

Abstract
The cAMP receptor protein (Crp) is a global transcriptional regulator that binds sequence-specific promoter elements when associated with cAMP. In the motile cyanobacterium Synechocystis sp. strain PCC 6803, intracellular cAMP increases when dark-adapted cells are illuminated. Previous work has established that Crp binds proposed Crp target sites upstream of slr1351 (murF), sll1874 (chlAII), sll1708 (narL), slr0442 and sll1268 in vitro, and that slr0442 is downregulated in a crp mutant during photoautotrophic growth. To identify additional Crp target genes in Synechocystis, 11 different Crp binding sites proposed during a previous computational survey were tested for in vitro sequence-specific binding and crp-dependent transcription. The results indicate that murF, chlAII and slr0442 can be added as ‘target genes of Sycrp1’ in Synechocystis. Promoter mapping of the targets revealed the same close association of RNA polymerase and Crp as that found in Escherichia coli class I and class II Crp-regulated promoters, thereby strongly suggesting similar mechanisms of transcriptional activation.
INTRODUCTION
The cAMP receptor protein (Crp, or ‘Sycrp1’ in Synechocystis where required for clarity) can act as a transcriptional regulator when bound to the cAMP ligand (Botsford & Harman, 1992). Intracellular cAMP levels change dynamically to control gene regulation (Cann, 2004; Hammer et al., 2006; Kolb et al., 1993; Ohmori & Okamoto, 2004; Sakamoto et al., 1991). Various environmental conditions signal Synechocystis to maintain low, moderate and high intracellular cAMP levels that can be defined accordingly. Following dark adaptation, intracellular cAMP levels are low, at 0.02–0.04 pmol cAMP [μg chlorophyll (Chl) a]−1 (Terauchi & Ohmori, 2004). During regular photoautotrophic growth, intracellular cAMP levels are moderate [0.14–0.20 pmol cAMP (μg Chl a)−1] (Ochoa de Alda et al., 2000; Terauchi & Ohmori, 1999). Following illumination with either blue or white light, dark-adapted cells increase intracellular cAMP to high levels [0.60– 0.80 pmol cAMP (μg Chl a)−1] (Masuda & Ono, 2004; Terauchi & Ohmori, 1998, 2004). These spectrum-specific photoresponses support the current view that the cAMP–Crp complex is ecologically beneficial for optimal positioning of motile cells relative to incident light (Bhaya et al., 2006; Masuda & Ono, 2004). Indeed, it has been shown that intracellular cAMP is necessary and sufficient to restore phototactic motility used by cells to escape from the confines of a colony during suboptimal illumination (Bhaya et al., 2006; Terauchi & Ohmori, 1999). Both Crp and cAMP are required for transcriptional activation of genes encoding type IV pilin biosynthesis proteins involved in motility, thereby strongly suggesting a role for regulation of motility by Crp (Yoshimura et al., 2002a, b).
To predict additional candidate genes for Crp regulation, a computational survey has previously proposed 11 different Crp binding target sequences based on the observation that Sycrp1 binds the Escherichia coli consensus ICAP Crp binding site (Ochoa de Alda & Houmard, 2000). Recently, a biochemical study by Omagari et al. (2008) demonstrated in vitro that systematic substitution of bases in ICAP could be used to fairly accurately predict the observed free energy change (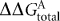 ) of Crp binding to any given DNA sequence (Omagari et al., 2008). The limit for detection of Crp binding in vitro was <3.1, and all intergenic sequences in the Synechocystis genome containing calculated <3.1 were bound by Crp. The study by Omagari et al. (2008) demonstrated Crp binding to three of the 11 target sequences (slr1351, sll1874 and sll1708) predicted by Ochoa de Alda & Houmard (2000). Most recently, an interspecies bioinformatic comparison of cyanobacterial genomes (Xu & Su, 2009) has been performed, based in part on probable Crp binding sites in the Synechocystis Crp transcriptome as identified by Yoshimura et al. (2002). Of the 53 target sequences that Xu & Su (2009) predicted for Synechocystis, seven (slr1732, slr1667, slr1351, sll1708, sll1874, slr0442 and sll1268) were bound by Crp in the Omagari et al. (2008) study, and three (slr1351, sll1708 and sll1874) were also predicted by Ochoa de Alda & Houmard (2000). These predictive and in vitro binding studies have not provided in vivo evidence, nor elucidated possible mechanisms of transcriptional activation by Crp (i.e. whether Sycrp1-dependent promoters demonstrate the same well-characterized promoter organization as in Escherichia coli).
) of Crp binding to any given DNA sequence (Omagari et al., 2008). The limit for detection of Crp binding in vitro was <3.1, and all intergenic sequences in the Synechocystis genome containing calculated <3.1 were bound by Crp. The study by Omagari et al. (2008) demonstrated Crp binding to three of the 11 target sequences (slr1351, sll1874 and sll1708) predicted by Ochoa de Alda & Houmard (2000). Most recently, an interspecies bioinformatic comparison of cyanobacterial genomes (Xu & Su, 2009) has been performed, based in part on probable Crp binding sites in the Synechocystis Crp transcriptome as identified by Yoshimura et al. (2002). Of the 53 target sequences that Xu & Su (2009) predicted for Synechocystis, seven (slr1732, slr1667, slr1351, sll1708, sll1874, slr0442 and sll1268) were bound by Crp in the Omagari et al. (2008) study, and three (slr1351, sll1708 and sll1874) were also predicted by Ochoa de Alda & Houmard (2000). These predictive and in vitro binding studies have not provided in vivo evidence, nor elucidated possible mechanisms of transcriptional activation by Crp (i.e. whether Sycrp1-dependent promoters demonstrate the same well-characterized promoter organization as in Escherichia coli).
In an attempt to elucidate possible mechanisms of trancriptional activation by Crp, sequence-specific Crp/DNA binding, transcriptional start sites, and Crp-dependent regulation of the slr1667–1668 operon have been demonstrated (Yoshimura et al., 2002a). Even though the results have not established a plausible mechanism (see discussion), this operon has subsequently been discussed in the context of Crp regulation (Dienst et al., 2008; Singh et al., 2008; Summerfield & Sherman, 2008). Furthermore, the Kazusa Cyanobase describes these genes as ‘target genes of Sycrp1’, based on data that demonstrated both (1) in vitro sequence-specific binding and (2) Crp-dependent gene expression. These two criteria will be referenced as such throughout this text. No other genes have been so annotated in the Synechocystis genome to date.
To identify additional ‘target genes of Sycrp1’, all Crp targets proposed by Ochoa de Alda & Houmard (2000) and a target (slr0442) proposed by Omagari et al. (2008) were studied using a motile Synechocystis strain capable of displaying large increases in intracellular cAMP following illumination. These proposed targets were tested in vitro for sequence-specific Crp/DNA binding, and expression was monitored in wild-type and crp cells to assess Crp-dependent regulation during a dark to light environmental change that causes a low to high intracellular cAMP change. The results indicate that slr1351 (murF), sll1874 (chlAII) and slr0442 can be classified as ‘target genes of Sycrp1’ in Synechocystis. Plausible Crp activation mechanisms of these cyanobacterial Crp targets are discussed based on transcriptional start sites mapped in Synechocystis and similar expression of promoter–reporter constructs derived from these targets and expressed in E. coli.
METHODS
Strains and growth conditions.
