

Abstract
An acute attack of gout is a paradigm of acute sterile inflammation, as opposed to pyogenic inflammation. Recent studies suggest that the triggering of IL-1β release from leucocytes lies at the heart of a cascade of processes that involves multiple cytokines and mediators. The NLRP3 inflammasome appears to have a specific role in this regard, but the biochemical events leading to its activation are still not well understood. We review the known mechanisms that underlie the inflammatory process triggered by urate crystals and suggest areas that require further research.
Introduction
Acute gout, also called podagra when it affects the first metatarsophalangeal joint, is due to the inflammatory reaction of joint tissues when monosodium urate (MSU) crystals form within the joint. The link between crystal deposition and joint inflammation was made by Garrod, who developed the thread-test assay of serum urate and stated that 'the deposited urate of soda may be looked upon as the cause, and not the effect, of the gouty inflammation'. Older clinical descriptions of acute gout, from Hippocrates to Charcot, detailed the very sudden and inflammatory character of an attack.
The initial symptoms of a gouty attack are sudden and violent, taking place nearly always at night. The patient feels a sudden onset of pain ... it often affects the big toe 1st MT joint, which becomes rapidly red and swollen; the veins of the leg can become dilated, and the leg can become purple and sometimes accompanied by bruising.
Charcot, Maladies des Vieillards, 1890
This account brings to life all of the cardinal manifestations of acute inflammation. After McCarty and Hollander published their observations that the crystals found in the synovial fluid were composed of MSU [1], there remained no doubt that urate crystals are the cause of this intense inflammation. These observations have led to detailed analyses of the inflammatory pathways triggered by urate crystals and the mechanisms that link crystal deposition to tissue inflammation.
During the past decade, the role of innate immune responses in the initiation of inflammatory and immune responses to injury and infection has gained prominence. In contrast to the acquired immune system, innate responses orchestrate the immediate and early phases of host defence to microbes as well as to injury, initiating the inflammatory reaction and recruiting cells of the acquired immune system to the site of inflammation. These advances bring our attention back to the interplay between cells of the innate immune system and the products of tissue damage and cell death - the so-called danger signals or danger-associated molecular patterns. MSU represents one of these signals that activate cellular defences. In the present review, we cover the recent developments in our under standing of MSU's effects, integrating this information with previous knowledge regarding the inflammatory response provoked by urate crystals.
Crystal factors
MSU crystallizes when its plasma concentration exceeds its solubility (around 7 mg/dl, or 420 μmol/l). Concentration may not be the only determinant, however, and other factors have been reported to affect urate solubility. We know from clinical observation that patients with hyperuricaemia can be asymptomatic, sometimes for long periods before the first declaration of gout, so there must be modifying factors present in biological fluids that affect urate solubility and their capacity to form crystals. Furthermore, patients can have known MSU deposits that are clinically quiescent - indicating that there is further regulation at the level of the tissue response to urate (see later).
The factor or factors predisposing certain hyperuricaemic persons and not others to develop gout are as yet unknown. In vitro, several factors have been described to influence urate solubility, including pH, temperature, ionic strength and the binding of urate to plasma macromolecules [2]. Katz and Schubert suggested that proteoglycans of connective tissue origin can increase urate solubility. Their experiments showed that increasing the concentration of a cartilage protein polysaccharide extract increased urate solubility threefold, from 6 to 17 mg/100 ml. How this may explain the site of deposition of urate crystals in vivo, however, is not yet clear [3]. Not only is the solubility of urate influenced by its binding to plasma proteins, but also its inflammatory potential. Crystals coated with IgG fragments were more inflammatory than naked crystals [4,5]. As inflammation subsides, apolipoprotein B displaces the IgG by competitively coating sites on crystals and could contribute in part to the resolution of acute gouty arthritis [6].
These findings indicate that crystal-bound proteins modify the cellular response, but the underlying mechanisms of these effects remain to be elucidated. As crystals are essentially foreign bodies, they have toxic effects on cells (see below). Surprisingly, the size of MSU crystals does not seem to be a major determinant of its inflammatory potential, as varying the mean crystal size between 2.0 and 11.8 μm did not affect TNF production by monocyte/macrophage cells [7].
