

Abstract
The matricellular protein osteonectin, secreted protein acidic and rich in cysteine (SPARC, BM-40), is the most abundant non-collagenous matrix protein in bone. Matricellular proteins play a fundamental role in the skeleton as regulators of bone remodeling. In the skeleton, osteonectin is essential for the maintenance of bone mass and for balancing bone formation and resorption in response to parathyroid hormone (PTH). It promotes osteoblast differentiation and cell survival. Mechanisms regulating the expression of osteonectin in the skeleton and in other tissues remain poorly understood. We found that the proximal region of the mouse osteonectin 3′ untranslated region (UTR) contains a well-conserved, dominant regulatory motif that interacts with microRNAs (miRs)-29a and -29c. Transfection of osteoblastic cells with miR-29a inhibitors increased osteonectin protein levels, whereas transfection of miR-29a precursor RNA decreased osteonectin. miR-29a and -29c were increased during osteoblastic differentiation in vitro. The up-regulation of these miRNAs correlated with decreased osteonectin protein during the matrix maturation and mineralization phases of late differentiation. In contrast, osteonectin transcript levels remained relatively constant during this process, implying repression of translation. Treatment of osteoblasts with LiCl induced miR-29a and -29c expression and decreased osteonectin synthesis. When cells were treated with Dickkopf-1 (Dkk-1), miR-29a and -29c expression was repressed. These data suggest that canonical Wnt signaling, which is increased during osteoblastic differentiation, induces expression of miR-29. Osteonectin and miR-29 are co-expressed in extra-skeletal tissues, and the post-transcriptional mechanisms regulating osteonectin in osteoblasts are likely to be active in other cell systems.
Keywords: SPARC, OSTEONECTIN, POST-TRANSCRIPTIONAL REGULATION, MICRORNA
Osteonectin, also called secreted protein acidic and rich in cysteine (SPARC) or BM-40, is a matricellular glycoprotein expressed in a variety of mammalian tissues including muscle, brain, adipose, testes, kidney, skin, bone, and cartilage [Sage et al., 1989]. Its expression is increased in areas of extracellular matrix remodeling such as wound healing, angiogenesis, tumor growth, and metastasis [Bradshaw and Sage, 2001]. The effects of osteonectin on cell behavior are highly tissue specific, and range from the regulation of collagen fibril assembly, to the modification of cell shape, migration, proliferation, differentiation, and survival [Motamed et al., 2002; Delany et al., 2003; Alford and Hankenson, 2006; Kunigal et al., 2006; Rentz et al., 2007]. It can also interact with growth factors and cell-associated molecules including vascular endothelial growth factor (VEGF), vascular cell adhesion molecule-1 (VCAM-1; CD106), and platelet derived growth factor BB (PDGF BB) [Raines et al., 1992; Chandrasekaran et al., 2007; Kelly et al., 2007].
Recently, osteonectin was shown to interact with β1 integrin and stimulate signaling from integrin-linked kinase [Weaver et al., 2008]. Knock down of osteonectin expression also decreases signaling from focal adhesion kinase, and we recently demonstrated attenuated coupling of Gs to adenylyl cyclase in an osteonectin-null osteoblastic cell line [Kessler and Delany, 2007; Shi et al., 2007]. Thus, osteonectin can modify signaling from selected membrane-associated effectors.
Osteonectin is the most abundant non-collagen extracellular matrix protein in bone [Robey and Boskey, 2006]. In the skeleton, osteonectin is critical for normal bone remodeling and the maintenance of bone mass. Osteonectin-null mice have increased marrow adiposity, develop profound low turnover osteopenia in the trabecular compartment, and have cortical bone with decreased mechanical properties and matrix quality [Delany et al., 2000; Boskey et al., 2003; Mansergth et al., 2007]. Osteonectin promotes osteoblast commitment at the expense of adipogenesis, and supports osteoblast survival in vitro and in vivo [Delany et al., 2003; Kessler and Delany, 2007; unpublished data]. Further, osteonectin levels modulate the balance of bone formation and resorption in response to intermittent parathyroid hormone (PTH) administration in vivo [Machado do Reis et al., 2008].
There is a rapidly growing body of literature describing the function of osteonectin/SPARC in multiple tissues and disease states. In contrast, there is a deficit in our knowledge of the mechanisms regulating the expression of osteonectin itself. A limited number of studies describe sequence motifs and trans-acting factors necessary for basal level transcription of osteonectin [Nomura et al., 1989; Dominguez et al., 1991; Hafner et al., 1995]. However, information on post-transcriptional regulation of osteonectin is quite sparse [Delany and Canalis, 1998].
One major mechanism mediating post-transcriptional gene regulation is control by microRNAs (miRNAs). miRNAs are a recently characterized class of small, non-coding RNAs that modulate the translation or stability of a target mRNA through interaction with its 3′ untranslated region (UTR), in a manner similar to that of small interfering RNA (siRNA). miRNAs are ~21–23 base single-stranded RNAs associated with a protein complex, the RNA-induced silencing complex (RISC). The RISC is a ubiquitous protein complex whose activity is targeted to specific transcripts by miRNAs, which are the limiting factors for regulation [Bartel, 2004; Bartel and Chen, 2004; Rana, 2007]. miRNA mode of action depends on the degree of homology to its target sequence. Perfect or near perfect pairing between the target and miRNA sequences results in cleavage of the mRNA. A lower degree of complementarity results in translational repression. Perfect pairing between target and miRNA sequence is rare in mammals, and translational repression appears to be the dominant regulatory mechanism in higher species. Complementary pairing between the “seed” region of the miRNA (bases 2–8) and the “box” region (3′ end) of the miRNA binding site in the target transcript is critical for function. A lesser degree of complementarity is allowed between the 5′ end of the target and 3′ end of the miRNA.
miRNAs provide a level of regulation that can be rapidly and reversibly deployed. Such post-transcriptional regulation may be more efficient, as RNAs of genes targeted by miRNAs could be more stable [Bartel and Chen, 2004]. This scenario fits well with what is known about osteonectin. The osteonectin transcript has a long half-life, >24 h under the conditions of transcriptional arrest [Delany and Canalis, 1998]. This implies that it would be difficult to rapidly silence osteonectin expression through transcriptional mechanisms. Thus, osteonectin appears to be an excellent target gene for miRNA regulation.
