

Abstract
THF solutions of a new iron(I) source, “[PhBPCH2Cy3]Fe” ([PhBPCH2Cy3] = [PhBP(CH2P(CH2Cy)2)3]−), effect the reductive cleavage of CO2 via O-atom transfer at ambient temperature. The dominant reaction pathway is bimetallic and leads to the formation of a structurally unprecedented diiron FeII(μ-O)(μ-CO)FeII core. X-ray data are also available to suggest that bimetallic reductive CO2 coupling to generate oxalate occurs as a minor reaction pathway. These initial observations forecast a diverse reaction landscape between CO2 and iron (I) synthons.
Direct O-atom transfer from CO2 is a difficult transformation to realize given the molecule’s thermodynamic and kinetic stability. Highly reducing early transition, lanthanide, and actinide metal complexes are known that facilitate reductive C-O cleavage of CO2.1 Later first row ions, while active for CO2 binding, do not typically display similar cleavage transformations.2,3 Nature, however, is presumed to exploit low-valent, later first row metal ions (e.g., Ni, Fe) to mediate CO2 reduction/CO oxidation in the C cluster of CODH enzymes.4
We describe herein an unusual iron(I) system that reacts readily with CO2 at ambient temperature to mediate its reductive cleavage. The dominant cleavage product is a structurally unprecedented bimetallic μ-carbonyl/μ-oxo core (i.e., Fe(μ-CO)(μ-O)Fe)). Structural evidence is also available for minor oxalate side products of the type Fe(μ-η2: η2-oxalato)Fe. This iron system is therefore able to mediate both the reductive cleavage 5 and coupling of CO2.5
Entry into the CO2 chemistry of present interest was realized using a new tris(phosphino)borate ligand, [PhB(CH2P(CH2Cy)2)3]− (abbreviated as [PhBPCH2Cy3]), featuring cyclohexylmethyl substituents at phosphorous. The yellow iron precursor [PhBPCH2Cy3]FeCl (1) was obtained in good yield from Tl[PhBPCH2Cy3] and FeCl2. XRD, combustion analysis, and a solution magnetic moment determination establish that 1 is a monomeric, pseudotetrahedral S = 2 species. When compound 1 is chemically reduced by Na/Hg in THF under N2, an intense lime-green solution is formed. This observation contrasts that of the Na/Hg reduction of its cousin [PhBPiPr3]FeCl under an N2 atmosphere, which gives rise to the red-brown dinitrogen-bridged dimer {[PhBPiPr3]Fe}2(μ-N2).6 We have no evidence for N2 uptake upon Na/Hg reduction of the [PhBPCH2Cy3]FeCl system under an N2 atmosphere. Combustion analysis data for the isolated reduction product confirms its empirical formula as “[PhBPCH2Cy3]Fe”, (2), and rules out the presence of nitrogen.
A sample of 2 in THF-d8 exhibits complicated solution NMR spectra indicative of both paramagnetic and diamagnetic components that are likely undergoing rapid exchange. For example, its 31P NMR spectrum features a single broad resonance that shifts from -29 ppm to 4 ppm when the temperature is varied from 60 to −60 °C. A 1H NMR spectrum of the sample contains broad, temperature dependent resonances ranging from −7 to 72 ppm, and sharp resonances in the diamagnetic region of the spectral window. Also, an axial EPR signal indicative of an S = ½ iron center is observed in a THF glass of 2 at 4 K. Scheme 1 shows two possible isomeric structures that would be consistent with these spectral data and the empirical formula “[PhBPCH2Cy3]Fe”. They include an Fe(III) alkyl-hydride wherein one of the cyclohexyl C-H bonds of the ligand is cyclometalated (A, S = 1/2), and an antiferromagnetically coupled dimer of such a structure with the hydride ligands in bridging positions (B, S = 0). Direct evidence for the presence of a metal hydride includes an IR stretch at 2058 cm−1 (KBr pellet) and the formation of CHCl3 (~ 40%, detected by 1H and 13C NMR) upon the addition of one equivalent of CCl4 in THF- d8.7
Scheme I.
