

Abstract
The melanocortin-4 receptor (MC4R) is a G-protein coupled receptor (GPCR) that is expressed in the central nervous system and has a role in regulating feeding behavior, obesity, energy homeostasis, male erectile response, and blood pressure. Since the report of the MC4R knockout mouse in 1997, the field has been searching for links between this genetic bio marker and human obesity and type 2 diabetes. More then 80 single nucleotide polymorphisms (SNPs) have been identified from human patients, both obese and non-obese controls. Many significant studies have been performed examining the pharmacological characteristics of these hMC4R SNPs in attempts to identify a molecular defects/insights that might link a genetic factor to the obese phenotype observed in patients possessing these mutations. Our laboratory has previously reported the pharmacological characterization of 40 of these polymorphic hMC4 receptors with multiple endogenous and synthetic ligands. The goal of the current study is to perform a similar comprehensive side-by-side characterization of 30 additional human hMC4R with single nucleotide polymorphisms using multiple endogenous agonists [α-, β, γ2-melanocyte stimulating hormones (MSH) and adrenocorticotropin (ACTH)], the antagonist agouti-related protein hAGRP(87-132), and synthetic agonists [NDP-MSH, MTII, and the tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9)]. These in vitro data, in some cases, provide a putative molecular link between dysfunctional hMC4R's and human obesity. These 30 hMC4R SNPs include R7H, R18H, R18L, S36Y, P48S, V50M, F51L, E61K, I69T, D90N, S94R, G98R, I121T, A154D, Y157S, W174C, G181D, F202L, A219V, I226T, G231S, G238D, N240S, C271R, S295P, P299L, E308K, I317V, L325F and 750DelGA. All but the N240S hMC4R were identified in obese patients. Additionally, we have characterized a double I102T/V103I hMC4R. In addition to the pharmacological characterization, the hMC4R variants were evaluated for cell surface expression by flow cytometry. The F51L, I69T, and A219V hMC4Rs possessed full agonist activity and significantly decreased endogenous agonist ligand potency. At the E61K, D90N, Y157S, and C271R hMC4Rs, all agonist ligands examined were only partially efficacious in generating a maximal signaling response (partial agonists) and possessed significantly decreased endogenous agonist ligand potency. Only the A219V, G238D, and S295P hMC4Rs possessed significantly decreased AGRP(87-132) antagonist potency. These data provide new information for use in GPCR computational development as well as insights into MC4R structure ad function.
The melanocortin system is involved in the regulation of a number of diverse physiological pathways including pigmentation (1), sexual function (2, 3), blood pressure (4, 5), and energy homeostasis (6, 7). The melanocortin system is comprised of five G-protein coupled receptors (GPCRs) (8-14), that stimulate the adenylate cyclase signal transduction pathway. The endogenous ligands are derived by post-translational processing of the proopiomelanocortin (POMC) protein by prohormone convertases PC1 and PC2 (15, 16). POMC is processed in the human brain to generate the endogenous melanocortin agonist peptides α-, β, γ2-melanocyte stimulating hormones (MSH) and adrenocorticotropin (ACTH) (17). The melanocortin pathway also has the only two known endogenous antagonists of GPCRs, agouti (18, 19) and agouti-related protein (AGRP) (20). AGRP is also expressed in the hypothalamus of the brain and projects to other regions of the brain that express the melanocortin-3 and -4 receptors (21, 22).
This study presented herein performs, for the first time for these 30 human MC4R polymorphisms, a side-by-side in vitro pharmacological comparison using multiple endogenous agonists, the antagonist hAGRP(87-132), as well as the synthetic agonists JRH887-9 (Ac-His-DPhe-Arg-Trp-NH2) (23-28), NDP-MSH (29), and MTII (30). We have previously reported characterization of 40 human MC4R polymorphisms (31) and extend the pharmacological profiling to include those presented herein. Since there are multiple putative endogenous melanocortin agonists (α-MSH, β-MSH, γ-MSH, and ACTH), it is hypothesized and supported by our previous studies (31), that while one endogenous ligand might result in modified potency at a hMC4 polymorphic receptor, other endogenous agonists or the antagonist AGRP might possess normal potencies. We have previously reported that the chimeric AGRP-melanocortin agonist AMW3-130 can restore a nM to sub nM ligand functional response of polymorphic hMC4Rs that did not respond potently to the endogenous agonist peptides (32). In this study, selected hMC4 receptors that possessed reduced endogenous agonist potencies and/or efficacies (F51L, E61K, I69T, D90N, Y157S, A219V, and C271R) were also examined with this agonist as well as the modified tetrapeptides JRH420-12 (24), JRH322-18 (25), the Ac-Mini-(His-DPhe-Arg-Trp)AGRP-NH2 AMW3-106 (33) and the small molecules THIQ (34) and JB25 (35) (Figure 1) to determine if any of these ligands could functionally rescue these receptors that did no respond normally to the endogenous ligands.
Figure 1.

Small molecule and amino acid sequences of the endogenous and synthetic melanocortin ligands examined in this study.
Materials and Methods
Peptides used in this study that were purchased from commercial sources include, α-MSH, NDP-MSH, MTII, ACTH(1-24), β-MSH, γ2-MSH (Bachem), and hAGRP(87-132) (Peptides International). The peptides that were synthesized as previously reported (32) include Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9) (24, 25), Ac-Anc-DPhe-Arg-Trp-NH2 (amino-2-naphthylcarboxylic acid, Anc, JRH420-12) (24), Ac-His-(pI)DPhe-Arg-Trp-NH2 (JRH322-18) (25), AMW3-130 (32), and AMW3-106 (33). The small molecule JB25 was obtained through a material transfer agreement with Professor Morton Meldal (Carlsberg Laboratory, Department of Chemistry, Gamle Carlsberg Vej 10, DK-2500 Valby, Denmark) (35). The THIQ small molecule was obtained through a material transfer agreement with Dr. Lex Van Der Ploeg at Merck Research Laboratories (34).
hMC4R In Vitro Receptor Mutagenesis
The human wild type N-terminally Flag tagged MC4R cDNA was generously provided by Dr. Robert Mackenzie (36), and was subcloned into the pBluescript plasmid (Stratagene) for subsequent mutagenesis. Mutant hMC4Rs were generated using a polymerase chain reaction (PCR) based mutagenesis strategy, as previously described by our laboratory (31, 32, 37). A complementary set of PCR primers were designed containing the reported nucleotide base pair changes resulting in the modified amino acid. After completion of the PCR reaction (95 °C 30 s, 12 cycles of 95 °C 30 s, 55 °C 1 min, 68 °C 9 min) the product was purified (Qiaquick PCR reaction, Qiagen) and eluted in water. Subsequently, the sample was cut with Dpn1 (Biolabs) to lineralize the wild type template DNA leaving only nicked circularized mutant DNA. This was transformed into competent DH5α e-coli. Single colonies were selected and the presence of the desired mutant was checked by DNA sequencing. The DNA containing the mutant was then excised and subcloned into the HindIII/XbaI restriction sites of the pCDNA3 expression vector (Invitrogen). Complete mutant hMC4R sequences were confirmed free of PCR nucleotide base errors by DNA sequencing (University of Florida sequencing core facilities). During sequencing, it was observed that the I102T plasmid also contained the V103I mutation. Due to the large frequency of occurrences of the V103I SNP, and unanticipated pharmacological profile, we decided to include its data.
Generation of Stable Cell Lines
HEK-293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS) and seeded 1 day prior to transfection at (1-2) × 106 cell/100-mm dish. Mutant and wild type DNA in the pCDNA3 expression vector (20 μg) were transfected using the calcium phosphate method (38). Stable receptor populations were generated using G418 selection (0.7-1 mg/mL) for subsequent bioassay analysis.
cAMP Based Functional Bioassay
HEK-293 cells stably expressing the mutant and wild type melanocortin receptors were transiently transfected with 4 μg CRE/β-galactosidase reporter gene as previously described (31, 32, 37, 39). Briefly, 5,000 to 15,000 post transfection cells were plated into collagen treated 96 well plates (Nunc) and incubated overnight. Forty-eight hours post-transfection the cells were stimulated with 100 μL peptide (10-5 - 10-12 M) or forskolin (10-4 M) control in assay medium (DMEM containing 0.1 mg/mL BSA and 0.1 mM isobutylmethylxanthine) for 6 hrs. The assay media was aspirated and 50 μL of lysis buffer (250 mM Tris-HCl pH=8.0 and 0.1% Triton X-100) was added. The plates were stored at −80°C overnight. The plates containing the cell lysates were thawed the following day. Aliquots of 10 μL were taken from each well and transferred to another 96-well plate for relative protein determination. To the cell lysate plates, 40 μL phosphate-buffered saline with 0.5% BSA was added to each well. Subsequently, 150 μL substrate buffer (60 mM sodium phosphate, 1 mM MgCl2, 10 mM KCl, 5 mM β-mercaptoethanol, 2 mg/mL ONPG) was added to each well and the plates were incubated at 37°C. The sample absorbance, OD405, was measured using a 96 well plate reader (Molecular Devices). The relative protein was determined by adding 200 μL 1:5 dilution Bio Rad G250 protein dye:water to the 10 μL cell lysate sample taken previously, and the OD595 was measured on a 96 well plate reader (Molecular Devices). Data points were normalized to the relative protein content. Ligand efficacy (ability to generate a maximum agonist response) and ligands that are reported as partial agonists were determined relative to the response of the same ligands at the wild type hMC4 control receptor. Fold changes in ligand potency [the effective concentration at 50% maximum response (EC50) for full agonists or for partial agonists, 50% of the maximal observed stimulation] are determined relative to the same ligand potency at the wild type hMC4 control receptor. The antagonistic properties of hAGRP(87-132) were evaluated by the ability of hAGRP(87-132) to competitively displace the MTII agonist (Bachem) in a dose-dependent manner, at up to 10 μM concentrations (37). The pA2 values were generated using the Schild analysis method (40).
Data Analysis
EC50 and pA2 values represent the mean of three or more independent experiments. EC50 and pA2 estimates, and their associated standard errors of the mean, were determined by fitting the data to a nonlinear least-squares analysis using the PRISM program (v4.0, GraphPad Inc.). Statistical analysis was performed using a student T-test compared to the wild type receptor values with statistical significance defined as *p<0.05.