The miR-29 family of miRNAs, consisting of miR-29a, -29b, and -29c, has been implicated in the control of extracellular matrix genes in cardiac fibroblasts and nasopharyngeal carcinomas [Sengupta et al., 2008; Van Rooij et al., 2008]. miR-29 binding sites are predicted within the osteonectin 3′ UTR. In this study, we validated osteonectin as a miR-29 target in osteoblasts, determined the role of miR-29 in the regulation of osteonectin expression, and characterized some mechanisms regulating miR-29 in bone cells.
MATERIALS AND METHODS
CELL CULTURE
The murine osteoblastic cell line MC3T3-E1 (MC3T3) was obtained from American Type Culture Collection (ATCC, Manassas, VA). MC3T3 cells were cultured in α-MEM (Invitrogen, Fredrick, MD) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, GA). Primary cultures of osteoblasts were established from cells isolated by the sequential collagenase digestion of neonatal (5–7 days old) C57Bl/6 mouse calvaria, as previously described [Delany et al., 2003]. For in vitro osteoblast differentiation studies, confluent osteoblastic cells were cultured in osteoblast differentiation medium (DMEM supplemented with 10% FBS, 100 μg/ml ascorbic acid, 20 mM HEPES, and 5 mM β-glycerolphosphate). Medium was changed twice a week for up to 3 weeks post-confluence. RNA was isolated from cells once in a week, always 3 days after their last medium change. These studies were approved by the Institutional Animal Care and Use Committee at the University of Connecticut Health Center. For some experiments, confluent cultures of wild-type mOb-I2 cells (an immortalized mouse calvarial osteoblast cell line) were serum-deprived over night and subsequently treated with vehicle, 50 ng/ml mouse Dickkopf-1 (Dkk-1) (R&D Systems, Minneapolis, MN), or 20 mM LiCl (Sigma, St. Louis, MO) [Delany et al., 2003].
CONSTRUCTS
PCR and appropriate primer sets were used to amplify osteonectin 3′ UTR of interest, using the mouse osteonectin cDNA as a template (provided by B. Hogan, Nashville, TN) [McVey et al., 1988]. UTR fragments were subcloned into the XbaI site of the vector pGL3Control (Promega, Madison, WI), so that the cloned UTRs could function as the 3′ UTR for the luciferase gene. The vector pGL3Control contains the CMV promoter to drive high level constitutive expression of luciferase, as well as the SV40 polyadenylation signal and enhancer. Site-directed mutagenesis was performed using “overlap extension.”
TRANSFECTIONS
MC3T3 cells were plated at 18,000 cells/cm2, and Fugene6 (Roche, Indianapolis, IN) (Fugene/DNA ratio 3:2) was used to co-transfect luciferase-osteonectin 3′ UTR constructs and a constitutively expressed β-galactosidase (β-gal) plasmid construct, as a control for transfection efficiency (Promega). Primary osteoblastic cells were plated at a similar density and were also transfected with Fugene6 (Fugene/DNA ratio 3:1).
Co-transfection of MC3T3 cells with luciferase-UTR constructs, β-gal expression plasmid, and 2-o-methyl RNAs complementary to the miRNAs of interest (antagomiRs, gifts from Dharmacon, Lafayette, CO) was accomplished using X-tremeGENE reagent (X-tremeGENE/nucleic acid ratio 5:1; Roche). The sequence of antagomiR for miR29a was 5′-UCUUCAACCGAUUUCAGAUGGUGCUAACCUU-3′; that for miR29c was 5′-UCUUCACCGAUUUCAAAUGGUGCUAACCUU-3′; and that for miR-96 was 5′-UCUUCGCAAAAAUGUGCUAGUGCCAAAACCUU-3′. A negative control antagomiR, the sequence of which was determined not to interact with any know miRNAs, was also provided by Dharmacon. The sequence was 5′-UCACAACCUCCUAGAAAGAGUAGA-3′.
48 h post-transfection, cells were serum-deprived over night, and cell lysates were prepared using Reporter Lysis buffer (Promega), according to the manufacturer’s instructions. Equal aliquots of cell lysate were used to determine luciferase activity (Luciferase assay system, Promega) and β-gal activity (Galacton chemiluminescent assay system; Tropix, Bedford, MA). Luciferase activity was expressed as a percentage of the β-gal activity. Transient transfections were performed using four to six replicates per experiment, and each experiment was performed at least three times. More than one DNA preparation for each construct was tested.
RNA ANALYSIS
RNA was isolated from cultured cells using Trizol (Invitrogen) and quantified spectroscopically. For qRT-PCR, aliquots of RNA were treated with RQ1 DNase I prior to analysis, to exclude any potential signal from DNA contamination.
To quantify miRNA levels in total RNA, the mirVana qRT-PCR miRNA Detection Kit (Ambion, Austin, TX) was used. Briefly, duplicate aliquots of RNA (50 ng for analysis of miR-29a or -29c; 5 ng for analysis of 5S) were reverse transcribed and subjected to PCR amplification using the BioRad real time PCR iQ hardware and software. The primers used are validated for amplification of the mature miR-29a, miR-29c, and 5S rRNA species. RNA levels were calculated using standard curves and miR-29 levels were expressed relative to those of 5S.