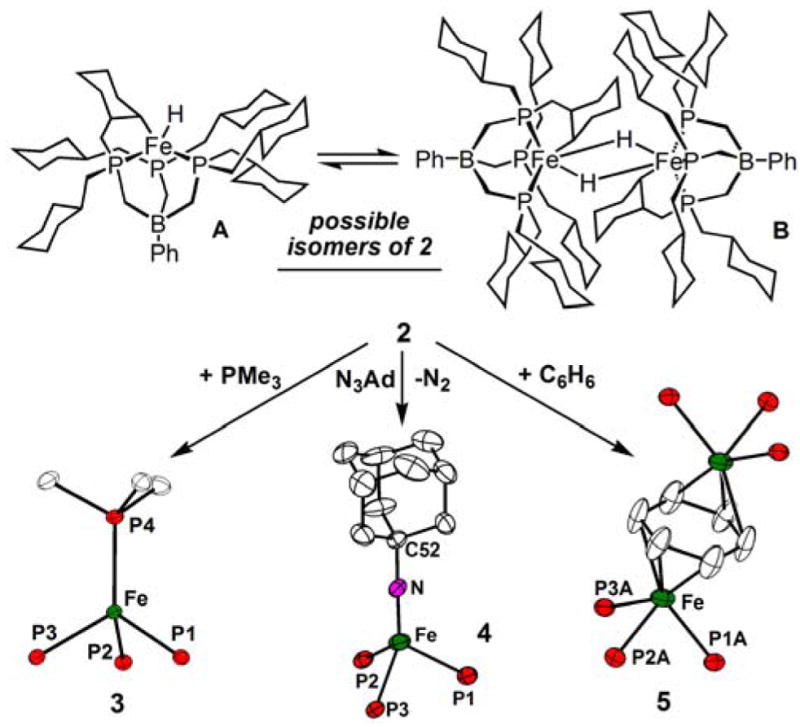
Regardless of its exact structure/s in THF solution, 2 behaves chemically as a very clean “[PhBPCH2Cy3]Fe(I)” source. For instance, the addition of PMe3 to a THF solution of 2 generates the d7 S = 3/2 complex [PhBPCH2Cy3]Fe(PMe3) (3). Also, the addition of one equivalent of 1-adamantyl azide to 2 triggers oxidative nitrene transfer to provide the S = ½ Fe(III) imide [PhBPCH2Cy3]Fe≡NAd (4).8 Both 3 and 4 are formed ca. quantitatively and have been structurally characterized (Scheme I). Additionally, reconstitution of a THF solution of 2 into benzene provides, ca. quantitatively, the dinuclear benzene complex {[PhBPCH2Cy3]Fe}2(μ-η3:η3-C6H6) 5. Given these observations it seem likely that the iron center in 2 is reversibly solvated by THF to produce an S = ½ iron(I) species such as [PhBPCH2Cy3]Fe(THF)2 in THF. Such a species could in fact account for the S = ½ signature of 2 in THF at 4 K, rather than the cyclometalated hydride A shown in Scheme I.
We next explored the reactivity of THF solutions of 2 with CO2 and found that an immediate though subtle color change occurs from lime-green to pine-green upon CO2 exposure. Inspection of the reaction solution in situ by 1H NMR spectroscopy indicates one major diamagnetic product (ca. 75% using 5 equiv of CO2, 5 runs). This product can be crystallized in analytically pure form (65% isolated yield) and exhibits a single peak in the 31P NMR spectrum at 51.9 ppm and an intense ν(CO) IR stretch at 1730 cm−1 (KBr, C6D6; 1734 cm−1, KBr pellet). This IR stretch represents a μ-CO ligand. Using 13C-labeled CO2, a 13C-NMR resonance for the μ-CO ligand at 289.8 ppm has been established. The ν(μ-CO) vibrations shifts to 1692 cm−1 (calcd: 1691 cm−1) upon isotopic substitution. Blue-green plate-like crystals of 6 can be grown from benzene/petroleum ether and have been examined by X-ray crystallography. As shown in Scheme II, the major product is {[PhBPCH2Cy3]Fe}2(μ-CO,μ-O), (6), indicating a net 2-electron reductive cleavage of CO2 to CO and O2−. The connectivity of 6 is very well-established, but of the various sets of crystals that have been examined by XRD each has suffered from problematic disorder, in part due to the floppy methylcyclohexyl substituents.9 An isotropic structure of 6 is therefore depicted in Scheme II. Its most striking structural feature pertains to its diiron μ-carbonyl/μ-oxo core. To our knowledge a bimetallic μ-oxo/μ-CO structure type had yet to be reported.10 Complex 6 features a very short Fe-Fe distance (2.35 Å). CV data in THF show a reversible 1-electron couple at − 0.2 V (vs Ag/AgNO3).
Scheme II.