The wild-type motile glucose-sensitive Synechocystis PCC sp. strain 6803 was obtained from the Pasteur Culture Collection of Cyanobacteria. All Synechocystis cells were pre-grown for 11 days in BG-11 medium (Stanier et al., 1971) containing 75.0 mM TES, pH 7.75, 10.0 mM bicarbonate, and were supplemented with 5.0 mM bicarbonate every 12 h in a manner that maintained exponential growth at pH 7.75 in an inorganic carbon-replete condition. Synechocystis cells were grown at 30 °C and illuminated with 30.0 μmol photons m−2 s−1 from cool white fluorescent lamps. Cultures in mid-exponential phase (OD730 0.6) were washed in fresh media and transferred to the dark 16 h prior to sampling. Samples for RNA extraction were rapidly chilled on ice water, pelleted in a prechilled rotor for 10 min at 4000 g, and flash-frozen immediately following sampling in the dark, 30 and 60 min following illumination. E. coli K-12 M182 Δlac wild-type (Casadaban et al., 1980; Casadaban & Cohen, 1980) and crp mutant (Busby et al., 1983) stock cultures were kindly provided by Stephen Busby (University of Birmingham), and were maintained in Luria–Bertani (LB) medium supplemented with 30.0 μg streptomycin ml−1 and 50.0 μg ampicillin ml−1, respectively. E. coli clones containing reporter plasmids were grown in LB with or without 3 % glucose in a roller drum at 37 °C for 48 h and assayed for green fluorescent protein (Gfp) signal as described below. All antibiotics were omitted from experimental cultures. E. coli DH5α MCR was used for plasmid amplification.
Molecular biology techniques.
Plasmid purifications, isolation, PCR, ligation reactions, Southern blotting and transformations were performed according to standard protocols (Ausubel et al., 2000) using commercial kits for DNA purification. Synechocystis sp. PCC 6803 genomic DNA was harvested as previously described (Summers et al., 1995).
sycrp1 mutant construction.
The sycrp1 gene (sll1371) was amplified from genomic DNA using primers ATTCAGAGTTTACTGAGCGT and CCTGAGTTGGCCACACTGA and cloned into pCR2.1 (Invitrogen). The cloned gene was then inactivated by insertion of a PvuII fragment of pZeo (Stevens et al., 1996), containing the ble zeocin-resistance gene, into the SmaI site within sll1371 to produce pKL2. This insertion was verified by sequencing at the California State University sequencing facility. The wild-type strain was subject to natural transformation with pKL2 and selection in zeocin to yield cAMP receptor protein sycrp1 : : ble mutants (crp). Zeocin-resistant crp mutant stock cultures were maintained in BG-11 supplemented with 6.0 μg zeocin ml−1.
Gfp reporter construction and quantification.
A novel SphI site was introduced into the multiple cloning site of the pIGA transcriptional reporter plasmid (Kunert et al., 2000) via a custom adaptor created by annealing GAGGGTACCGCATGCGGTACCTCA and TGAGGTACCGCATGCGGTACCCTC. KpnI digestion and ligation of the adaptor (KpnI-SphI-KpnI) located downstream of a strong T7 transcription terminator sequence, but upstream of gfp, into the KpnI site of pIGA created pIGS. The promoter region and 5′ N terminus of the indicated Synechocystis genes were amplified by PCR using gene-specific primer sets (Supplementary Table S1) that added restriction sites for KpnI or SphI. The PCR product was digested, and ligated into pIGA or pIGS. Primers flanking the multiple cloning site (Argueta et al., 2004) were used to sequence each insert, thereby confirming the proper orientation and absence of mutation. The slr0442 reporter construct was created by ligation of a partial Hsp92II Synechocystis genomic digest into pIGS and screening of E. coli DH5α clones with and without glucose. The identified clone contained the almost complete slr0442 intergenic region from chromosomal positions 2 080 200 to 2 080 940 (Kaneko et al., 1996). These resultant reporter plasmids were used to transform E. coli K-12 M182 Δlac wild-type and crp mutant strains via electroporation followed by selection in LB supplemented with 50.0 μg kanamycin ml−1.
E. coli M182 cells containing reporter constructs were washed twice in PBS and normalized to OD595 0.25 immediately prior to measuring Gfp fluorescence, as previously described (Argueta & Summers, 2005).
RNA isolation and reverse transcriptase-mediated quantitative PCR (RT-QPCR).
RNA was isolated as previously described (Kim et al., 2006) and visualized for integrity in a formaldehyde gel. Genomic DNA was removed by two rounds of RQ1 DNase (Promega) digestion and Zymoclean (Zymo Research) column purification according to the manufacturers' instructions. The absence of genomic DNA in the resultant RNA samples was confirmed by the absence of product following PCR using rnpB primers and a genomic control. RNA samples were normalized to 1.0 g l−1 and frozen once at −80 °C prior to RT-QPCR. Reverse transcription was performed with reverse primers (see Supplementary Table S1) and SuperscriptII (Invitrogen) according to the manufacturer's instructions using 1.0 μg RNA to generate cDNA. RT-QPCR was performed using 13.0 μl Power SYBR Green PCR Master Mix (Applied Biosystems), 2.0 μl of cDNA serial dilutions and gene-specific primer sets (Supplementary Table S1) at a final concentration of 300 nM each primer in a final volume of 26.0 μl. Temperature cycles in an ABI 7300 Real-Time PCR system were 10 min at 95 °C, 35 cycles of 15 s at 95 °C, 25 s at 51 °C, and 1 min at 72 °C, followed by a slow melt cycle to confirm specific product formation. Gel electrophoresis was also performed to confirm the absence of non-specific PCR products in experimental samples and controls. Six serial dilutions of each cDNA sample were used per target and reference sample, and the relative expression between wild-type and crp transcripts was calculated as described by Pfaffl (2001) using rnpB as an internal calibrator (Fernandez-Gonzalez et al., 1998). All PCR efficiencies calculated by serial dilution were within 10 % of expected doubling, and rnpB transcript levels yielded consistent PCR cycle fluorescence thresholds relative to total RNA for all samples.
Rapid amplification of cDNA ends (RACE).
The +1 start site of transcription was determined for selected genes using nested intragenic primers (Supplementary Table S1) and 1.0 μg total RNA for RACE analysis, as previously described (Argueta et al., 2006).
Protein purification and electrophoretic mobility shift assay (EMSA).
Masayuki Ohmori (Saitama University) kindly provided pCGA used to overexpress histidine-tagged His–Sycrp1. A method for purification, binding reactions and EMSA has been been described elsewhere (Yoshimura et al., 2000) and was essentially reproduced.
Following induction with IPTG, BL21(DE3) E. coli cells containing pCGA and pLysS were washed in lysis buffer (see below). Lysis and purification was performed at 4 °C. Cells were disrupted by sonication and the lysate was clarified by ultracentrifugation at 150 000 g for 45 min. Following loading and washing with four column volumes of lysis buffer, nickel and mono-Q columns were eluted using gradients to 400 mM imidazole (10 mM wash buffer) and 1.0 M NaCl, respectively, over 10 column volumes in 50 mM NaCl, 1.0 mM β-mercaptoethanol, 50.0 mM Tris, pH 8.0, and 10 % (v/v) glycerol. This solution was also used for between-column dialysis and as a lysis buffer. Homogeneity of the purified protein was confirmed by SDS-PAGE and Western blot detection using Tetra-His antibody. Binding activity specific to the purified protein was confirmed by visualization of the His–Sycrp1 in complex with slr1667 and not in complex with Rndm (see Table 1 for substrate sequences), using native PAGE (described below) and a Tetra-His antibody. The blocking reagent and Tetra-His antibody conjugated to horseradish peroxidase from Qiagen, and Supersignal CL-HRP visualization substrate from Novagen, were used according to the manufacturers' recommendations.