Competitive Displacement Binding Assays
NDP-MSH Iodination
125I-NDP-MSH was prepared using a modified chloramine-T method as previously described by Yang, et al (41). Using 50 mM sodium phosphate buffer pH 7.4 as the reaction buffer, 125I-Na (0.5 mCi, Amersham Life Sciences, Inc., Arlington Heights, IL) was added to 20 μg of NDP-MSH (Bachem, Torrance, CA) in 5 μL buffer. To initiate the reaction, 10 μL of a 2.4 mg/mL solution of chloramine T (Sigma Chemical Co., St. Louis, MO) was added for 15 seconds with gentle agitation. This reaction was terminated by the addition of 50 μL of a 4.8 mg/mL solution of sodium metabisulfite (Sigma Chemical Co.) for 20 seconds with gentle agitation. The reaction mixture was then diluted with 200 μL 10% bovine serum albumin and the resultant mixture layered on a Bio-Gel P2 (Bio-Rad Labs, Hercules, CA) column (1.0 × 30 cm Econocolumn, Bio-Rad Labs) for separation by size exclusion chromatography using 50 mM sodium phosphate buffer, pH 7.4 as column eluant. Fifteen drop fractions (approximately 500 μL) were collected into glass tubes containing 500 μL of 1% BSA. Each fraction was then counted on the Apex Automatic Gamma Counter (ICN Micromedic Systems Model 28023, Huntsville, AL with RIA AID software, Robert Maciel Associates, Inc., Arlington, MA) to determine peak 125I incorporation fractions.
Receptor Competitive Displacement Binding Studies
HEK-293 cells stably expressing the mutant and wild type MC4 receptors were maintained as described above. Preceding the experiment, 0.3 to 0.5 × 106 cells/well were plated into Primera 12 well plates (Falcon) and grown to confluence. For the assays, the media was removed and the cells were washed twice with a freshly prepared assay buffer (DMEM and 0.1% BSA). A 500μL sample of the NDP-MSH peptide concentration being tested and 150,000 cpms of 125I-radiolabeled NDP-MSH were added to the well and incubated at 37°C for 1 hr. The medium was subsequently removed, and each well was washed twice with assay buffer. The cells were lysed by the addition 0.5mL 0.1M NaOH and 0.5mL 1%Triton X-100. The mixture was left to lyse the cells for 10 min and the contents of each well were transferred to a labeled 16×150-mm glass tube and quantified on a Apex Automatic Gamma Counter. Dose-response curves (10-6 to 10-12 M) of NDP-MSH and IC50 values were generated and analyzed by a one-site competition nonlinear least squares analysis (42) and the PRISM program (v4.0, GraphPad Inc.). The percent total specific binding was determined based upon the non-specific values obtained using 10-6M NDP-MSH. Each experiment was performed using duplicate data points and repeated in at two independent experiments. The standard deviations were derived from the IC50 values from at least two independent experiments using the PRISM program (v4.0, GraphPad Inc.). Statistical analysis was performed using a student T-test compared to the wild type receptor values with statistical significance defined as *p<0.05.
Immunohistochemical Analysis of Wild type FLAG-Tagged hMC4R
Flow cytometric analysis (FACS) of intracellular FLAG-tagged wild type hMC4R was performed as described previously (31, 43). Briefly, cells were dissociated from monolayer culture dishes using cold Cell Dissociation buffer (Cellgro, Mediatech), centrifuged at 600×g for 5min, room temperature, and the pelleted cells were resuspended in sterile-filtered FACS buffer (1% BSA, 0.1% Na azide, in 1xPBS pH 7.2; Sigma Chemical, St Louis MO). The cells were distributed to multiple FACS tubes (Falcon, Fisher Scientific) at one million cells per tube. The cells were treated with 10mg/mL unconjugated mouse IgG (Upstate Biotech or Sigma) to block nonspecific antibody binding. To determine cell surface receptor protein expression, the cells were then incubated for 45 min at room temperature with anti-FLAG-PE (Prozyme, San Leandro, CA). To determine the total cellular receptor protein expression, the cells were fixed with 2% methanol free formaldehyde in 1xPBS (Ted Pella or EM Scientific, Fisher Scientific) for 10min, permeabilized for 20 min with Saponin Buffer [0.5% saponin (Sigma) in FACS buffer, pH 7.2], and subsequently washed with Saponin Buffer. After centrifugation (600×g, 5min), cell aliquots were conjugated with anti-FLAG-APC antibodies (Prozyme) for 1hr at room temperature to label the total (intracellular and surface) FLAG-tagged molecules. After the anti-FLAG antibody incubation, the labeled cells were washed 1mL of Saponin buffer 3 times prior to resuspension in FACS buffer for analysis. The PE- and APC-conjugated nonspecific antibodies (BD Biosciences-Pharmingen, CalTag, Burlingame, CA) served as isotype controls for the anti-FLAG antibody conjugates used in these analyses and were used to set the background for fluorescence staining detection on BD Biosciences FACS Calibur flow cytometers. Data was collected as both stained cell percentages (either surface or total detected) and as mean fluorescence per cell from a minimum of 10,000 collected events for each sample run. Receptor cell surface expression and total cellular expression (using permeabilized cells) were determined as summarized in Figure 2.
Figure 2.

Fluorescence cell activated sorting analysis (FACS) of the hMC4R polymorphisms in stably expressed in HEK-293. The total cell receptor expression levels were determined using permeabilized cells measuring both cell surface and intracellular protein expression. The cell surface expression levels were determined using non-permeabilized cells. Cell expression levels are presented relative to the wild type hMC4R control.
Results
The 30 human MC4 polymorphic receptors and the I102T/V103I hMC4R that were examined in this study are summarized in Tables 1-3. Table 1 summarizes the functional endogenous α-MSH, β-MSH, γ2-MSH, ACTH(1-24) and synthetic Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9) tetrapeptide agonist potency of the polymorphic hMC4Rs. Table 2 summarizes the synthetic melanocortin agonist NDP-MSH agonist potency and binding affinity in a competitive displacement assay using radiolabeled 125I-NDP-MSH. Table 3 summarizes the MTII agonist EC50 values and pA2 values of the hAGRP(87-132) hMC4R antagonist. Table 4 summarizes selected hMC4R's that did not respond normally to all the endogenous agonists (F51L, I69T, and A219V) as well as the selected hMC4R's that resulted in partial agonist responses (E61K, D90N, Y157S, and C271R), that were also tested with synthetic ligands (Figure 1) that have been reported previously to functionally rescue these types of receptors (32).
Table 1.
Summary of the endogenous melanocortin agonist and Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9) ligand pharmacology at the hMC4R polymorphisms.
| Mutation | TM | α-MSH EC50 (nM) | β-MSH EC50 (nM) | γ2-MSH EC50 (nM) | ACTH(1-24) EC50 (nM) | JRH887-9 EC50 (nM) |
|---|---|---|---|---|---|---|
| WT# | 1.34±0.5 | 0.30±0.05 | 107±32 | 1.63±0.3 | 0.57±0.05 | |
| R7H | N-Term | 0.82±0.23 | 0.18±0.04 | 77.2±26 | 3.77±2.5 | 0.63±0.13 |
| R18H | N-Term | 1.34±0.7 | 0.10±0.01 | 79.3±19 | 2.06±0.7 | 0.43±0.07 |
| R18L | N-Term | 0.87±0.6 | 0.21±0.05 | 100±54 | 2.25±0.8 | 0.47±0.08 |
| S36Y | N-Term | 0.88±0.3 | 0.51±0.19 | 196±74 | 6.86±3.3 | 0.86±0.19 |
| P48S | 1 | 3.27±1.4* | 0.74±0.16 | 150±11 | 4.50±1.3 | 3.60±0.26* |
| V50M | 1 | 1.72±0.5 | 0.54±0.09 | 128±33 | 4.38±0.1 | 0.68±0.08 |
| F51L | 1 | 17.1±4.3* | 7.71±1.7* | 1134±260* | 107±24* | 37.3±11.3* |
| E61K | 1 | 82±53* partial agonist | 43.7±15* partial agonist | 1450±860* partial agonist | 188±66* partial agonist | 149±25* partial agonist |
| I69T | 1 | 13.6±1.1* | 9.52±3.1* | 278±5.7* | 32.6±3.8* | 40.7±5.1* |
| D90N | 2 | 226±77* partial agonist | 66±36* partial agonist | 6875±1070* partial agonist | 240±26* partial agonist | 746±250* partial agonist |
| S94R | 2 | No stim | No stim | No stim | No stim | No stim |
| G98R | 2 | No stim | No stim | No stim | No stim | No stim |
| I102T/V103I | 2 | 19.2±2.9* | 3.90±1.5* | 548±153* | 42.6±7.5* | 16.1±3.6* |
| I121T | 3 | 1.06±0.48 | 0.95±0.30 | 256±112 | 7.42±1.6 | 4.32±0.35* |
| A154D | IL2 | 5.25±2.4* | 0.67±0.01 | 427±55* | 14.0±2.2* | 4.37±0.6* |
| Y157S | IL2 | 265±66* partial agonist | 168±42* partial agonist | 2540±170* partial agonist | 177±49* partial agonist | 1185±150* partial agonist |
| W174C | 4 | No stim | No stim | No stim | No stim | No stim |
| G181D | 4 | No stim | No stim | No stim | No stim | No stim |
| F202L | 5 | 0.74±0.68 | 0.85±0.78 | 240±180 | 3.74±1.64 | 1.59±0.84 |
| A219V | 5 | 15.4±1.13* | 4.36±0.37* | 594±65* | 79.3±4.95* | 9.78±1.12* |
| I226T | IL3 | 1.30±0.96 | 0.53±0.11 | 110±35 | 7.53±3.04 | 0.84±0.17 |
| G231S | IL3 | 1.62±1.32 | 0.41±0.11 | 164±73 | 3.11±0.49 | 1.88±0.28* |
| G238D | IL3/6 | 2.15±0.63 | 0.19±0.01 | 92.7±13.3 | 3.62±1.16 | 0.65±0.09 |
| N240S | IL3/6 | 1.06±0.71 | 0.35±0.11 | 189±108 | 1.39±0.47 | 0.76±0.06 |
| 750DELGA | 6 | No stim | No stim | No stim | No stim | No stim |
| C271R | 6/EX3 | 350±80.5* partial agonist | 342±131* partial agonist | 6480±1130* partial agonist | 487±85* partial agonist | 1600±1060* partial agonist |
| S295P | 7 | 1.10±0.21 | 0.69±0.009 | 160±130 | 5.96±2.5 | 2.77±0.12* |
| P299L | 7 | No stim | No stim | No stim | No stim | No stim |
| E308K | C-Term | 2.96±0.72 | 0.81±0.11 | 116±31 | 6.03±2.59 | 0.96±0.29 |
| I317V | C-Term | 1.42±0.45 | 0.60±0.24 | 140±47 | 4.78±2.14 | 1.42±0.28 |
| L325F | C-Term | 0.35±0.04 | 0.33±0.08 | 90±47 | 3.47±1.67 | 0.98±0.40 |
Indicates the average from seven independent experiments of HEK-293 cells stably expressing the wild type (WT) hMC4R. These WT values represent those obtained while running parallel experiments with the receptors reported in this study. The values indicated represent the mean of at least three independent experiments with the standard error of the mean indicated. No stim indicates that the endogenous agonists were unable to stimulate the receptor polymorphisms at up to 1 μM concentrations. Partial agonist indicates that some stimulatory agonist pharmacology resulted, but the maximal stimulation levels were less then the wild type control response. Statistical analysis was performed using a student T-test compared to the wild type receptor values with
p<0.05.