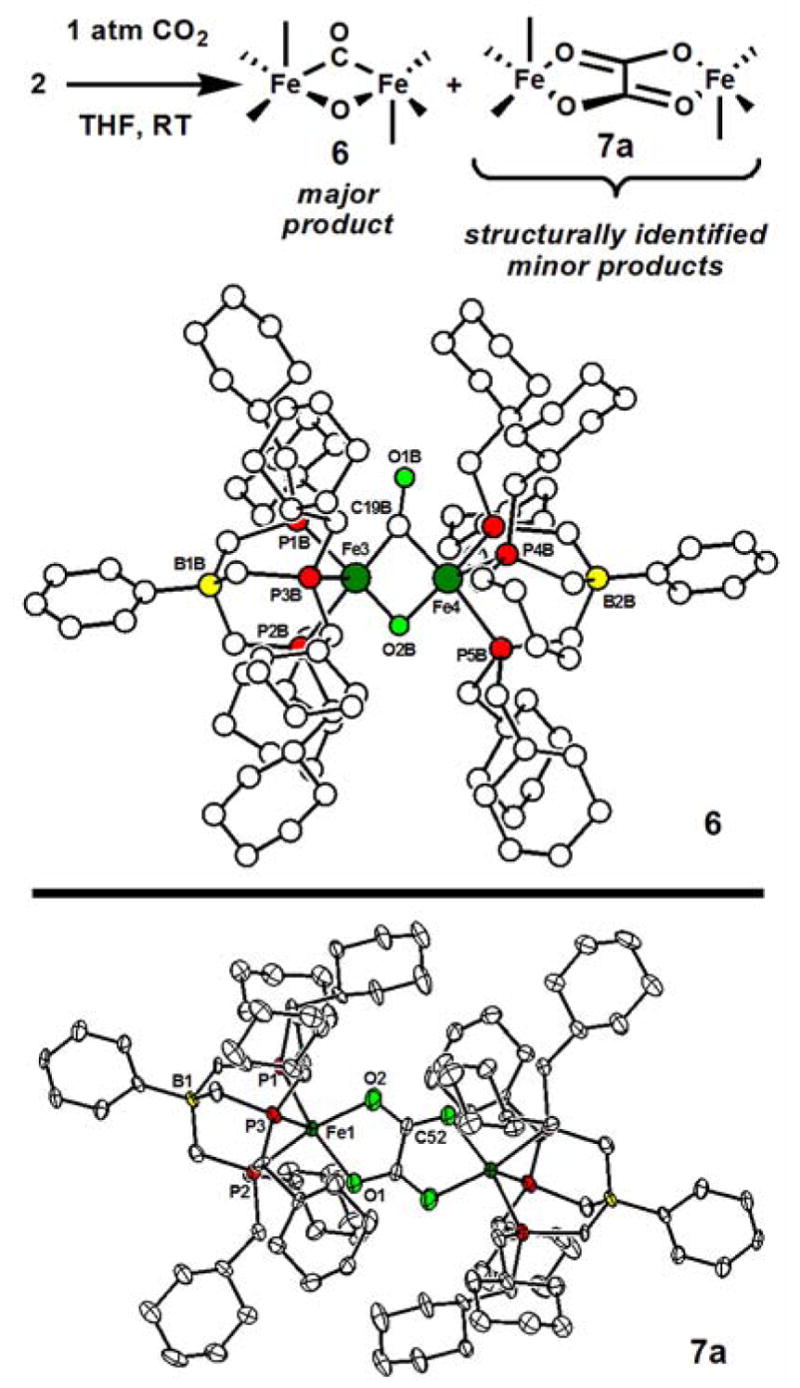
Varying the conditions of the CO2 reaction with 2 invariably leads to the same major product 6. This is true whether 0.5 equiv of CO2 is employed or a CO2 pressure of 10 atm. Moreover, 6 is the major product whether the reaction is carried-out at −41 ºC (complete in ca. 12 h), or at 22 ºC (complete in ca. 15 min.). The iron(I) phosphine adduct 3 also produces 6 as its major product upon exposure to CO2, albeit much more slowly.
During the course of these studies we have consistently observed a substantial secondary paramagnetic product by NMR spectroscopy (ca. 15–25% depending on exact conditions). By fractional crystallization of crude product mixtures we have been able to pick-out red-brown crystals for XRD analysis of the primary side product to establish its identity as {[PhBPCH2Cy3]Fe}2(μ-η2:η2-oxalato) 7a (See Scheme II). The IR spectrum of 7a shows a broad and intense vibration centered at 1647 cm−1 (KBr pellet) that shifts to 1598 cm−1 upon isotopic labeling with 13C-CO2. In one case, pale pink crystals also formed that were subjected to XRD analysis. For the crystals, the presence of a μ-oxalato ligand was also established, but in this case terminal CO ligands were also present ({[PhBPCH2Cy3]Fe(CO)}2(μ-η2:η2-oxalato); 7b). The structure of 7b is provided in the SI. Studies are now underway to try to control the selectivity of the CO2 reaction profile so as to favor the CO2 coupling product/s for further studies.