Cell types
The cells most studied in gouty inflammation are leucocytes - in particular, neutrophils and macrophages. Addition of MSU crystals to neutrophils results in cell death and release of lysosomal and cytoplasmic enzymes. In vitro studies showed that when native crystals were added to neutrophil preparations, ingested crystals induced the release of lysosomal contents by rupturing lysosomal membranes [8]. This pathway of cell death appears to be abrogated when crystals are coated with immunoglobulin, but release of lysosomal enzymes still took place [9]. The importance of neutrophils in the acute response is further supported by the effectiveness of colchicine, a potent inhibitor of neutrophil lysosomes as well as cell migration [10].
Neutrophils are attracted to the site of inflammation by chemotactic factors as well as cytokines and chemokines (see later) that act upon endothelial adhesion and transmigration. Among the factors known to be upregulated during gouty inflammation are IL-1β, IL-8, CXCL1 and granulocyte colony-stimulating factor [11].
The interactions between monocyte/macrophages with MSU do not appear to be so deadly, eliciting principally the secretion of proinflammatory cytokines. Both IL-1β and TNFα were secreted when monocytes were cultured in the presence of crystals, and their effects on cell proliferation were inhibited by specific blocking antibodies [12,13]. Macrophages do not appear to respond as well to MSU as to monocytes. When macrophages were differentiated in vitro, they became less proinflammatory in their response to MSU crystals, even though they retained their capacity for phagocytosis [7]. In a subsequent study by the same group, a potential role of transforming growth factor beta was suggested to account for this phenomenon [14]. The autoregulation of inflammation by phagocytes may explain the self-limiting nature of acute gout, as well as the fact that the presence of MSU crystals in a joint is not always accompanied by inflammatory signs and symptoms. The difference in the cellular response seems to be linked to the state of the macrophage, and as our understanding of the phenotypic variations of macrophages evolves, including the concept of M1 and M2 macrophages, we may gain a better understanding on how this impacts on inflammation. Of particular interest is the role of resident macrophages, as they have been reported to play a primordial part in initiating the tissue response to MSU in a mouse peritonitis model [15].
Among the other cell types that may participate in gouty inflammation, mast cells deserve a mention. In the air pouch model of gout, the density of mast cells increased in the lining layer rapidly after crystals were injected, and was correlated with the histamine content of the pouch liquid [16]. Using a microarray approach to analyse gene expression by lining cells of the air pouch, it was interesting to observe that the gene encoding histidine decarboxylase, the enzyme that synthesizes histamine, is rapidly upregulated after injection of MSU crystals [17]. In the peritonitis model of gout, depletion of peritoneal mast cells attenuated the neutrophil inflammatory response [18]. Finally, mast cells are able to release IL-1β on activation of the NALP3 inflammasome through lipopolysaccharide [4], but we have no information on whether MSU can also perform this function. These results suggest that mast cells participate in the acute response of tissue to MSU crystals and contribute to neutrophil recruitment.
IL-1 as a crucial mediator of gouty inflammation
Proinflammatory cytokines undoubtedly have a critical role in orchestrating the inflammatory reaction to MSU crystals. Recent attention has focused particularly on the role of IL-1. In murine models of crystal-induced inflammation, the injection of MSU into a preformed air pouch or within the peritoneal cavity elicited an influx of neutrophils that was accompanied by the local accumulation of chemoattractants and cytokines. Although care needs to be taken when extrapolating findings from these models to joint inflammation in gout, both the air pouch and the peritoneum have a lining that is morphologically similar to the synovium, and both models reconstitute the complex interplay that occurs between resident and infiltrating immune cells.
Experimental data from crystal-induced peritonitis, crystal-induced air pouch inflammation or urate crystals injected intra-articularly provide compelling evidence for the role of IL-1 in the pain and inflammation associated with gout. Two lines of study have been employed - a murine IL-1R1 knockout model, and the effects of IL-1 blockade (by antibodies against IL-1β, IL-1Rα or by mIL-1 Trap). These inhibitors of IL-1 were as effective as genetic deletion of IL-1R1 in reducing inflammation and hyperalgesia in these murine models [11,19,20]. Furthermore, the rapid clinical response of patients with acute gouty arthritis to IL-1 inhibition by a variety of agents (IL-1RA, IL-1Trap or an anti-IL-1β monoclonal antibody) validates the concept that this cytokine plays a key role in gouty inflammation [20-22].
The cellular target of IL-1β released by MSU crystals in the peritonitis model does not appear to be leucocytes. Using bone-marrow chimeras, Chen and colleagues demonstrated that MSU-induced inflammation was not modified when normal wild-type mice received IL-1RI-/- or MyD88-/- bone marrow, but inflammation was attenuated when IL-1RI-deficient or MyD88-deficient mice received normal bone marrow [19]. As both IL-1RI and MyD88 are essential for signal transduction on IL-1 binding, these results showed that nonhematopoieitic cells, most probably endothelial cells, are necessary to develop fully the inflammatory response to IL-1β.