For Northern blot analysis, 5 μg total RNA was denatured and subjected to electrophoresis through a 1% formaldehyde-agarose gel, and blotted onto a Gene Screen Plus membrane, as directed by the manufacturer (DuPont, Wilmington, DE). Triplicate cultures were analyzed. Restriction fragments containing portions of cDNA for bovine osteonectin (provided by M. Young, NIDCR, NIH, Bethesda, MD) and murine 18S rRNA (ATCC) were labeled with [α-32P]dCTP (3,000 Ci/mmol; NEN, PerkinElmer, Boston, MA) (Ready-to-Go labeling kit, Amersham, Piscataway, NJ) [Bolander et al., 1988; Oberbaumer, 1992] and used to probe the blots. Specific hybridization was detected by autoradiography and relative band densities were determined using Image J software (http://rsb.info.nih.gov/ij/download.html).
WESTERN BLOT ANALYSIS
The 24 h-conditioned media (serum-free) was harvested from differentiating cultures of primary osteoblasts once in a week.
To determine the effect of LiCl treatment on osteonectin protein levels, mObI-2 cells were serum-deprived over night and subsequently treated with LiCl or NaCl or DKK1 for the time points described. The treatment medium was then replaced with a fresh aliquot of serum-free medium. After 1 h, the conditioned media was harvested. Osteonectin protein secreted into the medium in that 1 h period was quantified by Western blot analysis.
To determine the effect of miRNAs on endogenous osteonectin levels, MC3T3 cells were transfected with antagomiRs using 6 μL Oligofectamine (Invitrogen) and 120 pM antagomiR. Transfections of miR-29a and negative control precursor RNA mimics (Ambion) were performed using 50 pM precursor and 6 μL oligofectamine. The 24 h-conditioned media (serum-free) was harvested at 48 or 72 hours post-transfection.
Conditioned media was precipitated with ½ volume 10% trichloroacetic acid (TCA) and resuspended in 1× reducing sample buffer (62.5 mM Tris pH 6.8, 10% glycerol, 2% SDS, 5% beta-mercaptoethanol and bromophenol blue). Cell layers were lysed in 1× sample buffer (62.5 mM Tris pH 6.8, 10% glycerol, 2% SDS) and homogenized. Protein content was quantified using the BioRad DC Protein Assay Kit. Equal amounts of cell layer protein or conditioned medium were subjected to electrophoresis through a 10.5% SDS–polyacrylamide gel under reducing conditions, and transferred to a PVDF membrane (Millipore, Billerica, MA). Membranes were blocked overnight in 3% BSA in Tris-buffered saline (TBST, 0.1% Tween), and were probed with a rabbit anti-bovine osteonectin primary antibody (BON-1; gift of L. Fisher, NIDCR, NIH), followed by goat anti-rabbit-horseradish peroxidase conjugated secondary antibody (Sigma) [Ingram et al., 1993]. Bands were visualized by chemiluminescence (Perkin-Elmer) and fluorography. Expression of osteonectin in the cell layer was normalized to that of actin, detected using a rabbit anti-actin antibody (Sigma). Triplicate cultures were analyzed. Relative band densities in scanned images were analyzed with Image J software.
DATA ANALYSIS
Data are presented as mean ± SEM. Data were analyzed by Student’s t-test or one-way ANOVA with Bonferroni post-hoc test as appropriate (KaleidaGraph, Synergy Software, Reading, PA).
RESULTS
OSTEONECTIN 3′ UTR ACTIVITY IN OSTEOBLASTS
The mouse osteonectin mRNA has a short 89 base 5′ UTR, a protein coding region of 908 bases, and a 3′ UTR of 1,080 bases (Fig. 1A). Sequence analysis (http://cbio.mskcc.org) suggests that the mouse osteonectin 3′ UTR has five potential miRNA binding sites: two for miR-29a, and one each for miR-29c, miR-204, and miR-212 (Fig. 1A). The osteonectin 3′ UTR has several regions that are well conserved across species, and one particularly conserved region lies within the first 220 bases of the UTR (Fig. 1B). Importantly, the three potential binding sites for the miR-29 family of miRNAs are clustered within this highly conserved proximal osteonectin 3′ UTR, and clustered miRNA binding sites mediate the most efficient repression of gene expression [Rana, 2007].
Fig. 1.

Osteonectin 3′ UTR activity in osteoblasts. A: Diagram of the mouse osteonectin mRNA. The relative locations of potential miRNA binding sites are indicated by arrows. The miR-29a binding site is located at cDNA bases 1,122–1,146, the miR-29a/-29c site at 1,153–1,159, the miR-204 site at 1,457–1,463, and the miR-212 site at 1,726–1,743. B: Mouse osteonectin cDNA bases 982–1,217 (the proximal region of the 3′ UTR). Bases conserved across mouse, rat, and human are in bold letters. A predicted overlapping binding site for miR-29 and -29c, deleted in later studies, is underlined and italicized. A second predicted miR-29a binding site is underlined. MC3T3 cells (C) or primary mouse calvarial osteoblasts (D) were transiently transfected with luciferase vector alone or luciferase-osteonectin 3′ UTR chimeric constructs as indicated. Luciferase activity is expressed as a percentage of β-gal activity. Mean ± SEM * = significantly different from vector alone, P ≤ 0.02.
