039/b716445a. [DOI] [PubMed] [Google Scholar]
- 60.Liu Q, Perreault S, Rovis T. J. Am. Chem. Soc. 2008;130:14066. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]