Role of NLRP3 (NALP3) inflammasome in gout
IL-1β is the prototypic inflammatory cytokine and has multiple actions on cells and tissues (reviewed in [23]). The cytokine is produced as an inactive pro-molecule by immune cells such as macrophages, monocytes and dendritic cells, and is then cleaved into the active p17 form of IL-1β to be secreted out of the cell. Cleavage of pro-IL-1β is catalysed by caspase-1 (also known as IL-1- converting enzyme). Caspase-1 is a member of the family of inflammatory caspases that include caspase-4, caspase-5, caspase-11 and caspase-12; in the context of IL-1β processing, caspase-1 accounts for the major activity that requires the formation of a molecular platform known as the inflammasome (reviewed in [24]). Other pathways of IL-1β processing independent of caspase-1 have been described, implicating neutrophilderived and mast cell-derived proteases [25,26].
The inflammasome is a cytoplasmic protein complex composed of a protein of the NLRP (or NALP) family, an adapter ASC protein as well as an inflammatory caspase. Fourteen NLRPs have been identified in humans, and they all have multiple domains that share common features. The ASC adapter contains a PYD domain that mediates interaction with a homologous domain on NLRP, as well as a CARD domain that interacts with caspase-1. For the NLRP3 inflammasome, a second adapter - the Cardinal protein - is needed to recruit a second molecule of caspase-1, so as to form a dimer with the caspase-1 recruited by ASC (Figure 1). Numerous danger-associated molecular patterns and pathogen associated molecular patterns have been reported to signal via the NLRP3 inflammasome, including bacterial cell wall components, bacterial RNA or bacteria themselves, such as Listeria monocytogenes or Staphylococcus aureus. In addition, NLRP3 can be activated by signals that induce potassium efflux, such as ATP, via the P2X7 receptor, or by toxins such as nigericin.
Figure 1.
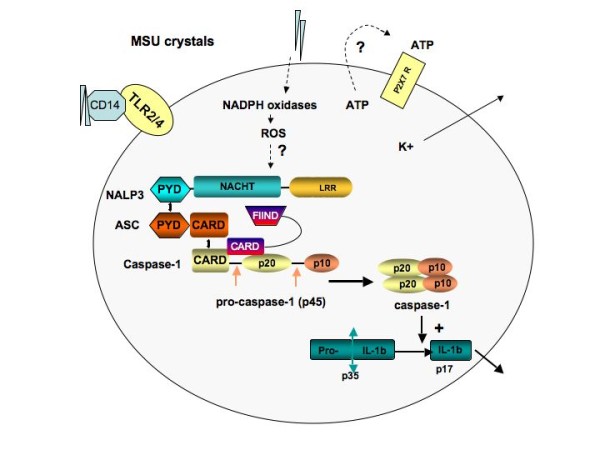
Composition of the NALP3 inflammasome and its activation by monosodium urate. Phagocytosis of monosodium urate (MSU) crystals leads to the generation of reactive oxygen species (ROS) through activation of NADPH oxidases. This event activates the NLRP3 inflammasome. MSU crystals may also induce the secretion of ATP, which in turn activates P2X7R. On activation of the P2X7 receptor, there is rapid exit of intracellular potassium that triggers the NLRP3 inflammasome. A rise in intracellular calcium is also required for the secretion of processed IL-1β. The macromolecular complex (inflammasome) consists of NLRP3, ASC and procaspase-1, and CARDINAL. Assembly leads to activation of caspase-1, which in turn cleaves pro-IL-1β to produce biologically active IL-1β. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD); FIIND, domain with function to find; LRR, leucine-rich repeat; MDP, muramyl dipeptide; NACHT, domain conserved in NAIP, CIITA, HET-E and TP1; NALP3, NACHT-containing, LRR-containing and PYD-containing protein; PYD, pyrin death domain.