We first defined the relative contribution of the proximal osteonectin 3′ UTR to the regulation of gene expression using constructs in which osteonectin 3′ UTR fragments were used as the 3′ UTR for the reporter gene luciferase. Constructs contained the full length 3′ UTR, consisting of sequence immediately after the osteonectin translation stop codon, but lacking the polyadenylation sequence (cDNA bases 997–2,048), the proximal UTR (cDNA bases 997–1,217), and the distal UTR (cDNA bases 1,217–2,048). The luciferase-3′ UTR constructs were transiently transfected into the murine osteoblastic cell line MC3T3. We found that luciferase activity was decreased by ~80% in the presence of the full length UTR. The proximal 997–1,217 UTR fragment also caused a strong decrease in luciferase activity. In contrast, the 1,217–2,048 UTR fragment had a more modest inhibitory effect on luciferase activity (Fig. 1C). Similar studies were performed in primary cultures of mouse osteoblastic cells, and a similar pattern of regulation was observed (Fig. 1D), indicating that this was not a cell line-specific phenomenon. These data suggest that the proximal region of the osteonectin 3′ UTR, which contains the miR-29a/c binding sites, plays a dominant role in the repression of gene expression in osteoblastic cells.
RECIPROCAL REGULATION OF miR-29 AND OSTEONECTIN DURING OSTEOBLASTIC DIFFERENTIATION IN VITRO
Osteoblasts differentiate from committed precursors to mature cells following a well-documented gene expression program with characteristic phases including cell proliferation, matrix deposition, maturation, and mineralization [reviewed in Stein and Lian, 1993]. During this differentiation process, osteonectin mRNA levels have been shown to remain fairly constant in both mouse and human cells [Dieudonne et al., 1999; Frank et al., 2002]. However, there is little information on osteonectin protein levels and miRNA expression during this process.
We determined the relative changes in the expression of osteonectin mRNA and protein, and that of miR-29a and -29c during osteoblastic differentiation using primary cultures of mouse osteoblastic cells. Cells were cultured under osteoblast differentiation conditions for up to 3 weeks post-confluence, and RNA and protein were isolated at 1 week intervals. We quantified osteonectin protein levels by Western blot analysis, osteonectin mRNA by Northern blot analysis, and the relative quantity of miR-29a and -29c by qRT-PCR. This study confirmed that steady-state osteonectin mRNA levels changed little during osteoblast differentiation (Fig. 2). In contrast, we observed accumulation of osteonectin in the cell layer at 1 week post-confluence, during the matrix deposition phase of the culture. Osteonectin levels in the cell layer remained high during the remainder of the culture, likely because of its affinity for type I collagen [Rentz et al., 2007]. However, we found that osteonectin protein secreted into the medium during the 24-h interval prior to sample harvest was greatest at confluence and at 1 week post-confluence. At 2 and 3 weeks post-confluence, as osteoblast maturation continued and the matrix became mineralized, osteonectin secretion was significantly decreased. These data suggested that osteonectin translation might be decreased during the later phases of osteoblast differentiation. We found that miR-29a and -29c levels were relatively low at confluence, and remained low at 1 week of differentiation. Then miR-29a and -29c levels increased in concert as osteonectin protein secretion decreased, suggesting that the rising levels of miR-29 could play a role in down-regulation of osteonectin.
Fig. 2.
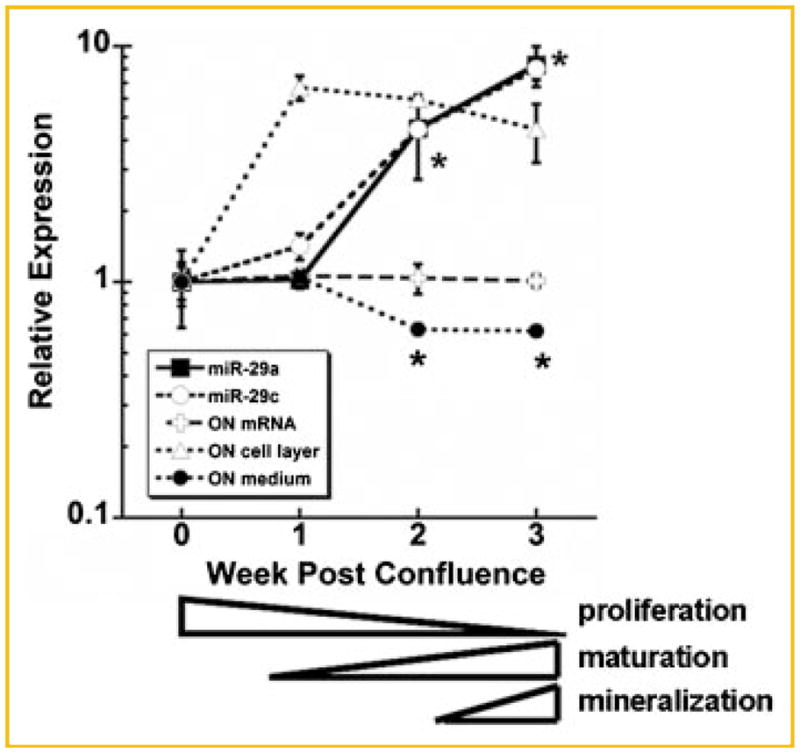
Regulation of miR-29 and osteonectin during osteoblast differentiation in vitro. Fold change in expression of osteonectin mRNA (✞), the relative quantity (vs. 5S rRNA) of miR-29a (□) and -29c (○), osteonectin protein in the cell layer (△) and 24 h-conditioned medium (●) in primary calvarial osteoblasts cultured for up to 3 weeks post-confluence under osteoblast differentiation conditions. Expression levels at confluence (week 0) were used as a base line. Note that curves for miR-29a and -29c are superimposable. Mean ± SEM * = significantly different from week 0, P ≤ 0.01. A general model illustrating the phases of osteoblastic phenotype development (cell proliferation, extracellular matrix maturation, and mineralization) was modified from [Stein and Lian, 1993].