Table 3.
Summary of the polymorphic hMC4R pharmacology of the endogenous C-terminal antagonist agouti-related protein ligand [hAGRP(87-132)] and the synthetic agonist MTII.
| Mutation | TM | MTII Agonist EC50(nM) | Antagonist pA2 hAGRP(87-132) | hAGRP(87-132) Inverse Agonist Activity Observed |
|---|---|---|---|---|
| WT# | 0.011±0.003 | 8.76±0.2 | Yes | |
| R7H | N-Term | 0.017±0.006 | 8.60±0.6 | Yes |
| R18H | N-Term | 0.011±0.007 | 8.79±0.8 | Not observed |
| R18L | N-Term | 0.006±0.002 | 8.88±0.1 | Not observed |
| S36Y | N-Term | 0.011±0.007 | 8.39±0.3 | Yes |
| P48S | 1 | 0.026±0.022 | 8.29±0.7 | Not observed |
| V50M | 1 | 0.015±0.006 | 8.04±0.1 | Yes |
| F51L | 1 | 0.028±0.013 | 8.72±0.2 | Yes |
| E61K | 1 | 0.28±0.0.08* partial agonist | 8.48±0.2 | Not observed |
| I69T | 1 | 0.071±0.001* | 8.35±0.2 | Not observed |
| D90N | 2 | 1.84±0.65* partial agonist | 8.61±0.3 | Not observed |
| S94R | 2 | No stim | ND | ND |
| G98R | 2 | No stim | ND | ND |
| I102T/V103I | 2 | 0.034±0.016 | 8.41±0.3 | Not observed |
| I121T | 3 | 0.010±0.005 | 8.58±0.2 | Not observed |
| A154D | IL2 | 0.038±0.004 | 8.04±0.2 | Not observed |
| Y157S | IL2 | 1.59±0.13* partial agonist | 8.31±0.1 | Not observed |
| W174C | 4 | No stim | ND | ND |
| G181D | 4 | No stim | ND | ND |
| F202L | 5 | 0.010±0.002 | 8.57±0.4 | Yes |
| A219V | 5 | 0.35±0.12* | 7.86±0.1* | Not observed |
| I226T | IL3 | 0.027±0.011 | 8.15±0.1 | Not observed |
| G231S | IL3 | 0.019±0.017 | 8.52±0.4 | Yes |
| G238D | IL3/6 | 0.029±0.013 | 7.74±0.2* | Yes |
| N240S | IL3/6 | 0.010±0.004 | 8.47±0.1 | Not observed |
| 750DELGA | C-Term | No stim | ND | ND |
| C271R | 6/EX3 | 1.62±0.99* partial agonist | 8.87±0.4 | Not observed |
| S295P | 7 | 0.065±0.005* | 7.88±0.3* | Yes |
| P299L | 7 | No stim | ND | ND |
| E308K | C-Term | 0.029±0.004 | 8.86±0.3 | Not observed |
| I317V | C-Term | 0.029±0.022 | 8.67±0.1 | Not observed |
| L325F | C-Term | 0.017±0.013 | 8.47±0.1 | Yes |
Indicates the average from seven independent experiments of HEK-293 cells stably expressing the wild type (WT) hMC4R. The MTII agonist EC50 values indicated represent the mean of at least three independent experiments with the standard error of the mean indicated. No stim indicates that the MTII agonist was unable to stimulate the receptor polymorphisms at up to 1 μM concentrations. Partial agonist indicates that some stimulatory agonist pharmacology resulted, but the maximal stimulation levels were less then the wild type control response. The antagonistic pA2 values were determined using the Schild analysis and the agonist MTII (Ki=-Log pA2). The indicated errors for the functional data (pA2) represent the standard error of the mean determined from at least three independent experiments. ND indicates that the pA2 value could not be determined since MTII was unable to potently stimulated the polymorphic receptor. Statistical analysis was performed using a student T-test compared to the wild type receptor values with
p<0.05.
Table 2.
Summary of the synthetic agonist NDP-MSH functional (EC50) and competitive displacement binding affinity studies (IC50) of the polymorphic hMC4Rs examined in this study. Non-labeled NDP-MSH was used to competitively displace 125I-NDP-MSH in a dose-response fashion.
| Mutation | TM | NDP-MSH EC50 (nM) | NDP-MSH Binding IC50 (nM) |
|---|---|---|---|
| WT# | 0.023±0.004 | 8.4±1.2 | |
| R7H | N-Term | 0.042±0.009 | 8.2±2.9 |
| R18H | N-Term | 0.015±0.004 | 6.0±0.8 |
| R18L | N-Term | 0.022±0.006 | 37.5±0.8* |
| S36Y | N-Term | 0.035±0.020 | 7.9±1.3 |
| P48S | 1 | 0.057±0.010 | 7.2±0.4 |
| V50M | 1 | 0.035±0.005 | 34.1±4.9* |
| F51L | 1 | 0.042±0.004 | 16.5±11.6 |
| E61K | 1 | 0.60±0.18* partial agonist | >1 |
| I69T | 1 | 0.50±0.18* | 18.2±12.7 |
| D90N | 2 | 2.47±0.89* partial agonist | 6.7±0.3 |
| S94R | 2 | No stim | >1 |
| G98R | 2 | No stim | >1 |
| I102T/V103I | 2 | 0.053±0.014 | 11.1±1.1 |
| I121T | 3 | 0.029±0.008 | 36.6±3.9* |
| A154D | IL2 | 0.060±0.007 | 36.9±1.2* |
| Y157S | IL2 | 8.60±4.3* partial agonist | 13.7±2.5 |
| W174C | 4 | No stim | >1 |
| G181D | 4 | No stim | >1 |
| F202L | 5 | 0.036±0.004 | 6.8±0.9 |
| A219V | 5 | 0.43±0.09* | 6.8±0.3 |
| I226T | IL3 | 0.18±0.11 | 9.1±3.1 |
| G231S | IL3 | 0.077±0.034 | 6.0±0.4 |
| G238D | IL3/6 | 0.23±0.19 | 7.1±0.4 |
| N240S | IL3/6 | 0.012±0.003 | 39.0±2.5* |
| 750DELGA | 6 | No stim | >1 |
| C271R | 6/EX3 | 3.73±2.4* partial agonist | 9.3±3.2 |
| S295P | 7 | 0.067±0.029 | 15.6±12.1 |
| P299L | 7 | No stim | >1 |
| E308K | C-Term | 0.31±0.17 | 9.1±3.0 |
| I317V | C-Term | 0.040±0.02 | 8.4±0.1 |
| L325F | C-Term | 0.022±0.004 | 41.2±0.5* |
Indicates the average from eight independent experiments of HEK-293 cells stably expressing the wild type (WT) hMC4R. No stim indicates that the NDP-MSH agonist was unable to stimulate the receptor polymorphisms at up to 1 μM concentrations. Partial agonist indicates that some stimulatory agonist pharmacology resulted, but the maximal stimulation levels were less then the wild type control response. >1 indicates that an IC50 could not be determined at up to 1 μM concentrations, or no competitive displacement binding of NDP-MSH was observed. Statistical analysis was performed using a student T-test compared to the wild type receptor values with
p<0.05.
Table 4.
Summary of the synthetic agonist ligand pharmacology at the selected polymorphic hMC4Rs.
| Mutation | JRH420-12 EC50 (nM) | JRH322-18 EC50 (nM) | AMW3-130 EC50 (nM) | THIQ EC50 (nM) | JB25 EC50 (nM) | AMW3-106 EC50 (nM) |
|---|---|---|---|---|---|---|
| WT | 0.54±0.02 | 0.31±0.02 | 0.11±0.07 | 0.16±0.11 | 182±27 | 0.27±0.08 |
| F51L | 18.6±5.3* | 63±17* partial agonist | 0.18±0.09 | 0.64±0.09* | 1700±273* | 1.19±0.24* |
| E61K | 182±55* partial agonist | >100 | 0.56±0.24 partial agonist | 109±40* partial agonist | 8390±1580* partial agonist | 30.0±8.2* partial agonist |
| I69T | 50.4±8.6* | 166±32* partial agonist | 0.74±0.49 | 3.78±0.26* | 5183±1415* | 13.8±2.8* |
| D90N | >100 | >100 | >100 | >100 | >100 | >100 |
| Y157S | 4220±1960* partial agonist | >100 | 5.05±0.92* partial agonist | 166±55* Full agonist | 20000±3850* partial agonist | 110±27* partial agonist |
| A219V | 11.4±4.1* | 75.4±14* partial agonist | 0.64±0.16 | 2.58±1.05* | 4330±1260* | 4.59±0.25* |
| C271R | 2154±1210* partial agonist | >100 | 4.28±0.26* partial agonist | 200±74* partial agonist | 22000±8310* partial agonist | 160±60* partial agonist |
The values indicated represent the mean of at least three independent experiments with the standard error of the mean indicated. >100 indicates that the ligand was unable to stimulate the receptor polymorphisms at up to 100 μM concentrations. Partial agonist indicates that some stimulatory agonist pharmacology resulted, but the maximal stimulation levels were less then the wild type control response. Statistical analysis was performed using a student T-test compared to the wild type receptor values with
p<0.05.
For comparative purposes, the wild type hMC4R values and pharmacological profiles observed are defined as “normal” in the experimental paradigm used herein. The criteria used to classify a hMC4 polymorphic receptor as similar to the wild type are that the polymorphic hMC4R does not result in statistically significant different experimental variables (EC50, pA2, IC50, total cell and cell surface expression) from the wild type hMC4R variables. The ability of AGRP to function as an inverse agonist (Table 3) is excluded from this comparison as this phenotype is only observable if the basal activities of the heterologously expressing cell lines are sufficient to detect a decrease in our assay readout. Thus, these observations, while worth noting, are at the limits of detection, and the author's do not wish to make further interpretations of the data.
hMC4R Polymorphic Receptors that Result in Decreased Agonist Ligand Efficacy and Potency
For this study, a 96-well β-galactosidase reporter gene assay is used (31, 32, 39) that responds to Gs and Gq G-protein coupled pathways. The hMC4R has been well established to signal via the cAMP second messenger pathway (13). Generally, when fully efficacious agonists are studied [i.e. NDP-MSH (29) and MTII (30)] at the wild type and most mutant melanocortin receptors, a maximal response is observed. Thus, the ligands are classified as full agonists. However, of the polymorphic receptors generated in this study four (E61K, D90N, Y157S, and C271R) hMC4Rs were only able to generate partial efficacy upon stimulation by all the melanocortin agonists examined in this study (Figure 4). Additionally, when comparing the potency of these same ligands, significant decreases were observed (Tables 1-4).