The group of Tschopp discovered that many inorganic particles, including MSU and calcium pyrophosphate dihydrate crystals, are capable of activating the NLRP3 inflammasome to process and secrete active IL-1β as well as IL-18 [27,28]. The list of NRLP3 triggers is currently ever expanding, and includes alum, hemozoin and DNA [29-31]. Macrophages deficient for components of the NLRP3 inflammasome were unable to secrete active IL-1β following stimulation with MSU and calcium pyrophosphate dihydrate crystals. Moreover, MSU-induced peritonitis was decreased in ASC-deficient or caspase-1-deficient mice. Colchicine, a drug commonly used in the treatment of acute gout, was found to block IL-1β maturation, probably by influencing crystal endocytosis and/or presentation of crystals to the inflammasome. The results show that MSU crystals initiate an inflammatory cascade, the starting point being the release of active IL-1β from monocytes and macrophages. These findings, however, also raise questions about the steps that connect cellular contact with crystals with inflammasome activation, processes that are still not completely understood. It may involve general mechanisms shared by other inflammasome activators, such as potassium efflux that is regulated by K+ channels like P2X7, or by the sensing of reactive oxygen species that are released during cell stress.
Other cytokines
Besides IL-1β, both IL-6 and TNFα are also upregulated when monocytes are in contact with MSU in vitro and with gouty tissues in vivo [11]. Although there is clear evidence that TNF is released at the site of inflammation in animal models, there has not been any systematic study into the effects of interfering with the TNF signalling pathway in gout. Unpublished data obtained by the authors showed that TNF blockade with an anti-TNFα antibody did not block neutrophil influx in the peritonitis model, arguing against a major role for TNF; but there are anecdotal data of chronic gout that responded to anti-TNF therapy in patients [32-34]. The role of IL-6 is even less clear, although its known proinflammatory properties on the acute phase response and on leucocytes and endothelium would suggest a role in amplifying inflammation.
There is a growing body of data showing that chemokines also play a central role in acute gouty inflammation, particularly in neutrophil recruitment. IL-8 (CXCL8) and closely related chemokines, such as GROα (CXCL1), bind to the receptor CXCR2 to promote neutrophil chemotaxis and can also affect angiogenesis. In this context, acute MSU crystal-induced neutrophilic inflammation (4 hours after crystal injection into a subcutaneous air pouch) was completely dependent on chemokine ligands of CXCR2 in mice, as demonstrated in experiments using CXCR2 knockout mice [35]. A role for IL-8 was also confirmed by neutralizing IL-8 with an antibody in a rabbit model of MSU-induced arthritis, resulting in inhibition of the delayed phase of neutrophil influx at 24 hours [36].
The evidence of a role for these and other cytokines is summarized in Table 1.
Table 1.
Cytokine and chemokines that play a role in gouty inflammation
| Cytokine | Actions contributing to gouty inflammation | Produced by | References |
|---|---|---|---|
| IL-1β | Endothelial adhesion | Monocyte, dendritic cells inflammatory macrophage | [11,12,20,25] |
| Endogenous pyrogen | |||
| TNFα | Cellular activation | Monocyte, macrophage, lining cell | [11,12] |
| Endothelial adhesion | |||
| Phagocytosis | |||
| IL-6 | ? | Monocyte, macrophage | [11,17] |
| Granulocyte colony-stimulating factor | Neutrophil survival and proliferation | Endothelium, macrophage | [11,17] |
| CXCL1 (KC, Groa) | Neutrophil chemotaxis | Macrophage, neutrophil | [11] |
| CXCL8 (IL-8) | Neutrophil chemotaxis | Macrophage, endothelium | [36,37] |
| CCL2 (monocyte chemoattractant protein-1) | Monocyte and dendritic cell chemotaxis | [11] | |
| Mast cell degranulation | |||
| CCL3 (macrophage inflammatory protein-1) | Neutrophil chemotaxis | Monocyte, macrophage | [11] |
Toll-like receptors in gout: are they important?
Toll-like receptors (TLRs) are pivotal sensors of infectious and danger signals on leucocytes, and are an integral part of the innate immune system [38] - and their role in the pathogenesis of inflammatory diseases has become increasingly clear [39]. MSU crystals may be a danger signal released from injured cells that trigger innate immune responses via TLRs, but the experimental evidence available to date provides no clear answer.
Using knockout mice in models of crystal-induced inflammation, macrophages from TLR2 and TLR4 knockout mice showed decreased expression of inflammatory cytokines such as IL-1β, TNFα and transforming growth factor beta-1 on exposure to MSU [40]. Furthermore, CD14 - an adaptor molecule that is shared by TLR2 and TLR4 to promote inflammatory signalling - appears to be an important component of the inflammatory response both in vitro and in vivo. Bone-marrow-derived macrophages from CD14 knockout mice showed reduced secretion of IL-1β in response to MSU, and leucocyte influx was reduced in the air pouch model in these same mice [41]. These experiments also suggest that the phagocytosis phase of the inflammatory response can be dissociated from the responder phase, when proinflammatory cytokines are produced and secreted. The absence of TLR2 or TLR4 reduced phagocytotic uptake as well as inflammation, but deficiency of CD14 reduced the production of IL-1β without any effect of crystal phagocytosis.