In summary, THF solutions of 2 provide an effective Fe(I) source for substrate binding and group transfer reactions. Such solutions effect the reductive cleavage of CO2 via O-atom transfer to provide a structurally unique Fe(μ-O)( μ-CO)Fe core. XRD studies reveal a reductive CO2 coupling process that is also kinetically competent to generate oxalate. These initial observations establish that Fe(I) participates in rich CO2 reaction chemistry motivate continued studies in this context.
Supplementary Material
Detailed experimental procedures and characterization data for [PhB(CH2P(CH2Cy)2)3]Tl, ligand precursors, and compounds 1–6. Crystallographic details for 1, 3–6, 7a, 7b are provided in CIF format. This material is available free of charge via the Internet at (http://pubs.acs.org)
Acknowledgments
We thank Larry M. Henling and Neal Mankad for crystallographic assistance. CTS is supported by an NSF graduate fellowship. We are grateful to the NIH (GM 070757) and BP (MC2 program) for financial support.
References
- 1.For representative stoichiometric reductive CO2 cleavage reactions see: Bryan JC, Geib SJ, Rheingold AL, Mayer JM. J Am Chem Soc. 1987;109:2826.Castro-Rodriguez I, Meyer K. J Am Chem Soc. 2005;127:11242. doi: 10.1021/ja053497r.Fachinetti G, Floriani C, Chiesi-Villa A, Guastini C. J Am Chem Soc. 1979;101:1767.Procopio LJ, Carroll PJ, Berry DH. Organometallics. 1993;12:3087.
- 2.Electrocatalytic CO2 reduction can be mediated by later first row metals: Simón-Manso E, Kubiak CP. Organometallics. 2005;24:96.Hammouche M, Lexa D, Momenteau M, Savéant JM. J Am Chem Soc. 1991;113:8455.Beley M, Collin JP, Ruppert R, Sauvage JP. J Am Chem Soc. 1986;108:7461. doi: 10.1021/ja00284a003.Fisher B, Eisenberg R. J Am Chem Soc. 1980;102:7361.Dubois DL, Miedaner A, Haltiwanger RC. J Am Chem Soc. 1991;113:8753–8764.
- 3.For Cu-catalyzed CO2 reduction using diborane reductants see: Laitar DS, Muller P, Sadighi JP. J Am Chem Soc. 2005;127:17196. doi: 10.1021/ja0566679.
- 4.(a) Ragsdale SW, Kumar M. Chem Rev. 1996;96:2515. doi: 10.1021/cr950058+. [DOI] [PubMed] [Google Scholar]; (b) Evans DJ. Coord Chem Rev. 2005;249:1582. [Google Scholar]
- 5.Well-defined reductive coupling reactions of CO2 to generate oxalate are extremely uncommon. See, for example: Evans WJ, Seibel CA, Ziller JW. Inorg Chem. 1998;37:770.
- 6.(a) Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (b) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Butts MD, Bergman RG. Organometallics. 1994;13:2668. [Google Scholar]; b Nolan SP, Hoff CD, Stoutland PO, Newman LJ, Buchanan JM, Bergman RG, Yang GK, Peters KG. J Am Chem Soc. 1987;109:3143. [Google Scholar]
- 8.For related Fe(III) imides: Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322. doi: 10.1021/ja028448i.Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913. doi: 10.1021/ja0453073.Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956. doi: 10.1021/ja0604358. Also see 6a.
- 9.Triclinic, monoclinic and tetragonal crystals of 6 were examined; each crystal form was problematic. The triclinic crystals diffracted poorly and contained large areas of highly disordered solvent (see Supporting Information). In the monoclinic crystals, 6 was very disordered in addition the disordered solvent molecules. The tetragonal crystal form yielded no atomic information.
- 10.There are a few examples of dimetal μ-sulfide/μ-CO core structures. See, for example: Kubiak CP, Woodcock C, Eisenberg R. Inorg Chem. 1980;19:2733.Balch AL, Catalano VJ, Olmstead MM. Inorg Chem. 1990;29:1638.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures and characterization data for [PhB(CH2P(CH2Cy)2)3]Tl, ligand precursors, and compounds 1–6. Crystallographic details for 1, 3–6, 7a, 7b are provided in CIF format. This material is available free of charge via the Internet at (http://pubs.acs.org)