Table 1.
Sequences of coding strand (complement not shown) of the double-stranded blunt-ended oligonucleotide DNA used in EMSA and calculated 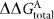 values for the consensus
values for the consensus
The putative Sycrp1 core binding consensus is indicated in bold type. ORF identifiers of genes demonstrating both sequence-specific binding and sycrp1-dependent transcriptional regulation are in bold underlined type. 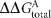 values <3.1 are in bold type. The value of was calculated by strict summation of position values given by the position-specific scoring matrix in Omagari et al. (2008), except that G and C substitution ΔG values at positions 9 and 14 were switched to accurately reflect the authors' intent.
values <3.1 are in bold type. The value of was calculated by strict summation of position values given by the position-specific scoring matrix in Omagari et al. (2008), except that G and C substitution ΔG values at positions 9 and 14 were switched to accurately reflect the authors' intent.
| Gene | Sequence | |
|---|---|---|
| slr1667 (target of Sycrp1) | ATACACAACAGTTGTGATCTGGGTCACAACCATTGAGTGA | 0.34*† |
| Rndm. (negative control) | AAGCCGTAAGACCTAATGTAGAAGTGCTCCAGAAGCTCAC | 19.89 |
| slr1991 cya1 | AGGCTCCCTGATGGGGACAGCGGTCACGGACCTTTACTTT | 5.85‡ |
| sll0065 ilvN | TTCCCTAACTCTAGTGAGGAATTTTGCAAAATGCAAGCTT | 8.04‡ |
| slr0194 rpiA | AACCGGAACTGTTCTGATAATGTTCGCACTGTAGAGATTT | 9.01‡ |
| slr1351murF/sll1247 hyp. | GCACCCATGGGAGGTGATCTAGATCACAGATAAAAATTGC | 0.67*†‡§ |
| slr1575 hyp. | GCGGAGACAAAATGGGAAATCACTCACGCCTCGTCTCAAT | 7.35‡ |
| sll1708 narL/slr1805 hik16 | CGGCACCCTTACCGTGATAGTAATCACCGATGAAGTACAA | 3.07*†‡§ |
| sll0682 pstA | GGCAGAACTGATTGTGAACAAAGTCAAAGCTAAATATTAG | 7.85‡ |
| sll0041 cheD | GACATTACCTGGTGTGAAACGGATCAAATTCAATCTCCCC | 6.84‡ |
| slr1200 livH | CAATGGCAACAATGTGATAATCCCCACACCTGCCCCCACG | 4.12‡ |
| sll1874at103,chlAII | CCTTCCACTGCTTGTGAGAATAATCACAGGCAGTTTTTTT | 2.62*†‡§ |
| slr1279 ndhC | CGGGCACCGAAATGTGAATCGTTTCAGAATTGGATTATTG | 3.49‡ |
| slr0442 hyp./sll1520 recN | TGGTTGGAGGGCTGTGATCCAGATCACATACGTGGGTTAA | 0.00*†§ |
| sll1268 hyp. | TCACCCAATAGTTGTGATCTAGATCACAGAGGGCCACGGC | 0.00*†§ |
*Sycrp1 binding sequences proposed by Xu & Su (2009).
†Oligonucleotide sequences demonstrating sequence-specific His–Sycrp1 binding in this work.
‡Sycrp1 binding sequences proposed by Ochoa de Alda & Houmard (2000).
§Sycrp1 binding sequences proposed by Omagari et al. (2008) to demonstrate His–Sycrp1 binding.
5′-Phosphate-free oligonucleotides were synthesized by Integrated DNA Technologies without HPLC purification, mixed 1 : 1 with their complement, annealed by boiling followed by slow cooling over 3 h, and gel-purified by UV-shadowing following native PAGE of DNA alone. This process allowed visualization of bands for excision and overnight extraction in 10 mM Tris, pH 8.0, which yielded pure 40-mer dsDNA for quantification. T4 polynucleotide kinase from Invitrogen was used following the manufacturer's instructions to label the blunt-ended double-stranded oligonucleotides indicated (Table 1) using 111 TBq (mmol [γ-32P]ATP)−1. Unincorporated label was removed with Bio-GelP-6 (#116561) from Bio-Rad as per the manufacturer's instructions.
For native PAGE gel-shift experiments, the binding reaction buffer contained 20 μM cAMP, 1.0 nM labelled dsDNA, His–Sycrp1 as indicated, 50 mM Tris/HCl, pH 7.5, 60 mM NaCl, 1.0 mM EDTA, 8.3 % (v/v) glycerol, and 0.1 mg acetylated BSA ml−1. Reactions were incubated at 22 °C for 25 min, then on ice for 15 min. The reactions were quickly and directly loaded onto the gel without loading buffer or dye. A voltage of 90 V was immediately applied for 10 min, and then increased to 200 V for an additional 35–45 min. The gel apparatus and buffers were pre-chilled and maintained at 4 °C. A voltage of 90 V was applied for at least 30 min prior to loading to remove mobile charged molecules. All running buffers were titrated to pH 8.0 to match the reaction buffer at 4 °C. All gels, running buffers and reaction buffers contained 20 μM cAMP (except where noted). Reagents were not filtered following addition of BSA or cAMP to maintain the indicated concentrations. The experiment-specific Tris-acetate-EDTA (TAE), Tris-borate-EDTA (TBE) and components that varied in the reaction buffer are indicated in the legend to Fig. 1. Gels were visualized by exposure to X-ray film at −80 °C.
Fig. 1.

EMSA demonstrating bound His–Sycrp1/DNA complexes and unbound DNA. The 40 bp radiolabelled dsDNA oligonucleotides surrounding previously proposed Crp binding sequences (Table 1) are indicated. (a) Side-by-side comparison of all targets predicted by Ochoa de Alda & Houmard (2000). Labelled slr1667 and Rndm. oligonucleotides are included as positive and negative controls, respectively. (b) Competitive binding to high-affinity and (c) low-affinity Crp binding sequences. Unlabelled competitor was added as indicated in addition to reaction buffers containing: (a, c) 500.0 nM His–Sycrp1, 20.0 μM cAMP, 500.0 nM unlabelled double-stranded Rndm. oligonucleotide and 1.0 nM of the labelled dsDNA indicated; (b) 100 nM His–Sycrp1, 20.0 μM cAMP and 1.0 nM of the labelled dsDNA indicated. Running buffers and 10 % acrylamide composition were: (a, c) 0.25× TBE, pH 8.0, at 4 °C, 20 μM cAMP and 50 : 1 (w/w) acrylamide : bis-acrylamide; (b) 1.0× TAE, pH 8.0, at 4 °C, 20 μM cAMP and 30 : 0.8 (w/w) acrylamide : bis-acrylamide.
Multiple sequence alignment (MSA).
MSAs were constructed using clustal w in Biology Workbench (http://workbench.sdsc.edu/) (Subramaniam, 1998).