The findings in favour of a role for TLRs, however, are put into question by results from another group. Using the peritonitis model, Chen and colleagues found that none of the nine TLR-deficient mouse strains available showed an impairment in neutrophil influx. To exclude the role of TLR5 or TLR8 (mice deficient in these TLRs are not available), transfectants were generated with all the known TLRs and the NF-κB response to MSU was measured. None of the 11 TLRs tested responded to MSU stimulation. Although these findings do not exclude a role for an unidentified co-receptor that may cooperate with TLRs, they strongly suggest that TLRs do not recognize MSU or calcium pyrophosphate dihydrate crystals and that TLR2 and TLR4 may not be crucial for MSU-induced inflammation as previously suggested [19].
Why these two sets of results are so different is still not understood, but one can invoke possibly different types of resident cells in the two models that have differential requirements for activation, or perhaps local modifying factors (such as proteins).
Events influencing the early phase of innate immune response in gouty inflammation include expression of the triggering receptor expressed on myeloid cells-1 (TREM-1). This molecule is a member of a family of surface receptors that act to amplify innate responses in infectious and inflammatory states [42]. TREM-1 expression was by murine resident peritoneal macrophage s, and infiltrating leucocytes were upregulated in the murine air pouch model of crystal-induced inflammation [17]. Interestingly, co-stimulation of resident peritoneal macrophages with MSU crystals and an anti-TREM-1 agonist antibody synergistically increased the production of both IL-1β and monocyte chemoattractant protein-1 compared with stimulation with crystals alone [43]. Little is known to date about the ligands that activate TREM-1, but TREM-1 blockade appears to attenuate inflammation in animal models.
Complement and soluble mediators of inflammation
MSU crystals also promote inflammation by indirect effects on activation of inflammatory cells, as exemplified by MSU crystal-induced activation of the classical pathway of complement in vitro [44]. This classical complement activation process does not require immunoglobulin, but is amplified by both C-reactive protein and IgG [45]. MSU crystals also activate the alternative pathway in vitro [46] and, in this process, direct cleavage of C5 to C5a and C5b is triggered by the formation of a stable C5 convertase on the MSU crystal surface [47].
The formation of leucotrienes and arachidonic acid metabolites induced by MSU has been extensively studied in the past, and is largely accounted for by the effects of MSU crystals on neutrophils and platelets recruited to the inflammatory site [48,49]. Pretreatment of animals with a nonsteroidal anti-inflammatory drug blocked the generation of inflammatory prostaglandins and exudate formation, but did not inhibit local neutrophil accumulation [50].
Unresolved questions
Although our understanding of how MSU crystals trigger inflammation has advanced considerably in the past 20 years, a number of fundamental and clinically relevant questions remain unanswered.
The first and foremost question concerns the exact mechanisms that lead to spontaneous resolution of inflammation in gout. We have already discussed the possible role of transforming growth factor beta secretion by mature macrophages, but is it possible that the inflammatory response of phagocytes to MSU may require multiple signals beyond those that are already mentioned? In in vitro experiments, triggering of the inflammasome to process IL-1β is a multistep process. In the absence of a first signal that induces production of pro-IL-1β, such as phorbol myristate acetate or ultrapure lipopolysaccharide, monocytes/macrophages do not spontaneously secrete mature IL-1β when cultured with MSU crystals (personal observation). Is it possible that such a first signal is also required in man? If this is the case, what may it be in the clinical situation? Could remission be related to the removal of such a first signal?
A second question that has vexed researchers in this field concerns the explanation for why crystals are not always inflammatory. In part, this could be linked to the mechanisms evoked in the paragraph above, but another possible explanation is the surface modification of crystals by proteins and other agents that may modify their phlogistic potential. A body of research from the 1990s showed that protein coating modified crystal-induced inflammation - but how does this work, and how do we integrate this with the more recent data on NLRP3 activation?
Finally, a mechanism to explain why urate-lowering therapies provoke acute flares needs to be found. This is a common clinical problem that may lead patients to abandon urate-lowering therapies. Although the strategy for prevention of gout flares is straightforward, a better understanding of its cause(s) will lead to more effective prevention.