Figure 4.

Illustration of the wild type hMC4R full agonist and partial agonist activity of the endogenous melanocortin agonists at the E61K, D90N, Y157S, and C271R hMC4Rs. The ligand efficacy is relative to non-receptor dependent maximal stimulation by the forskolin control compound which is defined as 1.0 on the Y-axis.
hMC4R Polymorphic Receptors that are expressed at the Cell Surface and Functionally Respond Differently to the Endogenous POMC Derived agonists
In this study, the three F51L, I69T, and A219V hMC4R polymorphic receptors possessed decreased wild type potency for all the endogenous melanocortin agonists examined [α-MSH, β-MSH, γ2-MSH, and ACTH(1-24)], Table 1. Only the P48S hMC4R possessed decreased α-MSH potency while retaining wild type equipotency for β-MSH, γ2-MSH, and ACTH(1-24). The A154D hMC4R possessed decreased α-MSH, γ2-MSH, and ACTH(1-24) potency values, yet was equipotent for β-MSH, as compared to the wild type receptor.
Functional Response to the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9)
Since all the endogenous melanocortin agonists contain a central conserved His-Phe-Arg-Trp sequence postulated to be important for melanocortin receptor-ligand molecular recognition and receptor stimulation, the nM Ac-His-Phe-Arg-Trp-NH2 tetrapeptide (23-28) was examined at the polymorphic receptors characterized in this study. The P48S, F51L, E61K, I69T, D90N, I121T, A154D, Y157S, A219V, G231S, C271R, and S295P hMC4Rs possessed significantly reduced potency for the JRH887-9 tetrapeptide, as compared to the wild type control. Interestingly, only at the I121T, G231S, and S295P hMC4Rs, was this tetrapeptide agonist the only ligand that possessed decreased wild type potency compared to the endogenous agonists shown in Table 1.
Pharmacological Characterization of the AGRP Endogenous Antagonist
Agouti-related protein (AGRP) was characterized as a competitive antagonist (20) as well as an inverse agonist (44, 45) at the MC4R. The full length 132 amino acid hAGRP contains five disulfide bridges and is not readily synthetically accessible by chemical means (46). The studies presented herein utilized the commercially available hAGRP(87-132) fragment that has been demonstrated to be equipotent to the full-length 132 amino acid hAGRP ligand (20). Figure 5 illustrates the hAGRP(87-132) antagonist pharmacology at the wild type hMC4R. Table 3 summarizes the antagonist pA2 values of the MC4R antagonist hAGRP(87-132) fragment, if AGRP(87-132) inverse agonist activity was observed, as well as the synthetic agonist MTII potency that was used to compete with AGRP(87-132) for the bioassay. The synthetic MTII agonist was selected for the antagonist Schild experiments (40) versus any of the endogenous agonists since it is more potent and has been observed to remain potent even at polymorphic and mutant MC4 receptors (31, 37). Agonist ligand potency is important when trying to perform competitive antagonist assays. For the Schild type of antagonist assay used in this study, at least three different concentrations of antagonist are needed to perform the analysis. If a receptor mutation decreases agonist potency (i.e. 100-fold), then the right shift in the dose-response curves critical for determining the antagonist potency may then be outside the detectable range of the assay, thus yielding inclusive data regarding the potency of an antagonist at the receptor being examined. This rational is demonstrated by the Y157S and C271R hMC4Rs, where the endogenous agonist potency (Table 1) are high nM to μM, where as the MTII potency is low nM resulting in the ability to evaluate antagonist potency (Figure 5).
Figure 5.

Illustration of the hAGRP(87-132) antagonist pharmacology at the wild type (WT), E61K, D90N, Y157S, and C271R hMC4Rs. The MTII ligand is a full agonist at the WT hMC4R and a partial agonist at the E61K, D90N, Y157S, and C271R hMC4Rs relative to non-receptor dependent maximal stimulation by the forskolin control compound which is defined as 1.0 on the Y-axis.
For the polymorphic receptors examined in this study, only the three A219V, G238D, and S295P hMC4Rs resulted in statistically decreased AGRP(87-132) antagonist potency, as compared to the wild type hMC4R. Decreased MTII agonist potency was observed for the E61K, D90N, Y157S, A219V, and C271R hMC4Rs.
Functional and Competitive Displacement Binding Characterization of the Synthetic Melanocortin Agonist NDP-MSH
The NDP-MSH ligand (29), a highly potent and chemically stable synthetic melanocortin receptor agonist that upon iodination, is the radioligand of choice for characterization of the melanocortin receptors. Table 2 summarizes the NDP-MSH ligand functional agonist potency EC50 values and competitive displacement binding affinity IC50 pharmacological results. Unlabeled “cold” NDP-MSH was used to competitively displace the 125I-NDP-MSH radioligand. Of the polymorphic hMC4 receptors examined in this study, only the six R18L, V50M, I121T, A154D, N240S, and L325F hMC4Rs possessed statistically significant decreases in NDP-MSH binding affinity compared to wild type hMC4R. Unanticipated however, there does not appear to be a corresponding significant decrease in NDP-MSH ligand potency. However, the E61K, I69T, D90N, Y157S, A219V, and C271R hMC4Rs did posses significantly decreased NDP-MSH potency, as compared to wild type hMC4R. Unexpectantly, for the E61K hMC4R, we were unable to generate a reliable NDP-MSH binding affinity IC50 value under the experimental conditions examined in this study. Functionally, this receptor possessed only partial agonist activity (Figures 4 and 5).
Radioactive label binding studies are also useful for characterizing cell line surface receptor expression levels. Since the label will bind to receptor proteins that are folded “properly” for molecular recognition at the cell surface, it is an experimental approach that can be used to detect receptor protein cell surface expression (Figure 3) and receptors that are expressed at the cell surface and that possess the required protein folding structure(s) important for ligand molecular recognition. These data are complementary to the FACS (Figure 2) data that determine protein cell surface expression levels, but do not provided an indication if the protein is properly folded for ligand molecular recognition or functional activity. Additionally, these data can aid in the assessment of receptor mutations to differentiate a molecular defect associated with molecular recognition and ligand binding versus a disability in ligand-receptor induced cell signaling. Figure 3 summarizes the total specific binding values for the hMC4R polymorphic and wild type hMC4R stably expressing HEK-293 cell lines generated in this study. These values represent the average total specific binding (total cmps – non-specific cpms) from two to five independent experiments. The S94R, G98R, W174C, and G181D hMC4R expressing cells did not bind to radiolabeled 125I-NDP-MSH above an average of 500 counts per minute (cpms), suggesting that these cell lines do not express very much of the correctly folded receptor protein at the cell surface or that the amino acid change in the receptor has changed the ability of 125I-NDP-MSH to interact with the putative binding pocket at the concentrations examined in this study.
Figure 3.

Summarizes the total specific binding counts (cpms) of 125I-NDP-MSH binding to the polymorphic hMC4R stable HEK-293 cell lines generated in this study. These data indicate that receptor protein expressed at the cell surface is properly folded to allow for ligand binding.
Cell Surface Expression of hMC4R Polymorphic Receptors in Stably Expressing HEK-293 Cells
One of the possible pharmacological differences between polymorphic hMC4R has been associated with poor cell surface expression levels (47-52). Figure 2 summarizes the percentage of total cellular receptor protein expression and receptor protein cell surface expression of the receptors stably expressed in HEK-293, relative to the wild type hMC4R. The engineered N-terminal FLAG-hMC4Rs (47) and Fluorescence Activated Cell Sorting (FACS) were used for comparative quantification. These data demonstrate that the total cell receptor expression for all of the polymorphic hMC4Rs generated in this study was at 80% of wild type levels. Using a combination of intracellular labeling on saponin-permeabilized cells and surface labeling on non-permeabilized cells, it was observed that the E61K, D90N, S94R, G98R, I121T, Y157S, W174C, G181D, C271R, P299L, I317V, L325F, and 750DelGA hMC4R cell surface expression levels are significantly decreased as compared to wild type. Interestingly however, while the S94R, G98R, W174C, G181D, P299Land 750DelGA hMC4Rs were not able to be stimulated by ligands (Tables 1-3), they did possess cellular expression levels comparable to the Y157S and C271R hMC4Rs that were able to be stimulated by ligands (albeit as partial agonists, Figures 4 and 5). The E61K, P299L, and 750DelGA hMC4Rs possessed detectable significant binding, indicative that some receptor protein did reach the cell surface and was able to bind radiolabeled ligand, albeit at extremely relatively low levels (Figure 3). The D90N and I317V hMC4Rs, while possessing significantly reduced cell surface protein expression levels, did possess significant binding values greater then 35,000 cpms (Figure 3), indicating a strong response to 125I-NDP-MSH ligand binding. Thus, for the S94R, G98R, W174C, and G181D hMC4Rs, these data suggest that failure of cell surface hMC4R protein expression may be a result of the inability to transport the polymorphic protein to the cell surface or alternatively, an unstable receptor conformation may result that might not be retained at the cell surface.
Functional Response of Selected Polymorphic hMC4Rs to Synthetic Ligands Postulated to Rescue Functional Activity
In a previous report by our laboratory (32), we examined the potential of synthetic and small molecule agonists (Figure 1) to “rescue” hMC4 polymorphic receptors that did not respond normally to the POMC derived endogenous ligands. Based upon preliminary results, the F51L, E61K, I69T, D90N, A219V, and C271R hMC4Rs were selected to be characterized using these synthetic agonist ligands (Table 4). The E61K, D90N, Y157S, and C271R were selected to determine if these ligands could “rescue” full agonist efficacy, as compared to the wild type control response. Interestingly, the THIQ small molecule was able to reach 100% maximal agonist efficacy, as compared to the wild type, at the Y157S hMC4R (Figure 6). At the E61K and C271R hMC4Rs, THIQ was able to increase agonist efficacy response to ca 95% and 89% that of wild type, respectively at μM concentrations. Surprisingly, the D90N hMC4R was not stimulated to even the agonist efficacy levels observed for the other ligands examined in this study (Figures 4-6). The tetrapeptide Ac-His-(pI)DPhe-Arg-Trp-NH2 (JRH322-18), which is a mMC3R antagonist with partial agonist activity (25, 53) and a full MC4R agonist (Figure 6) unexpectedly resulted in either partial agonist or no stimulatory activity at the polymorphic hMC4R's examined.
Figure 6.

Summary of the synthetic agonist peptide and small molecule agonist ligand pharmacology (Figure 2 and Table 4) at the wild type (WT) hMC4R and the F51L, E61K, I69T, D90N, Y157S, A219V, and C271R hMC4Rs. The ligands were full agonists at the wild type hMC4R and displayed different combinations of a) no stimulatory activity (up to 100 μM concentrations), b) partial agonist efficacy as normalized to the forskolin control (defined as 1.0 on the Y-axis), and c) to full agonist activity. Notable results include the THIQ small molecule to reach a full agonist response at the Y157S.