RESULTS
Crp sequence-specific binding was demonstrated
To demonstrate Crp binding in vitro, a positive binding control, slr1667, a randomly generated sequence, and all target sites proposed by Ochoa de Alda & Houmard (2000) were screened for Crp binding (Fig. 1a, Table 1). The chlAII, narL and murF target sites were bound by His–Sycrp1. These proposed target sites are the only loci common to all Omagari et al. (2008), Ochoa de Alda & Houmard (2000) and Xu & Su (2009) predictions. These results were in accord with the inability of Omagari et al. (2008) to detect binding to any proposed site that has a calculated >3.1. In Fig. 1(a), binding to narL was detected, =3.1; consequently, sensitivity similar to that achieved by Omagari et al. (2008) was demonstrated in this assay. The Kd of His–SyCrp1 from all proposed targets bound in Fig. 1 has been described (Omagari et al., 2008). In side-by-side experiments, all previously published interactions were detected and yet the test failed to identify any new interactions among targets proposed by Ochoa de Alda & Houmard (2000).
To demonstrate cAMP dependence for Crp/DNA binding in our in vitro binding conditions, all oligonucleotides listed in Table 1 were assayed exactly as in Fig. 1(a), except that cAMP was omitted from the reaction, running buffers and gels. Likely due to a combination of high affinity (even greater than that for the E. coli consensus ICAP) (Omagari et al., 2008) and cAMP carried over from E. coli expression, Crp binding to the proposed murF target was detectable. However, binding was severely reduced to <10 % bound as opposed to 100 % in the presence of 20 μM cAMP. Detectable binding was absent in all other instances (data not shown).
To further demonstrate reproducibility and sequence-specificity for these proposed binding sites, competition assays were performed (Fig. 1b, c). The putative Crp binding sites located upstream of slr0442 and sll1268 were also included. Expression of slr0442 is downregulated in a crp mutant (Yoshimura et al., 2002a); consequently, slr0442 was used as a positive control. The sll1268 target proposed by Omagari et al. (2008) was included because of the high degree of conservation between it and slr0442 in the intergenic and N-terminal coding regions. Our results demonstrated His–Sycrp1 sequence-specific binding to murF, narL, chlAII, slr0442 and sll1268 proposed targets via competition assays using the slr1667 target as a specific competitor (Yoshimura et al., 2002a) and a random 40-mer (Rndm.) as a non-specific competitor. In all cases, the unlabelled specific competitor titrated Crp away from the labelled complex in favour of the specific competitor, while unlabelled non-specific competitor did not. Crp does not bind the slr1667 target in the absence of cAMP in vitro as reported by Yoshimura et al. (2002a) and reproduced here (see above). Consequently, titration by competitors further demonstrated the presence of Crp/cAMP complex. Omagari et al. (2008) have established the Crp sequence-specificity to these proposed targets by correlation. Shown in Fig. 1(b, c) is the first verification of specificity by direct competition. These results confirmed that the murF, narL, chlAII, slr0442 and sll1268 intergenic sites described in Table 1 met sequence-specific binding criteria.
Owing to complex instability during electrophoresis at room temperature, as evidenced by smearing between bands in the work of Omagari et al. (2008) and reproduced in this work (data not shown), electrophoresis at 4 °C was performed. Despite the strong signal from labelled DNA, increased complex stability at 4 °C was demonstrated, because smearing between bands was minimal to absent. However, at 4 °C, Crp/DNA complexes precipitated in 0.25× TBE, which rendered them immobile by electrophoresis. Addition of 500 nM Rndm. completely restored solubility and allowed near 100 % binding, as shown by slr1667 and murF targets (Fig. 1a). As little as 0.5 mg l−1 double-stranded polydeoxyinosinic-deoxycytidylic acid (poly-dIdC) added to the reaction buffer also restored solubility, but reduced the fraction of Crp/DNA complex by 60 % (data not shown). Crp/DNA complexes were soluble in 1.0× TAE at 4 °C, but low-Crp-affinity targets ( >0.7) did not maintain Crp/DNA complexes in this running buffer. Consequently, electrophoresis of narL and chlAII was performed in 0.25× TBE. From these results, it is clear that the temperature and ionic strength of electrophoresis buffers greatly affect Crp/DNA complex detection by gel shift.
The sycrp1 mutant construction was gene-specific, and did not introduce polar effects
To allow examination of Crp-dependent functions, a crp mutant was constructed by insertional inactivation of sycrp1. Complete segregation was confirmed by PCR. Southern blotting further confirmed that recombination had occurred specifically in the sycrp1 locus and that sll1924 (sycrp2) or slr0593 homologues were not disrupted (data not shown). Additional evidence for gene inactivation was obtained by observing phototactic and crp non-motile phenotypes (Yoshimura et al., 2002b) (data not shown). To discount polar effects of genes surrounding the site of sycrp1 inactivation, transcript abundance of the two genes flanking sycrp1 (sll1370 and sll1372) was quantified in photoautotrophically growing cultures by RT-QPCR. The quantities of these transcripts in the wild-type did not differ detectably from those in crp mutant strains (Vasquez, unpublished results). To determine whether Crp function was absent in the crp mutant, wild-type and crp crude cell extracts were also assayed for binding to the slr1667 target. Sequence-specific binding was absent in the crp mutant crude extracts but present in wild-type samples (data not shown). Therefore, gene expression differences were ascribed specifically to inactivation of the sycrp1 locus and resultant protein inactivation rather than to polar effects or recombination at non-target sites.
The shift from dark to light environmental conditions stimulated Crp-dependent transcriptional activation
To confirm that intracellular cAMP increased under the experimental conditions described, it was quantified by a cell filtration method. Intracellular cAMP increased from 0.046 to 0.92 pmol cAMP (μg Chl a)−1 following a dark (low intracellular cAMP) to light (high intracellular cAMP) transition. These values were in good agreement with those previously reported (see introduction).
To quantify transcript levels, RNA samples were collected in the dark and at 30 and 60 min after illumination. Cultures were sampled over 1 h, because transcriptional profiles are most dynamic during this period (Gill et al., 2002). All target transcripts proposed by Ochoa de Alda & Houmard (2000) were quantified in a low-resolution screen. RNA was sampled from one culture of wild-type and one culture of crp cells (n=1) to focus effort on demonstrating the reproducibility of Crp-dependent transcription reported below. Primer pair amplification efficiencies were not considered in this low-resolution screen comparing relative expression; consequently, we employed a prudent twofold expression cut-off to differentiate between candidate Crp-dependent and Crp-independent expression. The slr0194 (rpiA) transcript was one of nine proposed targets that did not demonstrate Crp-dependent transcription in this low-resolution screen. Consequently, it was used as a negative, Crp-independent transcription control. Only murF and chlAII demonstrated more than twofold Crp-dependent expression out of the 11 targets proposed by Ochoa de Alda & Houmard (2000) tested in this low-resolution screen.