Conclusion
Gout is a paradigm for acute sterile inflammation that is triggered by interactions between MSU microcrystals and the local tissue environment. Our current understanding suggests that resident cells with such tissues, such as resident macrophages or monocytes, react to crystal deposition by uptake of crystals through phagocytosis. Through a series of steps that are still not well understood, these phagocytes activate the NLRP3 inflammasome, resulting in processing and secretion of IL-1β. The subsequent recruitment of inflammatory leucocytes to the site, mediated most probably by endothelial activation, can account for the subsequent release of inflammatory mediators and the recognized inflammatory manifestations of acute gout (Figure 2). Although multiple cytokines and chemokines are involved in the inflammatory process, there is a suggestion that these are produced in a hierarchical manner, secondary to IL-1β release. The understanding of these mechanisms offers us a novel opportunity to interfere with this inflammatory pathway, and may one day develop more effective therapies for gout.
Figure 2.
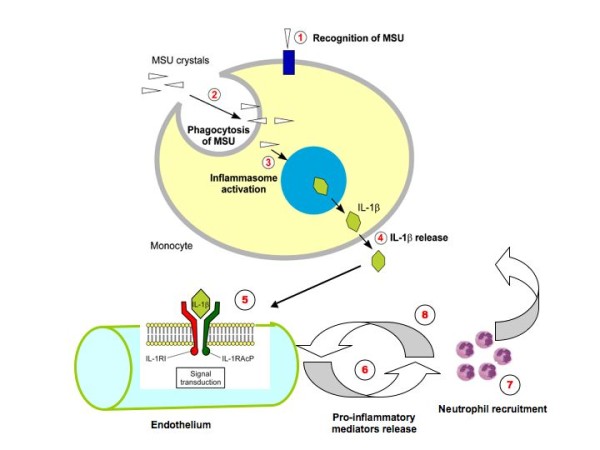
Multiple steps are needed to trigger inflammation in gout. (1) Recognition of monosodium urate (MSU) crystals by components of the innate immune system - Toll-like receptor (TLR) 2/TLR4, triggering receptor expressed on myeloid cells (TREM), and so forth. (2) Uptake of MSU by phagocytotic cells (for example, macrophages and eventually mast cells). (3) Activation of the NALP3 inflammasome by MSU (see details in Figure 1). (4) Release of IL-1β from the cell. (5) Endothelial IL-1β receptor type 1 (IL-1R) activation. (6) Proinflammatory mediators, including IL-8, release a potent chemokine for neutrophil recruitment. (7) Neutrophil recruitment into the site of inflammation. (8) Release of proinflamatory compounds by neutrophils, including more IL-1β.
Abbreviations
ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; IL: interleukin; MSU: monosodium urate; NALP3: NACHT-containing, LRR-containing and PYD-containing protein; NF: nuclear factor; TLR: Toll-like receptor; TNF: tumour necrosis factor; TREM-1: triggering receptor expressed on myeloid cells-1.
Competing interests
AS has received consultancy fees from Novartis and honoraria from Ipsen, Amgen and Novartis.
Note
This review is part of a series on Gout edited by Alex So.
Other articles in this series can be found at http://arthritis-research.com/series/gout
Contributor Information
Nathalie Busso, Email: nathalie.busso@chuv.ch.
Alexander So, Email: alexanderkai-lik.so@chuv.ch.