miR-29 REGULATION OF OSTEONECTIN
Deletion of the most conserved miR-29 motif, containing overlapping binding sites for miR-29a and -29c (underlined and italicized in the sequence shown in Fig. 1B; and shown in Fig. 3A; mouse osteonectin cDNA bases 1,122–1,146), in the context of the 997–1,217 proximal UTR fragment relieved the repression of luciferase activity by ~80%, indicating that the binding site was functional (Fig. 3B). To confirm that miR-29a and -29c act on the proximal osteonectin 3′ UTR, MC3T3 cells were co-transfected with the 997–1,217-luciferase construct and miRNA inhibitors or “antagomiRs” designed to interfere with the activity of miR-29a and miR-29c. As controls, we also transfected cells with antagomiR against miR-96, a miRNA with a seed-binding site that may theoretically interact with the 1,122–1,146 region of the osteonectin 3′ UTR (Fig. 3A), and a non-specific negative control. AntagomiRs against miR-29a and -29c relieved UTR-mediated repression of luciferase activity, whereas the miR-96 and negative control antagomiRs did not, indicating specific interaction of miR-29 with the UTR (Fig. 3C). qRT-PCR confirmed that miR-29a and -29c are expressed in serum-deprived cultures of MC3T3 cells. The relative quantity (vs. 5S rRNA) of miR-29a was 1.01 ± 0.03, and that of miR-29c was 0.56 ± 0.13 (mean ± SEM).
Fig. 3.

miR-29 negatively regulates osteonectin expression in osteoblasts. A: Sequence of a conserved miRNA binding site at 1,122–1,146 in the mouse osteonectin 3′ UTR, and predicted base pairing with miR-29a, -29c, and -96. Sequence homology of the miR-29 binding sites in the human and mouse 3′ UTRs. The miR-29 binding sites are underlined. B: MC3T3 cells were transiently transfected with luciferase vector alone, luciferase-997–1,217 3′ UTR construct (997–1,217), or the luciferase-997–1,217 3′ UTR construct with a deletion of the 1,122–1,146 miRNA binding site (underlined and italicized in Fig. 1B) (997–1,217 dmiR). Luciferase activity is expressed as a percentage of β-gal activity. Mean ± SEM * = significantly different from vector alone, P ≤ 0.02 (C). MC3T3 cells were transiently transfected with the luciferase-997–1,217 3′ UTR construct, in the presence or absence (none) of 50 nM antagomiR (miRNA inhibitor) directed against miR-29a, -29c, or -96, or a negative control inhibitor (control). Mean ± SEM * = significantly different from no inhibitor (none), P ≤ 0.01. D: Western blot analysis for osteonectin in conditioned medium from MC3T3 cells transfected with 120 pM antagomiR directed against miR-29a, miR-96, or the negative control inhibitor. Mean ± SEM * = significantly different from negative control, P ≤ 0.01. E: Western blot analysis for osteonectin in conditioned medium from MC3T3 cells transfected with 50 pM miR-29a precursor RNA or a negative control RNA. Mean ± SEM * = significantly different from negative control, P ≤ 0.01.
To confirm that miR-29 negatively regulates endogenous osteonectin expression, MC3T3 cells were transfected with antagomiR against miR-29a, miR-96, or the negative control antagomiR (Fig. 3D). We focused on miR-29a for these studies because its mature sequence differs from that of miR-29c by only one base, which is not within the seed-region of the miRNA. Thus, miR-29a and -29c are likely to be functionally redundant. Western blot analysis of 24 h-conditioned medium showed that endogenous osteonectin expression was increased in cells transfected with antagomiR against miR-29a compared to the negative control or miR-96 antagomiR. In contrast, osteonectin expression was decreased in cells transfected with miR-29a precursor RNA compared to a negative control RNA (Fig. 3E). Together, these data validate the regulation of endogenous osteonectin by miR-29 in osteoblasts.
miR-29A AND -29C ARE INDUCED BY CANONICAL WNT SIGNALING
Wnt signaling plays a critical role in osteoblastic differentiation [Macsai et al., 2008]. Therefore, we determined whether the canonical Wnt signaling pathway could modulate the expression of miR-29a and 29c in osteoblastic cells. Confluent cultures of immortalized osteoblasts (wild-type mOb-I2) were serum-deprived and treated for up to 12 h with 20 mM LiCl (Fig. 4A) [Delany et al., 2003]. LiCl treatment mimics canonical Wnt signaling by inhibiting the activity of glycogen synthase kinase 3, resulting in the stabilization of β-catenin [Case et al., 2008]. LiCl induced miR-29a expression as early as 1 h of treatment, and the effect was sustained throughout the 12-h time course (Fig. 4A). Conversely, treatment of cells with the Wnt signaling antagonist Dkk-1 caused an inhibition of miR-29a expression that was significant after 48 h of incubation (Fig. 4B). Similarly, LiCl induced miR-29c expression, with a peak in levels after 3 h of treatment (Fig. 4C), and Dkk-1 decreased miR-29c after 24 h of treatment (Fig. 4D). Together, these data suggest that miR-29 is induced by canonical Wnt signaling in osteoblasts.
Fig. 4.

Canonical Wnt signaling induces miR-29a and -29c expression. Relative quantity (vs. 5S rRNA) of miR-29a (A) and -29c (C) in wild-type mObI-2 cells treated with 20 mM LiCl or 20 mM NaCl (negative control) for up to 12 h. Relative quantity of miR-29a (B) and -29c (D) in wild-type mObI-2 cells treated with 50 ng/ml recombinant mouse Dkk-1 or vehicle control for 24 or 48 h. * = significantly different from control P ≤ 0.01. E,F: mObI-2 cells were treated with LiCl or NaCl for the indicated times, and then cultured in fresh serum-free medium for 1 h. Western blot analysis was used to quantify osteonectin protein secreted during that 1 h period. Mean ± SEM * = significantly different from negative control, P ≤ 0.05.