In terms of potency, the chimeric melanocortin-AGRP agonist AMW3-130 retained nM to sub-nM potency at the hMC4Rs examined. At the F51L, E61K, I69T, and A219V hMC4Rs, this ligand was the only agonist examined in this study that was not statistically different from wild type hMC4R potency at these receptors. However, at the Y157S, and C271R hMC4Rs, this AMW3-130 ligand did result only in partial agonist efficacy with significantly decreased potency compared to the wild type hMC4R. All the other ligands examined, resulted in significantly decreased agonist potency at the F51L, E61K, I69T, D90N, Y157S, A219V, and C271R hMC4Rs as compared to the wild type hMC4R (Table 4).
I102T/V103I double mutation
While the objective of this study was to generate and pharmacologically characterize the I102T polymorphic hMC4R which was observed in non-obese control patients (54-56), during our plasmid mutation sequencing verification, we found that the receptor also contained the common V103I mutation as well. For comparative purposes, we have previously reported the characterization of the I102S and V103I mutations using the same experimental paradigm presented in the currently study (31). That study found for the V103I hMC4R, normal pharmacological profiles for receptor expression levels, NDP-MSH ligand binding affinity, the endogenous and synthetic NDP-MSH and MTII agonists, as compared to the wild type hMC4R. However, decreased AGRP(87-132) antagonist potency was identified that was different between this V103I and wild type hMC4R. The I102S hMC4R however, possessed significantly reduced agonist potency at all the ligands examined, consistent with other reports (56, 57). A report by Tao et al. characterized the I102T hMC4R to possess normal NDP-MSH binding affinity and agonist potency, however 125I-NDP-MSH maximal binding values were only ca 20% of the wild type hMC4R (56). The I102T hMC4R was observed in both a non-obese (54) and an obese (55) subject, while the I102S was found only in an obese patient (58). The double I102T/V103 hMC4R resulted in significantly decreased agonist potencies for the endogenous and JRH887-9 tetrapeptide agonists in Table 1. However, the I102T/V103 hMC4R possessed MTII and NDP-MSH potencies equivalent to the wild type hMC4R as well as AGRP(87-132) antagonist potency and NDP-MSH binding affinity. Thus, these unanticipated pharmacological results appeared important to report, although the changes in pharmacology cannot be solely associated with the I102T mutation but may be due to the changes associated with the dual mutation.
Discussion
Obesity is becoming a significant health risk. Polymorphisms for the MC4R have been discovered in both obese and non-obese control human patients. Since the MC4R has been demonstrated in both mice (7) and humans to be involved in the regulation of feeding behavior and energy homeostasis, characterization of hMC4R SNPs at an in vitro molecular level may provide a link to explain the obese human phenotype. In vitro pharmacological characterization experiments provide an initial forum to identify if a polymorphic receptor may result in an unnatural response to endogenous ligands or be susceptible to improper cell surface trafficking. We previously reported the side-by-side in vitro pharmacological characterization of 40 hMC4R mutations reported to be identified in both obese and non-obese patient control groups (31). Since the initiation of those studies, a plethora of reports have been published identifying novel hMC4R polymorphisms and mutations. These reports also confirm the increasing abundance of several common hMC4R polymorphisms, with the most notable being V103I. An increasing number of receptor pharmacology characterizations have been reported with the discovery of novel hMC4R polymorphisms, but nonetheless, a uniform and consistent characterization with multiple endogenous agonists and the AGRP antagonist is not present in many reports. This study was undertaken to provide a comprehensive in vitro pharmacological profile of an additional 30 hMC4R SNPs with the multiple endogenous agonists [α-, β-, γ2-melanocyte stimulating hormones (MSH) and adrenocorticotropin (ACTH)], the antagonist agouti-related protein hAGRP(87-132), and synthetic agonists [NDP-MSH, MTII, and the tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 (JRH887-9)]. We have proposed the hypothesis that some hMC4R SNPs may respond differently to one or more endogenous agonist(s) and/or the AGRP antagonist (31) while retaining wild type hMC4R activity of others, and wanted to explore this concept with the 30 hMC4R SNPs examined herein to identify specific variants that fall into this category. Additionally, we have reported that some ligands can restore nM full agonist potency to polymorphic hMC4Rs that did not respond normally to endogenous agonists (32). Based upon this continuing theme, the F51L, E61K, I69T, D90N, Y157S, A219V, and C271R hMC4Rs in this study, were examined using these peptides and small molecules (Table 4) to identify possible ligands that could result in nM agonist potency and/or generate a maximal efficacy response for hMC4Rs with partial agonist profiles (E61K, D90N, Y157S, and C271R). If a common ligand could be identified that could restore wild type hMC4R profiles of hMC4R SNPs that do not respond normally to the endogenous agonists, then a potential therapeutic ligand lead could be followed-up to help these individuals obtain improved quality of life by decreasing their satiety and associated weight homeostasis consequences.
While an originating goal of this study was to gain mechanistic insights into naturally occurring hMC4R SNPs identified in obese humans and how these respond differently to endogenous and synthetic ligands in several pharmacological characterization bioassays, in some cases these studies failed to achieve this goal in that only normal wild type hMC4R pharmacology resulted, as discussed below. In other cases however, new and novel data was generated that allows for a speculative link between an hMC4R in vitro molecular defect and the observed obese patient phenotype reported (Figure 7 and Table 6). Additionally, these studies provide the identification of newly reported mutant GPCR tools that can be used to probe the consequences of endogenous ligand pharmacological profile changes in vitro or extended to in vivo studies, via the generation of genetic mouse models, to test specific molecular hypotheses such as ligand-receptor interactions and the rescue of functional defects via synthetic ligands.
Figure 7.
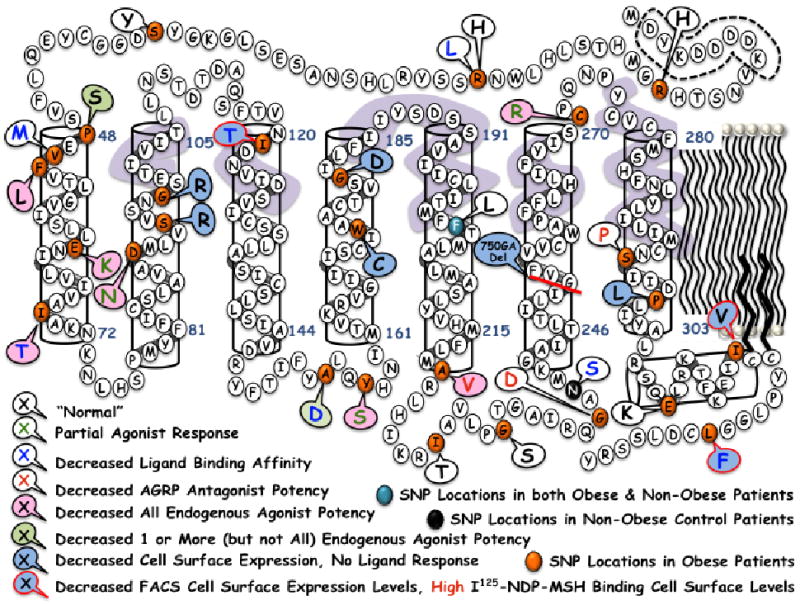
Summary of the 30 hMC4R polymorphisms examined in this study. The dashed line indicates the N-terminal FLAG sequence used for immunohistochemical cell expression studies. Changes in key receptor pharmacological profiles are indicated by either a change in font color and/or a colored callout circle, as indicated in the figure. The lavender color indicates hMC4R residues postulated to be involved in the putative ligand binding pocket.
Table 6.
Mechanistic insights into the putative in vitro molecular “defect” that may be extrapolated* to the obese human phenotype.
| hMC4R SNP | In Vitro Molecular Defect(s) | Putative Association with an Obese Patient Phenotype* | ||||
|---|---|---|---|---|---|---|
| Modified Properly Folded/Cell Surface Expression Levels | Modified NDP-MSH Ligand Binding Affinity | Modified Endogenous Agonist Potency | Agonists Not Maximally Efficacious | Endogenous AGRP Antagonist Potency Reduced | ||
| R7H | None, WT profiles | |||||
| R18H | None, WT profiles | |||||
| R18L | X | Inconclusive, perhaps decreased hMC4R basal activity proposed by Srinivasan et al. | ||||
| S36Y | None, WT profiles | |||||
| P48S | X | Reduced endogenous agonist potency | ||||
| V50M | X | Inconclusive | ||||
| F51L | X | Reduced endogenous agonist potency | ||||
| E61K | X | X | X | X | reduced cell surface expression, modified ligand binding, reduced agonist potency, and partial agonist stimulation | |
| I69T | X | Reduced endogenous agonist potency | ||||
| D90N | X | X | X | reduced cell surface expression, reduced agonist potency, and partial agonist stimulation | ||
| S94R | X | - | failure of cell surface hMC4R protein expression | |||
| G98R | X | - | failure of cell surface hMC4R protein expression | |||
| I121T | X | X | Inconclusive | |||
| A154D | X | X | Reduced endogenous agonist potency | |||
| Y157S | X | X | X | reduced cell surface expression, reduced agonist potency, and partial agonist stimulation | ||
| W174C | X | - | failure of cell surface hMC4R protein expression | |||
| G181D | X | - | failure of cell surface hMC4R protein expression | |||
| F202L | None, WT profiles | |||||
| A219V | X | X | Reduced endogenous agonist and AGRP antagonist potency | |||
| I226T | None, WT profiles | |||||
| G231S | None, WT profiles (except for 3-fold decreased JRH887-9 potency) | |||||
| G238D | X | Inconclusive | ||||
| N240S | X | Non-Obese Patient | ||||
| 750DELGA | X | - | failure of cell surface hMC4R protein expression | |||
| C271R | X | X | X | reduced cell surface expression, reduced agonist potency, and partial agonist stimulation | ||
| S295P | X | Inconclusive | ||||
| P299L | X | - | failure of cell surface hMC4R protein expression | |||
| E308K | None, WT profiles | |||||
| I317V | X | None, WT profiles | ||||
| L325F | X | X | Inconclusive | |||
These speculations only consider the associated link in attempts to relate the “artificial” HEK-293 in vitro system to the complex human obesity phenotype that additionally contains underlying psychological inputs.