References
- McCarty DJ, Hollander JL. Identification of urate crystals in gouty synovial fluid. Ann Intern Med. 1961;54:452–460. doi: 10.7326/0003-4819-54-3-452. [DOI] [PubMed] [Google Scholar]
- Kippen I, Klinenberg JR, Weinberger A, Wilcox WR. Factors affecting urate solubility in vitro. Ann Rheum Dis. 1974;33:313–317. doi: 10.1136/ard.33.4.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz WA, Schubert M. The interaction of monosodium urate with connective tissue components. J Clin Invest. 1970;49:1783–1789. doi: 10.1172/JCI106396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Kambe N, Saito M, Nishikomori R, Kim YG, Murakami M, Nunez G, Matsue H. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med. 2009;206:1037–1046. doi: 10.1084/jem.20082179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MH, Kozin F. Mechanisms of cellular interaction with monosodium urate crystals. IgG-dependent and IgG-independent platelet stimulation by urate crystals. Arthritis Rheum. 1978;21:896–903. doi: 10.1002/art.1780210805. [DOI] [PubMed] [Google Scholar]
- Ortiz-Bravo E, Sieck MS, Schumacher HR Jr. Changes in the proteins coating monosodium urate crystals during active and subsiding inflammation. Immunogold studies of synovial fluid from patients with gout and of fluid obtained using the rat subcutaneous air pouch model. Arthritis Rheum. 1993;36:1274–1285. doi: 10.1002/art.1780360912. [DOI] [PubMed] [Google Scholar]
- Landis RC, Yagnik DR, Florey O, Philippidis P, Emons V, Mason JC, Haskard DO. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum. 2002;46:3026–3033. doi: 10.1002/art.10614. [DOI] [PubMed] [Google Scholar]
- Hoffstein S, Weissmann G. Mechanisms of lysosomal enzyme release from leukocytes. IV. Interaction of monosodium urate crystals with dogfish and human leukocytes. Arthritis Rheum. 1975;18:153–165. doi: 10.1002/art.1780180213. [DOI] [PubMed] [Google Scholar]
- Kozin F, Ginsberg MH, Skosey JL. Polymorphonuclear leukocyte responses to monosodium urate crystals: modification by adsorbed serum proteins. J Rheumatol. 1979;6:519–526. [PubMed] [Google Scholar]
- Nuki G. Colchicine: its mechanism of action and efficacy in crystal-induced inflammation. Curr Rheumatol Rep. 2008;10:218–227. doi: 10.1007/s11926-008-0036-3. [DOI] [PubMed] [Google Scholar]
- Torres R, Macdonald L, Croll SD, Reinhardt J, Dore A, Stevens S, Hylton DM, Rudge JS, Liu-Bryan R, Terkeltaub RA, Yancopoulos GD, Murphy AJ. Hyperalgesia, synovitis, and multiple biomarkers of inflammation are suppressed by IL-1 inhibition in a novel animal model of gouty arthritis. Ann Rheum Dis. 2009;68:1602–1608. doi: 10.1136/ard.2009.109355. [DOI] [PubMed] [Google Scholar]
- Di Giovine FS, Malawista SE, Nuki G, Duff GW. Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1. J Immunol. 1987;138:3213–3218. [PubMed] [Google Scholar]
- di Giovine FS, Malawista SE, Thornton E, Duff GW. Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution. J Clin Invest. 1991;87:1375–1381. doi: 10.1172/JCI115142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2004;50:2273–2280. doi: 10.1002/art.20317. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60:281–289. doi: 10.1002/art.24185. [DOI] [PubMed] [Google Scholar]
- Schiltz C, Liote F, Prudhommeaux F, Meunier A, Champy R, Callebert J, Bardin T. Monosodium urate monohydrate crystal-induced inflammation in vivo: quantitative histomorphometric analysis of cellular events. Arthritis Rheum. 2002;46:1643–1650. doi: 10.1002/art.10326. [DOI] [PubMed] [Google Scholar]
- Pessler F, Mayer CT, Jung SM, Behrens EM, Dai L, Menetski JP, Schumacher HR. Identification of novel monosodium urate crystal regulated mRNAs by transcript profiling of dissected murine air pouch membranes. Arthritis Res Ther. 2008;10:R64. doi: 10.1186/ar2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getting SJ, Flower RJ, Parente L, de Medicis R, Lussier A, Woliztky BA, Martins MA, Perretti M. Molecular determinants of monosodium urate crystal-induced murine peritonitis: a role for endogenous mast cells and a distinct requirement for endothelial-derived selectins. J Pharmacol Exp Ther. 1997;283:123–130. [PubMed] [Google Scholar]
- Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, De Meulemeester M, Pikhlak A, Yücel E, Richard D, Murphy V, Arulmani U, Sallstig P, Schlesinger N. ACR Annual Meeting 2009. Philadelphia: Wiley; 2009. Canakinumab (ACZ885) vs. triamcinolone acetonide for treatment of acute flares and prevention of recurrent flares in gouty arthritis patients refractory to or contraindicated to NSAIDs and/or colchicine [abstract LB4] [Google Scholar]