Transcription of rpiA, murF, chlAII and slr0442 were further characterized to determine Crp-dependence following dark to light environmental changes (Fig. 2). Wild-type and crp cells were again cultured, this time in triplicate (n=3), to demonstrate reproducibility, and transcripts were quantified by RT-QPCR more accurately, taking amplification efficiency into account. The positive transcription control slr0442 was not activated in the wild-type either 30 min (grey bars) or 60 min (white bars) following illumination (Fig. 2a). The quantity of detectable transcript was constant for all time points versus the initial low intracellular cAMP condition, and resulted in an expression ratio of 1.0. Conversely, transcript levels in the mutant decreased following illumination (Fig. 2b). After 1 h illumination, wild-type expression of slr0442 was five times greater than that of the mutant (Fig. 2c) and almost twice that reported during moderate intracellular cAMP growth conditions (Yoshimura et al., 2002a). Consequently, it was inferred that the constant slr0442 expression in wild-type cells was due to a steady state achieved by simultaneous transcriptional activation by Crp and post-transcriptional mRNA degradation.
Fig. 2.

Relative gene expression during environmentally induced low to high intracellular cAMP in both wild-type (Wt) and crp mutant strains of Synechocystis and E. coli for indicated genes. (a–c) Synechocystis cells were acclimated to the dark for 16 h (low intracellular cAMP) and transferred to the light (high intracellular cAMP). Cells were collected for RNA quantification by RT-QPCR in the dark (black bars), 30 min (grey bars) and 60 min (white bars) following illumination. (d–f) Gfp fluorescence ratio of E. coli M182 cells containing the intergenic region of the indicated Synechocystis genes oriented to drive transcription of a promoterless gfp reporter were grown in either LB containing 3 % glucose (low intracellular cAMP, black bars) or LB alone (high intracellular cAMP, white bars). The black bars in (a), (b), (d) and (e) are calibrators presented for ease of reference only and do not contain error as defined previously (Pfaffl, 2001). The y axis labels in (a), (b) and (c) are as defined by Pfaffl (2001): ‘Expression ratio of a sample versus a control in comparison to a reference gene’=(Etarget)ΔCPtarget [(Ereference)ΔCPreference]−1, where ΔCP is the QPCR cycle threshold of the control−sample, target is the gene of interest, reference is rnpB, and E is PCR amplification efficiency. When Wt expression is the sample and Wt expression under the low intracellular cAMP condition (Wt low cAMP) is the control, the experimentally modified independent variable is time of illumination that causes high intracellular cAMP. The same holds for crp vs crp low cAMP. When Wt is the sample, and crp is the control, the experimentally modified independent variable is Crp. All samples were cultured in triplicate (n=3); error bars, ±sem. Note the log scales in (a), (b), (d) and (e), and the value break in (f).
This inference is supported by evidence that both crp and ssr3321 (hfq candidate) single mutants display striking similarity in expression of slr2015–2018, slr1667–1668 and slr0442, which are downregulated approximately four to five-, 40–48- and threefold, respectively, relative to wild-type cells during regular photoautotrophic growth (Dienst et al., 2008; Yoshimura et al., 2002a). Hfq is an RNA binding protein that acts to stabilize transcripts as a RNA chaperone or to facilitate the coupled degradation of sRNA–mRNA duplexes (Dienst et al., 2008). Although the mechanism of Hfq activity has not yet been demonstrated in cyanobacteria, it could explain post-transcriptional modification of slr1667 and slr0442. Under high-light stress, the 3′ mRNA of slr1667 is three to fourfold (clarified by personal communication with A. Singh) more abundant than the 5′ end (Singh et al., 2008). This finding clearly demonstrates strong post-transcriptional mRNA degradation that could be stabilized by a functional RNA chaperone.
Wild-type and mutant transcript levels for all genes in Fig. 2 were approximately equal in dark-adapted cells when intracellular cAMP was low. Transcripts of murF and chlAII were upregulated by illumination, but showed four and ten times, respectively, more transcript expression relative to the mutant following intracellular cAMP increase, thus demonstrating strong Crp dependence for transcription activation. In contrast, transcription from the negative transcription control rpiA did not exhibit Crp dependence, even though it was strongly induced following illumination (Fig. 2a, b). In sum, these results demonstrated that transcription from murF, chlAII and slr0442 met Crp-dependent expression criteria.
Expression driven by Sycrp1 ‘target’ promoters required Crp in E. coli
To determine whether the transcriptional machinery in E. coli is sufficient to stimulate Crp-dependent transcription from Crp ‘target’ promoters, all target promoters proposed by Ochoa de Alda & Houmard (2000) were oriented to drive transcription of a gfp reporter in wild-type and crp mutant strains of E. coli. Transcripts from cells grown in the high intracellular cAMP condition demonstrated Crp-dependent activation. The glucose effect is well documented (Kolb et al., 1993) and causes a drastic drop in intracellular cAMP. To decrease intracellular cAMP, glucose was added to the culture.
The positive transcription control slr0442 was strongly induced in the wild-type during high intracellular cAMP growth without glucose (Fig. 2d), but not in the mutant (Fig. 2e). Although wild-type expression was four times that of the mutant in the low intracellular cAMP growth condition with glucose (Fig. 2f), expression was 60 times greater in the high intracellular cAMP condition, thereby demonstrating a strong Crp activation dependence. Although not shown, it is interesting to note that the wild-type strain repressed narL transcription in the high intracellular cAMP condition 10 times more than in the crp mutant. The narL reporter was also independently isolated from the Hsp92II genomic reporter library due to similar expression characteristics (data not shown). Otherwise, in general, the E. coli reporter data paralleled results seen in Synechocystis, excepting chlAII. In this case, absolute fluorescence was indistinguishable from background fluorescence, indicating that the chlAII promoter did not drive transcription in E. coli. The background fluorescence between E. coli M182 wild-type and crp strains containing the gfp reporter plasmid but lacking the indicated intergenic regions was indistinguishable (data not shown). All other indicated constructs yielded signals well above this background. Consequently, these data demonstrated that the transcriptional elements in E. coli were sufficient to stimulate Crp-dependent transcription from slr0442 and murF intergenic regions.
Transcription start sites were determined
All presented transcription +1 start sites for murF, narL, chlAII, slr0442 and sll1268 were determined by RACE in this work (Fig. 3). These promoters are accordingly labelled in Fig. 3, and the most distal from the gene is assigned P1. We are unaware of any other studies mapping start sites for these genes. It should be noted that RACE requires much less transcript than primer extension due to its high sensitivity. However, RACE is not a quantitative method; consequently, the relative strengths of these promoters as affected by Crp activation were not inferred.
Fig. 3.

Promoter regions of crp target genes in Synechocystis. (a) Upstream regions of murF homologues in closely related cyanobacteria. CwatDRAFT_4119 and CY0110_02369 correspond to the indicated ORF identifiers in Crocosphaera watsonii and Cyanothece sp. CCY 0110, respectively. MSAs span conserved Crp sites to the conserved N-terminal amino acids belonging to protein homologues, thereby anchoring the alignment between two conserved regions. (b) Partial upstream region of chlAII. (c) Upstream regions of sll1268 and slr0442. A secondary low-affinity site proposed by Omagari et al. (2008) is boxed with dashes. Proposed Crp binding sites identified by competitive gel shift in Fig. 1 are boxed. All putative transcriptional start sites were determined by RACE mapping in this work, are labelled (gene Px), and are enclosed with small boxes. Suggested −10 sigma factor binding sites are underlined. Annotated translational start codons are double underlined. Tick marks are spaced 10.5 bases apart for reference. The murF P1 is not shown, but described in the text.