Further, Western blot analysis showed that treatment with LiCl decreased endogenous osteonectin secreted into the conditioned medium. The effect was seen as early as 1 h of treatment, indicating rapid regulation of actively synthesized osteonectin protein by canonical Wnt signaling (Fig. 4E,F). The degree of suppression was greatest after 3 h of LiCl treatment, when miR-29 levels are highest. However, using this assay system, we did not detect a significant change in endogenous osteonectin protein levels as a result of a 24- or 48-h Dkk-1 treatment (data not shown). This may be due to the more modest effects of DKK on miR-29 expression, or because other down stream effects of DKK, in addition to those on miR-29, may affect osteonectin synthesis with 24- or 48-h treatment.
DISCUSSION
This study provides novel insights into the mechanisms regulating the matricellular protein osteonectin, and regulation of the miR-29 family in osteoblasts. Although others reported the negative regulation of a luciferase–osteonectin 3′ UTR construct by miR-29c precursor RNA in transiently transfected HeLa cells [Sengupta et al., 2008], we demonstrated the regulation of endogenous osteonectin by miR-29a in osteoblasts, and mapped the sequence motifs involved (Fig. 3). Further, we found that miR-29 binding sites play a dominant role in the negative regulation mediated by the osteonectin 3′ UTR in osteoblasts (Figs. 1 and 3). In addition, we established that miR-29a and -29c are up-regulated during osteoblastic differentiation, implicating these miRNAs in the down-regulation of osteonectin, and likely the regulation of other proteins critical for osteoblast function. Lastly, we showed that miR-29a and -29c expression was induced by canonical Wnt signaling in osteoblasts.
The location and context of the miR-29 binding sites in the osteonectin 3′ UTR appear to be optimal for regulation by miRNAs [Lewis et al., 2003; Jing et al., 2005; Grimson et al., 2007]. This idea is supported by the finding that deletion of one miR-29a site could rescue reporter gene activity by ~80% (Fig. 3B). In addition, the miR-29 sites are within the proximal end of the UTR, and there is an A-rich region immediately down stream of these binding sites (Fig. 1B).
Database analysis suggests that other genes important for osteoblast function may also be targets for the miR-29 family of miRNAs (http://cbio.mskcc.org/cgi-bin/mirnaviewer). Recently, miR-29 was shown to regulate the 3′ UTR of the mRNAs for α1 and α2(I)collagen, α1(III)collagen, and fibrillin 1. Expression of these genes was up-regulated as miR-29 levels were down-regulated in response to myocardial infarction, implicating a role for miR-29 in controlling cardiac fibrosis [Van Rooij et al., 2008]. However, miR-29 levels were not thought to be the sole determinant of collagen regulation in this model. It will be important to define other regulatory elements important for post-transcriptional control of extracellular matrix gene expression.
It is well established that the synthesis of extracellular matrix and the expression of extracellular matrix genes is decreased as osteoblasts become more differentiated [Stein and Lian, 1993; Kalajzic et al., 2005]. Conversely, as osteoblasts differentiate, the expression of genes important for the activation of canonical Wnt signaling is increased [Kalajzic et al., 2005]. Our observations correlate with these gene expression patterns in that miR-29, a negative regulator of extracellular matrix gene expression, is lowest during the cell proliferation/matrix deposition phases of osteoblastic differentiation. Later, as osteoblasts become more mature and Wnt signaling increases, miR-29 levels increase, likely in part due to an increase in canonical Wnt signaling (Figs. 2 and 4).
Osteonectin-null mice are osteopenic and osteoblasts from osteonectin-null mice do not fully mature [Delany et al., 2000, 2003]. Although our data indicate that osteonectin plays a positive role in osteogenesis, it is likely that tight control of its expression is essential for normal osteoblastic differentiation and skeletal function. Mis-expression of osteonectin could impede osteoblastic differentiation, and osteonectin levels are controlled at multiple levels including transcription, mRNA stability, and translation [Nomura et al., 1989; Dominguez et al., 1991; Hafner et al., 1995; Delany and Canalis, 1998] (Fig. 4). The osteonectin transcript is quite stable, with a half life of >24 h under conditions of transcription arrest [Delany and Canalis, 1998]. Thus, it would be difficult to rapidly down-regulate osteonectin expression by transcriptional mechanisms. Treatment of osteoblasts with LiCl increases miR-29 expression and significantly decreases osteonectin synthesis within 1 h. Our data indicate up regulation of miR-29 could provide a mechanism for prompt regulation of osteonectin protein levels in these cells.
The expression of miR-29a and -29c was increased during the later phases of osteoblast differentiation in vitro. We hypothesize that miR-29 positively regulates osteoblast differentiation by controlling the expression of osteonectin and likely other targets. Indeed, miR-29 was shown to function as an enhancer of muscle cell differentiation and as a tumor suppressor in rhabdomyosarcoma [Wang et al., 2008]. In the course of our studies, we attempted to stably over express miR-29a and -29c in osteoblastic cells using a retroviral approach. However, despite multiple attempts, we were unable to generate such cell lines. Other investigators reported a similar experience when attempting to stably over express miR-29 in pre-B cells, suggesting that over expression of miR-29 could impose a strong negative selection against cell growth in vitro [Chang et al., 2008]. Although miR-29 family members were recently shown to up-regulate p53 levels and induce apoptosis, we did not detect differences in proliferation or apoptosis in MC3T3 cells that are transiently transfected with miR-29a inhibitors or precursor RNAs (data not shown) [Chang et al., 2008].