-Indicates that no stimulated was observed at up to μM concentrations of ligands.
hMC4R Polymorphisms that possess normal wild type hMC4R in vitro pharmacological profiles
The R7H, R18H, S36Y hMC4Rs located within the N-terminal domain, the F202L (TM5) hMC4R, the I226T (IL3) hMC4R, the G231S (IL3) hMC4R, the E308K (C-Term) hMC4R, and the I317V (C-term) hMC4R possessed wild type cell surface expression levels (Figure 2) and total 125I-NDP-MSH specific binding counts (Figure 3). These data indicate that the receptor protein is expressed at the cell surface at the same, or at slightly higher levels (Figure 2) as compared to the wild type hMC4R, and that they are properly folded for melanocortin ligand molecular recognition and binding. These SNPs possess NDP-MSH binding affinity and potency, endogenous agonist potency, JRH887-9 tetrapeptide potency, MTII potency, and AGRP(87-132) potency equivalent to the wild type hMC4 receptor values (Tables 1-3), with the exception of G231S that possesses 3-fold reduced JRH887-9 potency. Thus, in all assays examined in this study, it appears that these hMC4R side chain modifications do not modify the in vitro pharmacology in a fashion that might associate a particular molecular dysfunction of this SNP with the obese human phenotype (Table 6).
Hypothesized links between these in vitro studies and obese human patients
One of the goals of the current study was to perform a side-by-side in vitro pharmacological characterization of the 30 polymorphic hMC4Rs with multiple ligands and assays to identify pharmacological changes that result and link the in vitro tissue culture molecular defects as a causative factor for the obese patients manifesting the hMC4R SNP (Table 6). While human obesity is a complex phenotype that involves central and peripheral signaling networks to regulate energy homeostasis, emotional processes and states of stress, anxiety, and depression are also contributing factors. Nonetheless, herein we speculate about the in vitro findings that were observed in this study and possible consequences and links to the obese human patient(s) possessing hMC4R SNPs.
The thirteen E61K, D90N, S94R, G98R, I121T, Y157S, W174C, G181D, 750DelGA, C271R, P299L, I317V, and L325F hMC4R polymorphic proteins were all expressed at the surface in the cell lines generated at significantly reduced levels (FACS, Figure 2), as compared to the wild type hMC4R. However, only the six E61K, S94R, G98R, W174C, G181D, and P299L hMC4R cell lines also resulted in less then 1000 cpms of the total specific binding cpms (Figure 3), and only the six S94R, G98R, W174C, G181D, 750DelGA, and P299L hMC4R polymorphic cell lines were not able to be stimulated by any endogenous or synthetic agonist examined in this study (Tables 1-3). If one were to speculate, based upon these in vitro data reported herein, a molecular defect that might explain the obese human phenotype for the four S94R, G98R, W174C, and G181D hMC4Rs (Table 5, Figure 7) is reduced, or lack of, cell surface expression (Table 6). The four E61K, D90N, Y157S, and C271R polymorphic hMC4Rs possessed reduced protein cell surface expression levels but resulted in partial agonist activity (Figures 4-6) as well as significantly reduced agonist ligand potency. Thus, for these specific polymorphic hMC4Rs it could be postulated that a combination of reduced cell surface expression, reduced potency, and partial stimulation by endogenous agonists are the in vitro molecular defects that could be correlated with an obese human phenotype (Table 6).
Table 5.
Summary of the hMC4R polymorphism and if they were observed in obese versus non-obese patients.
| Mutation | Genotype | Non-Obese Patient | Obese Patient | Ref |
|---|---|---|---|---|
| R7H | Heterozygous | X | (71) | |
| R18H | Unable to determine | X | (52) | |
| R18L | Unable to determine | X | (71) | |
| S36Y | Unable to determine | X | (55) | |
| P48S | Heterozygous | X | (72) | |
| V50M | Heterozygous | X | (58) | |
| F51L | Heterozygous | X | (73) | |
| E61K | Heterozygous | X | (74) | |
| I69T | Heterozygous | X | (74) | |
| D90N | Heterozygous | X | (66) | |
| S94R | Unable to determine | X | (75) | |
| G98R | Heterozygous & Homozygous | X | (76) | |
| I121T | Unable to determine | X | (75) | |
| A154D | Heterozygous | X | (77) | |
| Y157S | Homozygous | X | (78) | |
| W174C | Heterozygous | X | (79) | |
| G181D | Heterozygous | X | (55, 75) | |
| F202L | Heterozygous | X% | X# | %(54)#(74) |
| A219V | Unable to determine | X | (55) | |
| I226T | Heterozygous | X | (80, 81) | |
| G231S | Heterozygous | X | (82) | |
| G238D | Heterozygous | X | (74) | |
| N240S | Heterozygous | X | (54) | |
| 750DELGA | Homozygous | X | (75) | |
| C271R | Homozygous | X | (74) | |
| S295P | Heterozygous | X | (77) | |
| P299L | Heterozygous | X | (80) | |
| E308K | Heterozygous | X | (83) | |
| I317V | Heterozygous | X | (75, 79) | |
| L325F | Unable to determine | X | (55) |
A change in agonist molecular recognition and ligand binding affinity of a polymorphic hMC4R has been postulated to cause human obesity by several laboratories. This study has identified seven (R18L, V50M, E61K, I121T, A154D, N240S and L325F) hMC4Rs that possessed statistically significant decreases in NDP-MSH IC50 binding affinities as compared to the control receptor (Table 2). However, if a mutation in the receptor modified ligand binding is to be a causative pharmacological effect, a correlation in the change in ligand functional potency should also be observed (i.e. 10-fold change in ligand binding and a 10-fold change in functional potency). In the hMC4R SNPs examined in this study, only the E61K hMC4R that we were unable to determine a binding IC50 value for, also possessed a significantly reduced functional NDP-MSH agonist potency value (Table 2). In addition to reduced agonist potency, this E61K hMC4R possessed only partial agonist activity for all the ligands examined. The R18L, V50M, I121T, A154D, N240S, and L325F hMC4Rs all resulted in NDP-MSH functional agonist potency that was not statistically different from the wild type hMC4R values. While the A154D hMC4R did not demonstrate reduced NDP-MSH agonist potency, reduced endogenous agonist potency was observed for α-MSH, γ2-MSH, and ACTH(1-24) (Table 1). These findings however, do not eliminate the potential molecular defect of ligand-hMC4R molecular recognition since the high affinity 125I-NDP-MSH ligand used in this study has been shown previously at the mMC4R's to respond differently at some receptor mutations than the radiolabeled antagonists SHU9119 and AGRP(83-132) as well as the radiolabeled agonist MTII (37). Additionally, in a study of the hMC1R where radiolabeled NDP-MSH was used in conjunction with the unlabeled α-MSH, NDP-MSH, MTII, and γ-MSH (59), differences in the unlabeled ligand binding affinity profiles were observed. Thus, in this study the E61K hMC4R results in a change in NDP-MSH binding affinity that can is supported by decreased functional NDP-MSH agonist potency. The other six polymorphic hMC4Rs that possessed significantly decreased NDP-MSH ligand binding affinity did not possess reduced NDP-MSH ligand potency, thus it is difficult to speculate if this modified binding affinity is an associative mechanism that might explain the obese phenotype observed in patients with these specific SNPs without further experimental studies.
In this study, the five P48S, F51L, I69T, A154D, A219V hMC4Rs were expressed at the cell surface and possessed full agonist efficacies similar to the wild type hMC4R, but resulted in significantly reduced endogenous agonist potency (Table 1). Of these five, the P48S and A154D hMC4Rs possessed significant differences for one or more, but not all of the endogenous agonists examined in this study. These two hMC4Rs could become valuable in vitro tools to study the different endogenous agonist-hMC4R interactions at a basic science level. These data also provide supporting experimental evidence that polymorphic hMC4Rs may respond differently to the endogenous agonists (31, 32). In terms of linking a molecular defect with human obesity, the inability of the endogenous ligands to signal through a polymorphic hMC4R that has reduced potency could be postulated to modify the satiety homeostasis resulting in the desire to eat more.
The melanocortin system also responds to endogenous antagonists. The AGRP ligand has been demonstrated to be both a competitive antagonist and inverse agonist at the MC4R (20, 44). In this study, only the three A219V, G238D, and S295P hMCRs possessed decreased antagonist potency of AGRP as compared to the wild type control (Table 3). At both the G238D and S295P hMC4Rs, the inverse activity of the AGRP ligand was also observed. For these two proteins, the decreased AGRP potency was the only putative molecular defect observed in the in vitro studies performed herein (Table 6). The physiological consequences of decreased AGRP potency remain to be identified. The AGRP antagonist has been demonstrated to be involved in the feedback loop in the arcuate nucleus of the hypothalamus (43, 60) regulating the endogenous agonist POMC expressing neurons (presumably via the MC3R). Based upon these reports, it might be speculated that decreased AGRP(87-132) antagonist potency could potentially alter the hypothalamic POMC agonist expression levels and thus indirectly result in an obese phenotype. Granted, this is stretching these results to wildly speculate about a molecular link between these in vitro data and the obese human patient. Nonetheless, it is an interesting concept to consider that indirectly a hMC4R polymorphism could be affecting endogenous melanocortinergic system tone and thus altering energy homeostasis. Further studies would need to be performed to provide experimental evidence either way in regard to this speculative mechanism.
At the conclusion of such studies, it could be hoped that a “larger picture” might emerge regarding mechanistic insights, generalizations, and trends in SNPs and general receptor function (Figure 7). Examination of the 30 hMC4R SNPs generated in this study in terms of possible global structure-function modifications (Figure 7) and associations between particular SNPs and the obese human phenotype observed in patients (Table 5) resulted in three general insights. The first being that to the author's knowledge, no hMC4R SNPs have been identified that are located in the putative binding pocket (Figure 7, lavender coloration) identified by GPCR homology molecular modeling and/or designed receptor mutagenesis studies. Yet a large number of polymorphic hMC4R SNPs modify ligand-receptor interactions in terms of endogenous agonist potency and/or ability to generate a maximal signaling response, relative to the wild type hMC4R. The second and unanticipated observation is that the hMC4R TM1 domain appears to be important in modifying endogenous agonist potency. The third unanticipated observation is that the TM2 hMC4R SNPS examined in this study (S94R and G98R) putatively located towards the extracellular half of the transmembrane domain resulted in significantly reduced cell surface expression and very minimal 125I-NDP-MSH ligand binding. Thus, the charged R side chain modifies cell surface expression as well as the putative ligand binding domain.
Selected hMC4R polymorphisms that do not respond the same as the wild type hMC4R that have been characterized with additional synthetic ligands (Table 4)
In this study, the F51L (TM1), I69T (TM1), and A219V (TM5) hMC4Rs were identified based upon their inability to respond potently to the endogenous agonists (Table 1). The E61K (TM1), D90N (TM2), Y157S (IL2), and C271R (TM6/EX3) hMC4Rs were selected based upon the ability of both endogenous and synthetic agonists to result in only partial agonist responses (Figure 4). Table 4 and Figure 6 summarizes the synthetic ligands and resulting pharmacology at these selected polymorphic hMC4Rs.