- Schumacher HR, Sundy J, Terkeltaub R, Knapp HR, Mellis S, Soo Y, King-Davis S, Weinstein S, Radin A. ACR Annual Meeting 2009. Philadelphia: Wiley; 2009. Placebo-controlled study of rilonacept for gout flare prophylaxis during intiation of urate-lowering therapy [abstract 1096] [Google Scholar]
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60:3642–3650. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, Pham CT, Meer JW van der, Dinarello CA, Berg WB van den. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1β. Arthritis Rheum. 2009;60:3651–3662. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O'Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- Santos Moreno P, Latore C. Treatment of severe gouty arthritis with etanercept [abstract] Ann Rheum Dis. 2009;68(Suppl 3):680. [Google Scholar]
- Tausche AK, Richter K, Grassler A, Hansel S, Roch B, Schroder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis. 2004;63:1351–1352. doi: 10.1136/ard.2003.015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn C, Zeier M. Successful treatment of chronic tophaceous gout with infliximab (Remicade) Rheumatol Int. 2006;26:274–276. doi: 10.1007/s00296-005-0617-7. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W. The murine homolog of the interleukin-8 receptor CXCR-2 is essential for the occurrence of neutrophilic inflammation in the air pouch model of acute urate crystal-induced gouty synovitis. Arthritis Rheum. 1998;41:900–909. doi: 10.1002/1529-0131(199805)41:5<900::AID-ART18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Nishimura A, Akahoshi T, Takahashi M, Takagishi K, Itoman M, Kondo H, Takahashi Y, Yokoi K, Mukaida N, Matsushima K. Attenuation of monosodium urate crystal-induced arthritis in rabbits by a neutralizing antibody against interleukin-8. J Leukoc Biol. 1997;62:444–449. doi: 10.1002/jlb.62.4.444. [DOI] [PubMed] [Google Scholar]
- Hachicha M, Naccache PH, McColl SR. Inflammatory microcrystals differentially regulate the secretion of macrophage inflammatory protein 1 and interleukin 8 by human neutrophils: a possible mechanism of neutrophil recruitment to sites of inflammation in synovitis. J Exp Med. 1995;182:2019–2025. doi: 10.1084/jem.182.6.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev. 2009;227:248–263. doi: 10.1111/j.1600-065X.2008.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52:2936–2946. doi: 10.1002/art.21238. [DOI] [PubMed] [Google Scholar]
- Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006;177:6370–6378. doi: 10.4049/jimmunol.177.9.6370. [DOI] [PubMed] [Google Scholar]
- Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Akahoshi T, Hayashi I, Endo H, Kawai S, Inoue M, Kondo H, Kitasato H. Induction of triggering receptor expressed on myeloid cells 1 in murine resident peritoneal macrophages by monosodium urate monohydrate crystals. Arthritis Rheum. 2006;54:455–462. doi: 10.1002/art.21633. [DOI] [PubMed] [Google Scholar]
- Giclas PC, Ginsberg MH, Cooper NR. Immunoglobulin G independent activation of the classical complement pathway by monosodium urate crystals. J Clin Invest. 1979;63:759–764. doi: 10.1172/JCI109360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell IJ, Papaioannou C, McDuffie FC, MacIntyre S, Kushner I. Effect of IgG and C-reactive protein on complement depletion by monosodium urate crystals. J Rheumatol. 1983;10:425–433. [PubMed] [Google Scholar]
- Fields TR, Abramson SB, Weissmann G, Kaplan AP, Ghebrehiwet B. Activation of the alternative pathway of complement by monosodium urate crystals. Clin Immunol Immunopathol. 1983;26:249–257. doi: 10.1016/0090-1229(83)90143-5. [DOI] [PubMed] [Google Scholar]
- Russell IJ, Mansen C, Kolb LM, Kolb WP. Activation of the fifth component of human complement (C5) induced by monosodium urate crystals: C5 convertase assembly on the crystal surface. Clin Immunol Immunopathol. 1982;24:239–250. doi: 10.1016/0090-1229(82)90235-5. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Lundberg U, Weissmann G, Samuelsson B. Formation of leukotrienes and hydroxy acids by human neutrophils and platelets exposed to monosodium urate. Prostaglandins. 1984;27:563–581. doi: 10.1016/0090-6980(84)90092-3. [DOI] [PubMed] [Google Scholar]
- Rae SA, Davidson EM, Smith MJ. Leukotriene B4, an inflammatory mediator in gout. Lancet. 1982;2:1122–1124. doi: 10.1016/S0140-6736(82)92785-4. [DOI] [PubMed] [Google Scholar]
- Forrest MJ, Zammit V, Brooks PM. Inhibition of leucotriene B4 synthesis by BW 755c does not reduce polymorphonuclear leucocyte (PMNL) accumulation induced by monosodium urate crystals. Ann Rheum Dis. 1988;47:241–246. doi: 10.1136/ard.47.3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]