Our data suggest that, in osteoblasts, miR-29a is expressed at relatively higher levels compared with miR-29c (Fig. 4). miR-29a is co-expressed on the same pri-miRNA as miR-29b1, whereas miR-29c is co-expressed from the same pri-miRNA as miR-29b2. These pri-miRNAs are transcribed from different genetic loci, and their putative promoter regions have some notable differences [Chang et al., 2008]. Interestingly, miR-29b is rapidly degraded in cycling cells, and is found predominantly in the nucleus. In contrast, miR-29a is degraded more slowly and localized primarily in the cytoplasm. It is suggested that miR-29b may regulate the transcription or splicing of target transcripts, rather than inhibit translation [Hwang et al., 2007].
Since the 3′ UTR can control mRNA targeting, stability, and translation, mutations or polymorphisms in this region have the potential to regulate gene expression [Conne et al., 2000]. We recently demonstrated that haplotypes consisting of three single nucleotide polymorphisms (SNPs) in the 3′ UTR of the osteonectin gene were associated with idiopathic osteoporosis and bone density in Caucasian men [Delany et al., 2008]. We identified haplotypes associated with higher bone density and those associated with lower bone density at multiple skeletal sites. The location and sequence of the miR-29 binding sites in the mouse osteonectin 3′ UTR is conserved in the human gene, and this site does not contain any SNPs in humans. It is possible that the regulation mediated by the miR-29 binding sites is critical for appropriate gene expression in multiple tissues, and alterations in these elements could be evolutionarily disadvantageous. Characterization of the human osteonectin 3′ UTR is ongoing in our laboratory.
In conclusion, osteonectin is a gene contributing to bone mass in mouse models and is associated with bone mass in at least one subset of osteoporosis patients. Post-transcriptional regulation of osteonectin, mediated in part by the miR-29 family, is a vital mechanism modulating gene expression during osteoblast differentiation. These data have important implications with regard to both normal bone remodeling and fracture repair.
Acknowledgments
Grant sponsor: National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS); Grant number: AR44877.
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, AR44877 (A.M. Delany). We thank Jonathan Shubert-Coleman and Dr. Henry Furneaux for technical advice on experiments and comments on the manuscript. We thank Dr. Larry Fisher for providing osteonectin antiserum, Dr. Marian Young for providing the bovine osteonectin cDNA, Dr. Bridget Hogan for providing the mouse osteonectin cDNA, and Dharmacon for providing the antagomiRs.
References
- Alford AI, Hankenson KD. Matricellular proteins: Extracellular modulators of bone development, remodeling, and regeneration. Bone. 2006;38:749–757. doi: 10.1016/j.bone.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Bolander ME, Young MF, Fisher LW, Yamada Y, Termine JD. Osteonectin cDNA sequence reveals potential binding regions for calcium and hydroxyapatite and shows homologies with both a basement membrane protein (SPARC) and a serine proteinase inhibitor (ovomucoid) Proc Natl Acad Sci USA. 1988;85:2919–2923. doi: 10.1073/pnas.85.9.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. J Bone Miner Res. 2003;18:1005–1011. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107:1049–1054. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case N, Ma M, Sen B, Xie Z, Gross TS, Rubin J. B-catenin levels influence rapid mechanical responses in osteoblasts. J Biol Chem. 2008;283:29196–29205. doi: 10.1074/jbc.M801907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran V, Ambati J, Ambati BK, Taylor EW. Molecular docking and analysis of interactions between vascular endothelial growth factor (VEGF) and SPARC protein. J Mol Graph Model. 2007;26:775–782. doi: 10.1016/j.jmgm.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conne B, Stutz A, Vassalli JD. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat Med. 2000;6:637–641. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- Delany AM, Canalis E. Basic fibroblast growth factor destabilizes osteonectin mRNA in osteoblasts. Am J Physiol (Cell Physiol) 1998;274:C734–C740. doi: 10.1152/ajpcell.1998.274.3.C734. [DOI] [PubMed] [Google Scholar]
- Delany AM, Amling M, Priemel M, Howe C, Baron R, Canalis E. Osteopenia and decreased bone formation in osteonectin-deficient mice. J Clin Invest. 2000;105:915–923. doi: 10.1172/JCI7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E. Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology. 2003;144:2588–2596. doi: 10.1210/en.2002-221044. [DOI] [PubMed] [Google Scholar]
- Delany AM, McMahon DJ, Powell JS, Greenberg DA, Kurland ES. Osteonectin/SPARC polymorphisms in Caucasian men with idiopathic osteoporosis. Osteoporos Int. 2008;19:969–973. doi: 10.1007/s00198-007-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieudonne SC, Kerr JM, Xu T, Sommer B, DeRubeis AR, Kuznetsov SA, Kim IS, Robey PG, Young MF. Differential display of human marrow stromal cells reveal unique mRNA expression patterns in response to dexamethasone. J Cell Biochem. 1999;76:231–243. doi: 10.1002/(sici)1097-4644(20000201)76:2<231::aid-jcb7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Dominguez P, Ibaraki K, Gehron Robey P, Hefferan TE, Termine JD, Young MF. Expression of the osteonectin gene potentially controlled by multiple cis- and trans-acting factors in cultured bone cells. J Bone Miner Res. 1991;10:1127–1136. doi: 10.1002/jbmr.5650061015. [DOI] [PubMed] [Google Scholar]