The F51L hMC4R possessed slightly increased total and cell surface expression and total specific 125I-NDP-MSH binding levels as compared to the control hMC4R, demonstrating wild type cell surface expression levels. Interestingly, this variant possessed decreased endogenous agonist and JRH887-9 potencies as compared to the wild type hMC4R values, but did not result in decreased potency for the synthetic NDP-MSH and MTII agonists. The binding affinity of NDP-MSH as well as the endogenous AGRP(87-132) antagonist potency values were not statistically different from the wild type hMC4R values and AGRP inverse agonist activity was observed. These data clearly implicate the aromatic Phe side chain at the 51 position of the hMC4R as important for endogenous agonist potency. However, a comparison of the ligand amino acid sequences (Figure 1) does not provide any key ligand structural insights. One might speculate that all the endogenous agonists contain a LPhe7 whereas the synthetic agonists contain a DPhe7 and that this structural difference might be important at this mutation, however the fact that the JRH887-9 tetrapeptide contains the DPhe7 and possesses 65-fold decreased potency abates this conclusion. Attempts to further probe the postulated ligand Phe7 using the tetrapeptide template and the JRH322-18 tetrapeptide containing a (pI)DPhe7 (Figure 1, Table 4) resulted in the conversion of this ligand from a full agonist at the wild type hMC4R to a partial agonist with 200-fold decreased potency. The AMW3-130 ligand which is a chimeric AGRP-melanocortin ligand possessing the core His-DPhe-Arg-Trp melanocortin domain retained equipotent full agonist activity as compared with the wild type hMC4R. However, the larger chimeric AMW3-106 AGRP-melanocortin agonist in which the AGRP Arg-Phe-Phe core residues were substituted with the agonist His-DPhe-Arg-Trp resides also retained full agonist activity, albeit with 4-fold decreased potency at the F51L hMC4R versus the wild type hMC4R. These data might lead to the speculation that this F51L variant can be important for the differentiation of endogenous agonist versus the AGRP antagonist ligand-receptor structure and function correlation. Further examination by the small molecules THIQ and JB25 resulted in decreased potency for both these ligands indicating they cannot rescue normal wild type potency/activity at the F51L hMC4R. However, there are clearly key structural differences between the endogenous and synthetic agonists that might be teased out in further studies to pin point a specific molecular hypothesis to account for these data. Additionally, it is unclear from these data if the Phe side chain is directly involved in the putative ligand binding pocket or if this receptor side chain is important for receptor function and/or endogenous ligand-induced conformational changes important for signal transduction.
The E61K hMC4R SNP located towards the center of TM1 is expressed at similar levels as the wild type hMC4R cells, yet has significantly reduced cell surface and 125I-NDP-MSH total specific binding levels as compared to the wild type control values. Interestingly, at up to 1 μM concentrations of unlabeled NDP-MSH, no IC50 could be determined in these studies. This might be linked to the fact that all the agonist ligands examined only resulted in partial agonist activity with significantly reduced potencies (Figure 4). However, AGRP(87-132) antagonist potency remained unchanged from the wild type hMC4R value. Inverse activity of AGRP(87-132) was not observed and is attributed to the decreased basal activity of this cell line and the limits of detection of the assay. Characterization of this receptor with other synthetic tetrapetpdies, chimeric AGRP-melanocortin ligands, as well as the THIQ and JB25 small molecule (Table 4) revealed consistent partial agonist activity and reduced agonist potency. However, the AMW3-130 ligand still possessed sub nM potency, albeit 5-fold decreased as compared to the wild type hMC4R. Unexpectantly, the JRH322-18 (pI)DPhe7 containing tetrapeptide was unable to generate any stimulatory response at up to 100 μM concentrations. These data are consistent with previous reports by others for the E61K hMC4R (51), and clearly indicate that this E61K hMC4R possess multiple in vitro molecular defects including reduced cell surface expression, decreased agonist potencies, as well as only partial agonist activities.
The I69T hMC4R in TM1 possessed cell surface expression levels at ca 80% of the wild type hMC4R levels, but was not statistically different. Interestingly however, evaluation of the total specific 125I-NDP-MSH binding resulted in ca 12-fold decreased counts (38,000 vs. 3,200 cpms respectively) indicating that while the I69T hMC4R is expressed at the cell surface at levels consistent with the wild type hMC4R, it does not recognize and/or bind 125I-NDP-MSH at control levels. Based upon these data, it might be postulated that the I69T hMC4R protein folded conformation(s) at the cell surface are not similar to the wild type hMC4R. Since this study utilizes stably expressing cell lines, and this I69T variant is postulated to be positioned towards the intracellular domain of TM1 (Figure 7), and the hydrophobic Ile residue is changed to a hydrophilic Thr residue, it is possible that a modification in the TM1-extracellular domain has occurred that affects global receptor structure, receptor phosphorylation sites, and/or changes the equilibrium rates of the formation of the GPCR ternary complex structures (61, 62) important for shifting receptor equilibrium towards the “agonist-induced active conformation.” This speculation would need to be experimentally verified using crystallographic techniques (63, 64) however. In support of this hypothesis, all the endogenous agonists, JRH887-9, NDP-MSH, and MTII possessed decreased agonist potency compared with the control hMC4R values. NDP-MSH binding affinity for the I69T hMC4R was slightly reduced, and consistent with a report by Tan et al (51). However, the antagonist AGRP(87-132) potency was the same as at the wild type hMC4R and the chimeric AGRP-melanocortin agonist AMW3-130 potency was not statistically different from the control value as well. These data support a hypothesis that this I69T hMC4R mutation might be affecting the endogenous agonist induced “active” form of this receptor versus the endogenous antagonist induced “inactive” receptor population form. We have postulated that the AMW3-130 ligand possessing the melanocortin agonist His-DPhe-Arg-Trp amino acids in the antagonist AGRP template possesses distinct ligand structure(s) compared with either AGRP or the melanocortin based agonist ligands (33, 65) and it could be these structural differences that might explain how this chimeric ligand can access a structural alternative of the endogenous agonist induced hMC4R “active GPCR” conformation. Thus, based upon these data it could be speculated that as judged by our in vitro system, this I69T hMC4R SNP changes the hMC4R conformational equilibrium important for endogenous agonist “activation” by modifying the global 3D receptor structure, and thus resulting in the obese human patient phenotype.
The D90N hMC4R variant, located within the TM2 domain, possessed statistically reduced cell surface expression levels from the control hMC4R, yet interestingly possessed total specific 125I-NDP-MSH binding levels equivalent to the wild type hMC4R. These data might lead to the extrapolation that the receptor conformation population that is at the cell surface could be shifted into the “active” receptor conformation in response to the presence agonist. However, the endogenous agonists, JRH887-9, NDP-MSH, MTII ligands resulted in significantly reduced agonist potency as well as partial agonist activity as compared to the hMC4R control. These data are consistent with previous reports by others (66). As might be anticipated from the total specific binding results, 125I-NDP-MSH binding affinity was not different from the wild type hMC4R. The endogenous AGRP(87-132) antagonist potency was also similar as the control hMC4R, however inverse activity was not observed and can be linked with decreased basal activity of the cell lines. Remarkably, at this D90N hMC4R none of the synthetic agonists, or small molecules, including AMW3-130, was able to generate a stimulatory response at up to 100 μM concentrations. Additionally, this is an interesting receptor mutation that appears to increase 125I-NDP-MSH induced cell surface levels similar to the wild type hMC4R while affecting the ability of ligands to induce a receptor conformation that results in normal wild type hMC4R signal transduction properties.
The Y157S mutation located within the second intracellular domain was expressed in the cells at wild type hMC4R values but possessed cell surface expression levels only ca 30% that observed for the control levels. Total specific 125I-NDP-MSH binding values were also low (2,800 cpms), implicating poor cell surface expression levels. The binding affinity of NDP-MSH was not significantly changed from wild type control values. All the peptide agonist ligands examined resulted in partial agonist efficacy with significantly reduced potency values as compared to the control hMC4R. Interestingly, the small molecule THIQ was able to generate a full agonist response at this Y157S hMC4R (Figure 6), albeit with reduced potency. The endogenous antagonist AGRP(87-132) possessed normal potency and inverse agonist activity was not observed due to low cellular basal activity. These data are consistent with previous reports by others (67), and there appears to be several in vitro molecular defects associated with this mutant.
The A219V SNP located at the TM5-intracellular loop interface, is expressed in the cells, at the cell surface, and has 125I-NDP-MSH total specific binding levels supporting the conclusion that this A219V hMC4R is expressed at the cell surface at nearly wild type hMC4R levels. The NDP-MSH ligand binding affinity is not statistically different then the control hMC4R value. All the endogenous and synthetic agonists examined in this study resulted in significantly reduced potency values (with the exception of AMW3-130), and the AGRP(87-132) antagonist potency was also decreased as compared to the wild type hMC4R values. Interestingly, the JRH322-18 tetrapeptide was only able to generate a partial agonist response and the AMW3-130 chimeric AGRP-melanocortin agonist ligand possessed sub nM potency values that were not statistically different from the wild type hMC4R value. Larson et al. reported for the A219V hMC4R, wild type NDP-MSH binding affinity but reduced MTII efficacy and functional potency (55).
The C271R SNP located proximal to the third extracellular domain and the extracellular portion of TM6, is expressed in the stably expressing cell lines at levels equivalent to the wild type hMC4R control levels. However, significantly decreased cell surface expression levels are observed in both the FACS and 125I-NDP-MSH total specific binding assays. The agonist NDP-MSH binding affinity is not changed from the wild type hMC4R, yet all the endogenous and synthetic agonists, as well as the small molecules examined in this study, resulted in only partial agonist efficacy and reduced ligand potency. The JRH322-18 tetrapeptide agonist was unable to stimulate this mutant hMC4R at up to 100 μM concentrations. The AGRP(87-132) antagonist potency remained similar to the wild type hMC4R however. Thus, these in vitro data imply the molecular defects of poor cell surface expression levels and agonist ligand reduced potency and efficacy as links to explain the human obese phenotype. Structurally, it has been postulated that this Cys271 hMC4R to be important for forming a disulfide bridge with an adjacent Cys residue within the putative third extracellular domain (68), and thus has both receptor structural as well as putative ligand interaction implications.