RNA from both wild-type and crp strains experiencing both low and high intracellular cAMP all yielded the same start sites, although ±1 base chatter between samples was observed at murF P2 (Fig. 3a). P1 TGGTAAGATACACCCTG (transcriptional start site in italicized bold type) is not shown and lies 136 bases upstream of P2. Crocosphaera watsonii and Cyanothece sp. CCY 0110 MurF whole-protein blast scores were 9e-136 and 1e-129, respectively, relative to Synechocystis MurF. The intergenic and non-conserved N-terminal murF regions from these closely related cyanobacteria were aligned to highlight other conserved elements because of an apparent conservation of the proposed Crp site. A total of 30 and 31 base gaps were observed for Crocosphaera and Cyanothece, respectively in the region between the conserved Crp core binding sequence and the strongly conserved protein-coding region. These deletions corresponded almost exactly to three 10.5 bp turns of the α-helix. In the alignments shown, a 12 bp gap that corresponds to approximately one helical turn is located between P3 and the putative Crp binding site.
The intergenic chlAII region contained only one putative transcriptional start site (Fig. 3b). Similar sequence alignments of intergenic regions from chlA orthologues of these closely related cyanobacteria were uninformative due to low sequence homology and the absence of readily identifiable Crp site core sequences. Consequently, an alignment is not shown.
The slr0442 and sll1268 intergenic sequences were aligned, because the encoded proteins of these genes bear 75 % identity within the first 58 N-terminal amino acids, and the proposed Crp sites were roughly equidistant from the common, not the annotated, ATG start codon (Fig. 3c). Only one base-spacing difference was observed between the transcription start and the proposed Crp sites for these two gene promoters.
DISCUSSION
Some proposed ‘target genes of Sycrp1’ may not have been detected
The utility of dark to light conditions for elucidating Crp-dependent activation as proposed by Omagari et al. (2008) has been demonstrated by the results reported here. Predicted targets were upregulated in a Crp-dependent manner only when predictions made by Ochoa de Alda & Houmard (2000), Xu & Su (2009), and Omagari et al. (2008) overlapped (bold and underlined type in Table 1). In all cases, excluding narL, those intergenic regions containing Crp core binding sites <3.1 were bound by His–Sycrp1 in vitro and demonstrated Crp-dependent activation in vivo. Consequently, murF, chlAII and slr0442 meet the ‘target gene of Sycrp1’ criteria used to annotate the Kazusa Cyanobase.
It is, however, conceivable that the low-resolution expression screen overlooked subtle expression differences such as those of the divergently transcribed regulatory genes narL and slr1805 (hik16 subunit). The possible protein–protein interactions suggested by yeast two-hybrid experiments in which NarL interacts with Hik16 and MurC (Sato et al., 2007) suggest protein-level regulation in the first step of the peptidoglycan biosynthetic pathway three enzymic reactions upstream of MurF-catalysed ligation. Such regulation is expected to be involved in modulating the balance of intracellular carbon and nitrogen (Singh et al., 2008).
Consistent with the only microarray data, to our knowledge, published using the motile glucose-sensitive Synechocystis exposed to similar environmental conditions (Gill et al., 2002), the target transcripts in Fig. 2 that were proposed by Ochoa de Alda & Houmard (2000) were strongly upregulated following illumination. Such upregulation is also consistent with the functions of these gene products. Specifically, murF upregulation is expected, because its protein product is essential for the peptidoglycan synthesis required for cell division during periods of growth (Malakhov et al., 1995). Also, chlAII upregulation upon illumination is expected, because cultures that are dark-adapted for prolonged periods and attain oxygen equilibrium with the air are micro-oxic relative to actively photosynthesizing cultures. This micro-oxic state is achieved via the same mechanisms that cause the diurnal dissolved oxygen cycles observed in lakes and cyanobacterial mats (Jorgensen et al., 1979). Under micro-oxic illuminated conditions, chlAII is transcribed in a putative operon containing ho2 (Sugishima et al., 2005; Xu & Su, 2009; Zhang et al., 2005) and hemN1, which catalyse three steps in the chlorophyll biosynthetic pathway (Minamizaki et al., 2008). The Crp-dependent chlAII activation observed suggests coordination between motility and photosynthetic acclimation, but requires further characterization of both pilin and chlAII Crp-dependent expression at these promoters.
Analysis of elements in Sycrp1 class I and II promoters
Although mutagenesis was not used to demonstrate that the proposed Crp binding sites in Fig. 3 are required for gene activation, in vivo evidence exists to support such a conclusion. First, the conserved spacing of proposed Crp binding and +1 transcriptional start sites between these promoters and extensively characterized promoters of E. coli was observed. Second, in vivo transcriptional activation under conditions stimulating high intracellular cAMP required Crp in both E. coli and Synechocystis, thus demonstrating that transcription elements in E. coli are sufficient to stimulate Crp-dependent transcription from murF and slr0442 intergenic regions (Fig. 2). Together these data indicate that the E. coli Crp mechanisms can be compared with those in Synechocystis.
E. coli Crp promoters are classic model systems that have been thoroughly reviewed recently (Borukhov & Lee, 2005) and in the past (Busby & Ebright, 1999). By definition, Crp and RNA polymerase (RNAP) must be on the same side of the DNA strand to make contacts that stimulate transcription via the readily describable mechanisms of class I, class II and class III promoters. Consequently, intervals of 10.5 bp α-helical turns must be maintained from the middle of the −10 sigma factor binding site to the middle of the Crp site for Crp to contact RNAP. Five or more turns is defined as class I, and three turns as class II. Four-turn spacing does not occur, because several Crp–RNAP interactions would be impeded. In Synechocystis, transcription start site mapping of the slr1667–1668 operon has revealed that the proposed Crp binding site is 15.5 helical turns upstream from the middle of the −10 region, thereby placing Crp on the opposite side of the DNA strand relative to RNAP (Yoshimura et al., 2002a). In this case, Crp cannot contact RNAP via the readily describable mechanisms outlined here. Consequently, the Crp activation mechanism at this locus is unclear. As opposed to class I and class II promoters, class III promoters require two or more activator molecules and RNAP for full transcription activation. A major difference between E. coli and cyanobacterial promoters is the frequent absence of a −35 sigma factor binding site (Curtis & Martin, 1994); however, the −10 region TATAAT is conserved and TANNNT is most frequently observed (Su et al., 2005; Vogel et al., 2003). The proposed class I and class II promoters described below are inferred based on this spacing, until the involvement of an additional element is demonstrated to define class III organization.
The murF P3 contains class I promoter spacing relative to the transcriptional start site
The murF P3 bears class I promoter structure only in that the proposed Crp binding site is 7.0 α-helical turns from the transcriptional start site (Fig. 3a). However, the −10 region is not readily discernible within four to seven bases of the transcriptional start. The only conserved TNNNNT sequence places Crp centred 5.7 helical turns away in suboptimal positioning but close to the same side of the DNA strand as the proposed sigma factor binding site. However, the spacing of these elements is not conserved among these freshwater cyanobacteria. Instead, deletions totalling three helical turns seem to have occurred independently because the deletions are not of identical lengths. One is 30, the other 31 bp long. This keeps the elements that are retained on either side of the deletions in similar helical orientation and on same side of the DNA strand in the Crocosphaera and Cyanothece sequences shown. Therefore, the observed conservation of helical spacing may be significant for regulation. Xu & Su (2009) predicted a −10 region, TAACAT, located 32 bp downstream from the proposed Crp binding site. This −10 region is not properly positioned to initiate transcription from any of the +1 sites identified by RACE. Retention of the proposed Crp core binding sequences in these closely related cyanobacteria suggests that Crp regulation of murF is also conserved.