- Frank O, Heim M, Jakob M, Barbero A, Schafer D, Bendik I, Dick W, Herver M, Martin I. Real-time quantitative RT-PCR analysis of human bone marrow stromal cells during osteogenic differentiation in vitro. J Cell Biochem. 2002;85:737–746. doi: 10.1002/jcb.10174. [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KKH, Johnson WK, Garret-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Zimmermann K, Pottgiesser J, Krieg T, Nischt R. A purine-rich sequence in the human BM-40 gene promoter region is a prerequisite for maximum transcription. Matrix Biol. 1995;14:733–741. doi: 10.1016/s0945-053x(05)80016-2. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- Ingram RT, Clarke BL, Fisher LW, Fitzpatrick LA. Distribution of noncollagenous proteins in the matrix of adult human bone: Evidence of anatomic and functional heterogeneity. J Bone Miner Res. 1993;8:1019–10122. doi: 10.1002/jbmr.5650080902. [DOI] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-Rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Kalajzic I, Staal A, Yang WP, Wu Y, Johnson SE, Feyen JH, Krueger W, Maye P, Yu F, Zhao Y, Kuo L, Gupta RR, Achenie LE, Wang HW, Shin DG, Rowe DW. Expression profile of osteoblast lineage at defined stages of differentiation. J Biol Chem. 2005;280:24618–24626. doi: 10.1074/jbc.M413834200. [DOI] [PubMed] [Google Scholar]
- Kelly KA, Allport JR, Yu AM, Sinh S, Sage EH, Gerszten RE, Weissleder R. SPARC is a VCAM-1 counter-ligand that mediates leukocyte transmigration. J Leuko Biol. 2007;81:748–756. doi: 10.1189/jlb.1105664. [DOI] [PubMed] [Google Scholar]
- Kessler CB, Delany AM. Increased notch 1 expression and attenuated stimulatory G protein coupling to adenylyl cyclase in osteonectin-null osteoblasts. Endocrinology. 2007;148:1666–1674. doi: 10.1210/en.2006-0443. [DOI] [PubMed] [Google Scholar]
- Kunigal S, Gondi CS, Gujrati M, Lakka SS, Dinh DH, Olivero WC, Rao JS. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. Int J Oncol. 2006;29:1349–1357. [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih I, Jonse-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin-null mice. Bone. 2008;43:264–273. doi: 10.1016/j.bone.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macsai CE, Foster BK, Xian CJ. Roles of Wnt signaling in bone growth, remodeling, skeletal disorders and fracture repair. J Cell Physiol. 2008;215:578–587. doi: 10.1002/jcp.21342. [DOI] [PubMed] [Google Scholar]
- Mansergth FC, Wells T, Elford C, Evans SL, Perry MJ, Evans MJ, Evans BA. Osteopenia in Sparc (osteonectin)-deficient mice: Characterization of phenotypic determinants of femoral strength and changes in gene expression. Physiol Genomics. 2007;32:64–73. doi: 10.1152/physiolgenomics.00151.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey JH, Nomura S, Kelly P, Mason IJ, Hogan BL. Characterization of the mouse SPARC/osteonectin gene. Intron/exon organization and an unusual promoter region. J Biol Chem. 1988;263:11111–11116. [PubMed] [Google Scholar]
- Motamed K, Funk SE, Koyama H, Ross R, Raines EW, Sage EH. Inhibition of PDGF-stimulated and matrix-mediated proliferation of human vascular smooth muscle cells by SPARC is independent of changes in cell shape or cyclin-dependent kinase inhibitors. J Cell Biochem. 2002;84:759–771. doi: 10.1002/jcb.10095. [DOI] [PubMed] [Google Scholar]
- Nomura S, Hashmi S, McVey JH, Ham J, Parker M, Hogan BLM. Evidence for positive and negative regulatory elements in the 5′-flanking sequence of the mouse sparc (osteonectin) gene. J Biol Chem. 1989;264:12201–12207. [PubMed] [Google Scholar]
- Oberbaumer I. Retroposons do jump: A B2 element recently integrated in an 18S rDNA gene. Nucleic Acids Res. 1992;20:671–677. doi: 10.1093/nar/20.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines EW, Lane TF, Iruela-Arispe ML, Ross R, Sage EH. The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc Natl Acad Sci USA. 1992;89:1281–1285. doi: 10.1073/pnas.89.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: Understanding the structure and function of small RNAs. Nature Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem. 2007;282:22062–22071. doi: 10.1074/jbc.M700167200. [DOI] [PubMed] [Google Scholar]
- Robey PG, Boskey AL. Extracellular matrix and biomineralization in bone. In: Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society for Bone and Mineral Research; 2006. pp. 12–19. [Google Scholar]
- Sage H, Vernon RB, Decker J, Funk S, Iruela-Arispe ML. Distribution of the calcium-binding protein SPARC in tissues of embryonic and adult mice. J Histochem Cytochem. 1989;37:819–829. doi: 10.1177/37.6.2723400. [DOI] [PubMed] [Google Scholar]
- Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulated mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci USA. 2008;105:5874–5878. doi: 10.1073/pnas.0801130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN. Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene. 2007;26:4084–4094. doi: 10.1038/sj.onc.1210181. [DOI] [PubMed] [Google Scholar]
- Stein GS, Lian JB. Molecular mechanisms mediating proliferation/ differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev. 1993;14:424–442. doi: 10.1210/edrv-14-4-424. [DOI] [PubMed] [Google Scholar]
- Van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, Croche CM, Guttridge DC. NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–381. doi: 10.1016/j.ccr.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver MS, Workman G, Sage EH. Functional mapping of SPARC: Peptides from two distinct Ca(++)-binding sites modulate cell shape. J Biol Chem. 2008;283:22826–22837. [Google Scholar]