The chlAII promoter class is unclear
The chlAII promoter class is unclear because the proposed Crp site is very distant and on the opposite face of the DNA strand relative to the transcriptional start site (Fig. 3b). The proposed Crp site is 28.6 helical turns from the transcriptional start site. Furthermore, a plausible −10 region is not apparent; thus, we cannot support the validity of this proposed transcriptional start site by relation to other conserved elements. Xu & Su (2009) predicted that a −10 region, TCGATT, is 29 bp downstream of the proposed Crp site; however, no +1 sites were identified by RACE in this region.
The slr0442 P2 contains class II promoter spacing
The slr0442 P2 bears class II promoter structure maintaining the characteristic three α-helical turn spacing between the centre of the near-consensus P2 −10 region TAAAAT and the proposed Crp site (Fig. 3c). Crp binding would repress transcription from P1 via steric hindrance of RNAP, thereby switching most initiation to P2. The proposed intimate proximity with RNAP strongly suggests interactions between Crp and RNAP; however, the analysis of activating regions 2 and 3 described for E. coli (see reviews cited above) by primary sequence alignment is insufficient to address the possibility of these interactions. The perfect alignment of the proposed Crp and −10 sites in both slr0442 and sll1268 was also predicted by Xu & Su (2009), and strongly suggests conservation of function as class II Crp promoter regulation. A second Crp site that was proposed by Omagari et al. (2008) (boxed with dashes in Fig. 3c) has not been confirmed by binding studies. It is optimally positioned on the same side as RNAP, thereby potentially implicating class III promoter structure.
The genes slr0442 and sll1268 are homologous within the N-terminal domain. This homology defines a large set of hitherto uncharacterized cyanobacterial proteins. The significance of this conserved region suggests coordinated regulation of slr0442 and sll1268 by Crp.
As has been discussed above for murF and slr0442, promoter mapping revealed the same well-characterized class I and class II promoter organization in Synechocystis as in E. coli. When intergenic regions containing these promoters were oriented to drive gfp transcription in E. coli, the results paralleled the regulatory effects observed in Synechocystis. These results thereby illustrate structure and function associations in vivo (Fig. 2), and strongly suggest that cyanobacterial Crp-dependent promoter mechanisms can function similarly to those in E. coli. Furthermore, we provide the first experimental evidence, to our knowledge, to support the validity of bioinformatic predictions based on class II spacing of −10 and Crp site elements in cyanobacteria (Xu & Su, 2009).
Acknowledgments
The authors thank those who provided materials listed in the text, Cindy Malone for critical reading of the manuscript, and students who participated in the introductory molecular microbiology course and constructed reporter plasmids. This study was supported by grants from the NSF MCB 0099327, NIH 2 SO6 GM048680, to M. L. S., and a University Corporation project award for academic research, California State University, Northridge, to J. H.
Abbreviations
EMSA, electrophoretic mobility shift assay
MSA, multiple sequence alignment
RACE, rapid amplification of cDNA ends
RT-QPCR, reverse transcriptase-mediated quantitative PCR
RNAP, RNA polymerase
Footnotes
A supplementary table, listing primers used in this study, is available with the online version of this paper.
References
- Argueta, C. & Summers, M. L. (2005). Characterization of a model system for the study of Nostoc punctiforme akinetes. Arch Microbiol 183, 338–346. [DOI] [PubMed] [Google Scholar]
- Argueta, C., Yuksek, K. & Summers, M. (2004). Construction and use of GFP reporter vectors for analysis of cell-type-specific gene expression in Nostoc punctiforme. J Microbiol Methods 59, 181–188. [DOI] [PubMed] [Google Scholar]
- Argueta, C., Yuksek, K., Patel, R. & Summers, M. L. (2006). Identification of Nostoc punctiforme akinete-expressed genes using differential display. Mol Microbiol 61, 748–757. [DOI] [PubMed] [Google Scholar]
- Ausubel, F., Brent, R., Kingston, R., Moore, D., Seidman, J., Smith, J. & Struhl, K. (2000). Current Protocols in Molecular Biology. New York: Wiley.
- Bhaya, D., Nakasugi, K., Fazeli, F. & Burriesci, M. S. (2006). Phototaxis and impaired motility in adenylyl cyclase and cyclase receptor protein mutants of Synechocystis sp. strain PCC 6803. J Bacteriol 188, 7306–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov, S. & Lee, J. (2005). RNA polymerase structure and function at lac operon. C R Biol 328, 576–587. [DOI] [PubMed] [Google Scholar]
- Botsford, J. L. & Harman, J. G. (1992). Cyclic AMP in prokaryotes. Microbiol Rev 56, 100–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby, S. J. & Ebright, R. H. (1999). Transcription activation by catabolite activator protein (CAP). J Mol Biol 293, 199–213. [DOI] [PubMed] [Google Scholar]
- Busby, S., Kotlarz, D. & Buc, H. (1983). Deletion mutagenesis of the Escherichia coli galactose operon promoter region. J Mol Biol 167, 259–274. [DOI] [PubMed] [Google Scholar]
- Cann, M. J. (2004). Signalling through cyclic nucleotide monophosphates in cyanobacteria. New Phytol 161, 23–34. [Google Scholar]
- Casadaban, M. J. & Cohen, S. N. (1980). Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138, 179–207. [DOI] [PubMed] [Google Scholar]
- Casadaban, M. J., Chou, J. & Cohen, S. N. (1980). In vitro gene fusions that join an enzymatically active β-galactosidase segment to amino-terminal fragments of exogenous proteins: Escherichia coli plasmid vectors for the detection and cloning of translational initiation signals. J Bacteriol 143, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, S. E. & Martin, J. A. (1994). The transcription apparatus and the regulation of transcription initiation. In The Molecular Biology of Cyanobacteria, pp. 613–639. Edited by D. A. Bryant. Norwell, MA: Kluwer Academic Publishers.
- Dienst, D., Duhring, U., Mollenkopf, H. J., Vogel, J., Golecki, J., Hess, W. R. & Wilde, A. (2008). The cyanobacterial homologue of the RNA chaperone Hfq is essential for motility of Synechocystis sp. PCC 6803. Microbiology 154, 3134–3143. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gonzalez, B., Martinez-Ferez, I. M. & Vioque, A. (1998). Characterization of two carotenoid gene promoters in the cyanobacterium Synechocystis sp. PCC 6803. Biochim Biophys Acta 1443, 343–351. [DOI] [PubMed] [Google Scholar]
- Gill, R. T., Katsoulakis, E., Schmitt, W., Taroncher-Oldenburg, G., Misra, J. & Stephanopoulos, G. (2002). Genome-wide dynamic transcriptional profiling of the light-to-dark transition in Synechocystis sp. strain PCC 6803. J Bacteriol 184, 3671–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, A., Hodgson, D. R. & Cann, M. J. (2006). Regulation of prokaryotic adenylyl cyclases by CO2. Biochem J 396, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, B. B., Revsbech, N. P., Blackburn, T. H. & Cohen, Y. (1979). Diurnal cycle of oxygen and sulfide microgradients and microbial photosynthesis in a cyanobacterial mat sediment. Appl Environ Microbiol 38, 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, T., Sato, S., Kotani, H., Tanaka, A., Asamizu, E., Nakamura, Y., Miyajima, N., Hirosawa, M., Sugiura, M. & other authors (1996). Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res 3, 109–136. [DOI] [PubMed] [Google Scholar]
- Kim, B.-H., Oh, H.-M., Lee, Y.-K., Choi, G.-G., Ahn, C.-Y., Yoon, B.-D. & Kim, H.-S. (2006). Simple method for RNA preparation from cyanobacteria. J Phycol 42, 1137–1141. [Google Scholar]
- Kolb, A., Busby, S. J., Buc, H., Garges, S. & Adhya, S. (1993). Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem 62, 749–795. [DOI] [PubMed] [Google Scholar]
- Kunert, A., Hagemann, M. & Erdmann, N. (2000). Construction of promoter probe vectors for Synechocystis sp. PCC 6803 using the light-emitting reporter systems Gfp and LuxAB. J Microbiol Methods 41, 185–194. [DOI] [PubMed] [Google Scholar]
- Malakhov, M. P., Los, D. A., Wada, H., Semenenko, V. E. & Murata, N. (1995). Characterization of the murF gene of the cyanobacterium Synechocystis sp. PCC 6803. Microbiology 141, 163–169. [DOI] [PubMed] [Google Scholar]
- Masuda, S. & Ono, T. A. (2004). Biochemical characterization of the major adenylyl cyclase, Cya1, in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett 577, 255–258. [DOI] [PubMed] [Google Scholar]
- Minamizaki, K., Mizoguchi, T., Goto, T., Tamiaki, H. & Fujita, Y. (2008). Identification of two homologous genes, chlAI and chlAII, that are differentially involved in isocyclic ring formation of chlorophyll a in the cyanobacterium Synechocystis sp. PCC 6803. J Biol Chem 283, 2684–2692. [DOI] [PubMed] [Google Scholar]
- Ochoa de Alda, J. A. & Houmard, J. (2000). Genomic survey of cAMP and cGMP signalling components in the cyanobacterium Synechocystis PCC 6803. Microbiology 146, 3183–3194. [DOI] [PubMed] [Google Scholar]
- Ochoa de Alda, J. A., Ajlani, G. & Houmard, J. (2000). Synechocystis strain PCC 6803 cya2, a prokaryotic gene that encodes a guanylyl cyclase. J Bacteriol 182, 3839–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori, M. & Okamoto, S. (2004). Photoresponsive cAMP signal transduction in cyanobacteria. Photochem Photobiol Sci 3, 503–511. [DOI] [PubMed] [Google Scholar]
- Omagari, K., Yoshimura, H., Suzuki, T., Takano, M., Ohmori, M. & Sarai, A. (2008). ΔG-based prediction and experimental confirmation of SYCRP1-binding sites on the Synechocystis genome. FEBS J 275, 4786–4795. [DOI] [PubMed] [Google Scholar]
- Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, T., Murata, N. & Ohmori, M. (1991). The concentration of cyclic AMP and adenylate cyclase activity in cyanobacteria. Plant Cell Physiol 32, 581–584. [Google Scholar]
- Sato, S., Shimoda, Y., Muraki, A., Kohara, M., Nakamura, Y. & Tabata, S. (2007). A large-scale protein–protein interaction analysis in Synechocystis sp. PCC6803. DNA Res 14, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A. K., Elvitigala, T., Bhattacharyya-Pakrasi, M., Aurora, R., Ghosh, B. & Pakrasi, H. B. (2008). Integration of carbon and nitrogen metabolism with energy production is crucial to light acclimation in the cyanobacterium Synechocystis. Plant Physiol 148, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier, R. Y., Kunisawa, R., Mandel, M. & Cohen-Bazire, G. (1971). Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol Rev 35, 171–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, D. R., Rochaix, J. D. & Purton, S. (1996). The bacterial phleomycin resistance gene ble as a dominant selectable marker in Chlamydomonas. Mol Gen Genet 251, 23–30. [DOI] [PubMed] [Google Scholar]
- Su, Z., Olman, V., Mao, F. & Xu, Y. (2005). Comparative genomics analysis of NtcA regulons in cyanobacteria: regulation of nitrogen assimilation and its coupling to photosynthesis. Nucleic Acids Res 33, 5156–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam, S. (1998). The Biology Workbench – a seamless database and analysis environment for the biologist. Proteins 32, 1–2. [PubMed] [Google Scholar]
- Sugishima, M., Hagiwara, Y., Zhang, X., Yoshida, T., Migita, C. T. & Fukuyama, K. (2005). Crystal structure of dimeric heme oxygenase-2 from Synechocystis sp. PCC 6803 in complex with heme. Biochemistry 44, 4257–4266. [DOI] [PubMed] [Google Scholar]
- Summerfield, T. C. & Sherman, L. A. (2008). Global transcriptional response of the alkalitolerant cyanobacterium Synechocystis sp. strain PCC 6803 to pH 10. Appl Environ Microbiol 74, 5276–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers, M. L., Wallis, J. G., Campbell, E. L. & Meeks, J. C. (1995). Genetic evidence of a major role for glucose-6-phosphate dehydrogenase in nitrogen fixation and dark growth of the cyanobacterium Nostoc sp. strain ATCC 29133. J Bacteriol 177, 6184–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi, K. & Ohmori, M. (1998). An adenylate cyclase, CyaD, mediates the signal of blue light in the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol 39, s153. [DOI] [PubMed] [Google Scholar]
- Terauchi, K. & Ohmori, M. (1999). An adenylate cyclase, Cya1, regulates cell motility in the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol 40, 248–251. [DOI] [PubMed] [Google Scholar]
- Terauchi, K. & Ohmori, M. (2004). Blue light stimulates cyanobacterial motility via a cAMP signal transduction system. Mol Microbiol 52, 303–309. [DOI] [PubMed] [Google Scholar]
- Vogel, J., Axmann, I. M., Herzel, H. & Hess, W. R. (2003). Experimental and computational analysis of transcriptional start sites in the cyanobacterium Prochlorococcus MED4. Nucleic Acids Res 31, 2890–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M. & Su, Z. (2009). Computational prediction of cAMP receptor protein (CRP) binding sites in cyanobacterial genomes. BMC Genomics 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, H., Hisabori, T., Yanagisawa, S. & Ohmori, M. (2000). Identification and characterization of a novel cAMP receptor protein in the cyanobacterium Synechocystis sp. PCC 6803. J Biol Chem 275, 6241–6245. [DOI] [PubMed] [Google Scholar]
- Yoshimura, H., Yanagisawa, S., Kanehisa, M. & Ohmori, M. (2002a). Screening for the target gene of cyanobacterial cAMP receptor protein SYCRP1. Mol Microbiol 43, 843–853. [DOI] [PubMed] [Google Scholar]
- Yoshimura, H., Yoshihara, S., Okamoto, S., Ikeuchi, M. & Ohmori, M. (2002b). A cAMP receptor protein, SYCRP1, is responsible for the cell motility of Synechocystis sp. PCC 6803. Plant Cell Physiol 43, 460–463. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Migita, C. T., Sato, M., Sasahara, M. & Yoshida, T. (2005). Protein expressed by the ho2 gene of the cyanobacterium Synechocystis sp. PCC 6803 is a true heme oxygenase. Properties of the heme and enzyme complex. FEBS J 272, 1012–1022. [DOI] [PubMed] [Google Scholar]