Summary
In conclusion, we have pharmacologically characterized 30 hMC4R polymorphisms identified in human patients in a side-by-side comparison for receptor cellular expression levels, multiple endogenous melanocortin agonists [α-MSH, β-MSH, γ2-MSH, and ACTH(1-24)] and antagonist [hAGRP(87-132)] potency as well as synthetic agonists NDP-MSH, MTII, and the JRH887-9 tetrapeptide. Additionally, for selected polymorphic receptors (F51L, E61K, I69T, D90N, Y157S, A219V, and C271R) six other synthetic peptide and small molecule agonists were evaluated in attempts to stimulate maximal efficacy for receptors resulting in partial agonist activity as well as potentially identify if the AMW3-130 chimeric AGRP-melanocortin ligand could functional rescue agonist potency similar to the wild type hMC4R. For the eight R7H, R18H, S36Y, F202L, I226T, G231S, and E308K, and I317V hMC4R's, the pharmacological profiles of receptor cell surface expression and endogenous ligand potency do not provide any insights in a molecular defect(s) that could allow for the postulation of a link between these in vitro studies and an obese human phenotype. The six S94R, G98R, W174C, G181D, 750DelGA, and P299L hMC4Rs examined in this study possessed significantly reduced cell surface expression levels in two assays (FACS and total 125I-NDP-MSH specific binding) and were not able to be stimulated by any ligand examined in this study. Interestingly, the three I121T, I317V, and L325F hMC4Rs possessed significantly reduced cell protein expression levels determined in the FACS assay, however total specific binding of 125I-NDP-MSH indicated relatively high levels of 125I-NDP-MSH bound to properly folded cell surface hMC4Rs. This pharmacological profile was also previously observed for the I301T and I316S hMC4Rs (31) and appears to be unique for these five hMC4R SNPs characterized by our laboratory out of a total of 70. While the first intuitive hypothesis for these SNPs is to link in vitro decreased cell surface expression levels as a major defect that could be associated with obesity in humans, the observation that 125I-NDP-MSH binds at relatively high levels (ca I121T hMC4R 22,700 cpms, I317V hMC4R 36,100 cpms, L325F hMC4R 25,500 cpms, wild type hMC4R 37,500 cpms) suggests that a receptor bound melanocortin agonist might be able to shift the cell surface hMC4R equilibrium to favor an “active” conformation. If this hypothesis is correct for the endogenous ligands, other than the synthetic NDP-MSH, then the previously clear link between poor cell surface expression levels of these five hMC4R SNPs and human obesity becomes muddied if the ligand can correct or up-regulate cell surface expression levels similar to the wild type hMC4R. Clearly additional experiments need to be performed to support these concepts, but nonetheless, these I121T, I301T, I316S, I317V, and L325F hMC4Rs provide a unique tool set to study such general “ternary complex” GPCR phenomena. The four E61K, D90N, Y157S, and C271R hMC4Rs possessed partial agonist activity as well as decreased agonist potency, while retaining wild type hMC4R like AGRP(87-132) antagonist potency. The three A219V, G238D, and S295P hMC4Rs were the only receptors in this study to result in decreased AGRP(87-132) antagonist potency. The F51L, I69T, and A219V hMC4Rs possessed decreased potency for all the endogenous agonists examined in this study. The P48S and A154D hMC4Rs possessed reduced agonist potency for one or more of the endogenous agonists examined, but not all. These latter data support our hypothesis that the hMC4R polymorphisms may respond normally to some putative endogenous melanocortin ligands and differently to others. Detailed in vitro characterization of individual hMC4R polymorphisms is critical for understanding the possible functional consequences of individual SNPs and the implication of receptor structural and functional consequences and human obesity. In most cases, these data reported herein present in vitro pharmacological profiles that can provide testable hypotheses to further support a molecular defect(s) concept that could theoretically be linked with the obese human patients from which these SNPs were identified. These data also provide the necessary foundation for computational three-dimensional homology molecular modeling studies (51, 69) as well as the development of general GPCR algorithms and programs (70) for in silico mutagenesis and predictive approaches for the discovery of those receptor residues that are important for ligand recognition, agonist and antagonist function, GPCR activation and G-protein coupling.
Acknowledgments
Funding Information: This work has been supported by NIH grants RO1DK063974, RO1DK57080, RO1DK064250 (CHL).
The abbreviations used are
- ACTH
Adrenocorticotropin Hormone
- AGRP
Agouti-related Protein
- CA
constitutively active
- cAMP
cyclic Adenosine Monophosohate
- CRE
cyclic Adenosine Monophosohate Response Element
- DMEM
Dulbecco's Modified Eagle's Medium
- FACS
Fluorescence Activated Cell Sorting
- GPCR
G-Protein coupled Receptor
- HEK-293
Human Embryonic Kidney-293
- hMC4R
human Melanocortin-4-Receptor
- MSH
Melanocyte Stimulating Hormone
- MTII
Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2
- NDP-MSH
4-Norleucine-7-D-Phenylalanine
- ONPG
o-Nitrophenyl-β-D-Galactopyranoside
- POMC
pro-opiomelanocortin
- SNP
single nucleotide polymorphism
- and TM
Transmembrane Domain
Footnotes
Dedication: This work is dedicated to Mr. Herbert H. Haskell (lived between February 16, 1918 and April 9, 2009).
References
- 1.Lerner AB, McGuire JS. Effect of Alpha- and Beta-Melanocyte Stimulating Hormones on the Skin Colour of Man. Nature. 1961;189:176–179. doi: 10.1038/189176a0. [DOI] [PubMed] [Google Scholar]
- 2.Bertolini A, Vergoni W, Gessa GL, Ferrari W. Induction of Sexual Excitement by the Action of Adrenocorticotrophic Hormone in Brain. Nature. 1969;221:667–669. doi: 10.1038/221667a0. [DOI] [PubMed] [Google Scholar]
- 3.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine N. Synthetic Melanotropic Peptide Initiates Erections in Men with Psychogenic Erectile Dysfunction: Double-blind, Placebo Controlled Crossover Study. J Urol. 1998;160:389–393. [PubMed] [Google Scholar]
- 4.Lindner E, Scholkens B. ACTH and alpha-MSH: cardiovascular and antiarrhythmic properties. Arch Int Pharmacodyn Ther. 1974;208:19–23. [PubMed] [Google Scholar]
- 5.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of Blood Pressure by Central Melanocortinergic Pathways. N Engl J Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 6.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity Syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 7.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Smith FJ, Kesterson RA, Boston BA, Fang Q, Berkemeir LR, Gu W, Cone RD, Campfield LA, Lee F. Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 8.Chhajlani V, Wikberg JES. Molecular Cloning and Expression of the Human Melanocyte Stimulating Hormone Receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 9.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The Cloning of a Family of Genes that Encode the Melanocortin Receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 10.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a Receptor for γ Melanotropin and Other Proopiomelanocortin Peptides in the Hypothalamus and Limbic System. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the Melanocortin-4 Receptor (MC4-R) in Neuroendocrine and Autonomic Control Circuits in the Brain. Mol Endo. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 12.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular Cloning of a Novel Melanocortin Receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 13.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular Cloning, Expression, and Gene Localization of a Fourth Melanocortin Receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 14.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular Cloning, Expression, and Characterization of a Fifth Melanocortin Receptor. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 15.Eipper BA, Mains RE. Structure and Biosynthesis of Pro-ACTH/Endorphin and Related Peptides. Endocrin Rev. 1980;1:1–26. doi: 10.1210/edrv-1-1-1. [DOI] [PubMed] [Google Scholar]
- 16.Smith AI, Funder JW. Proopiomelanocortin Processing in the Pituitary, Central Nervous System and Peripheral Tissues. Endocrin Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- 17.Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin Processing in the Hypothalamus: Impact on Melanocortin Signalling and Obesity. J of Endocrinology. 2002;172:411–421. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- 18.Bultman SJ, Michaud EJ, Woychick RP. Molecular Characterization of the Mouse Agouti Locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- 19.Miller MW, Duhl DM, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning Of The Mouse Agouti Gene Predicts A Secreted Protein Ubiquitously Expressed In Mice Carrying The Lethal Yellow Mutation. Genes & Development. 1993;7:454–67. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 20.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 21.Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The Neuropeptide Y/Agouti Gene-related Protein (AGRP) Brain Circuitry In Normal, Anorectic, And Monosodium Glutamate-treated Mice. Proc Natl Acad Sci U S A. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the Neuroanatomical Distribution of Agouti Related Protein (AGRP) Immunoreactivity in the Rhesus Monkey and the Rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- 23.Haskell-Luevano C, Holder JR, Monck EK, Bauzo RM. Characterization of Melanocortin NDP-MSH Agonist Peptide Fragments at the Mouse Central and Peripheral Melanocortin Receptors. J Med Chem. 2001;44:2247–2252. doi: 10.1021/jm010061n. [DOI] [PubMed] [Google Scholar]
- 24.Holder JR, Bauzo RM, Xiang Z, Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors: I Modifications at the His Position. J Med Chem. 2002;45:2801–2810. doi: 10.1021/jm0104872. [DOI] [PubMed] [Google Scholar]
- 25.Holder JR, Bauzo RM, Xiang Z, Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors: Part 2 Modifications at the Phe Position. J Med Chem. 2002;45:3073–3081. doi: 10.1021/jm010524p. [DOI] [PubMed] [Google Scholar]
- 26.Holder JR, Xiang Z, Bauzo RM, Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors: Part 4 Modifications at the Trp Position. J Med Chem. 2002;45:5736–5744. doi: 10.1021/jm020296e. [DOI] [PubMed] [Google Scholar]
- 27.Holder JR, Xiang Z, Bauzo RM, Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors: Part 3 Modifications at the Arg Position. Peptides. 2003;24:73–82. doi: 10.1016/s0196-9781(02)00278-4. [DOI] [PubMed] [Google Scholar]
- 28.Holder JR, Marques FF, Xiang Z, Bauzo RM, Haskell-Luevano C. Characterization of Aliphatic, Cyclic, and Aromatic N-Terminally “Capped” His-DPhe-Arg-Trp-NH2 Melanocortin Tetrapeptides at the Melanocortin Receptors. Eur J Pharmacol. 2003;462:41–52. doi: 10.1016/s0014-2999(03)01322-0. [DOI] [PubMed] [Google Scholar]
- 29.Sawyer TK, Sanfillippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-Phenylalanine-α-Melanocyte-Stimulating Hormone: A Highly Potent α-Melanotropin with Ultra Long Biological Activity. Proc Natl Acad Sci USA. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Obeidi F, Castrucci AM, Hadley ME, Hruby VJ. Potent and Prolonged Acting Cyclic Lactam Analogues of α-Melanotropin: Design Based on Molecular Dynamics. J Med Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 31.Xiang Z, Litherland SA, Sorensen NB, Proneth B, Wood MS, Shaw AM, Millard WJ, Haskell-Luevano C. Pharmacological Characterization of 40 Human Melanocortin-4 Receptor Polymorphisms with the Endogenous Proopiomelanocortin-Derived Agonists and the Agouti-Related Protein (AGRP) Antagonist. Biochemistry. 2006;45:7277–7288. doi: 10.1021/bi0600300. [DOI] [PubMed] [Google Scholar]
- 32.Xiang Z, Pogozheva ID, Sorenson NB, Wilczynski AM, Holder JR, Litherland SA, Millard WJ, Mosberg HI, Haskell-Luevano C. Peptide and Small Molecules Rescue the Functional Activity and Agonist Potency of Dysfunctional Human Melanocortin-4 Receptor polymorphisms. Biochemistry. 2007;46:8273–8287. doi: 10.1021/bi7007382. [DOI] [PubMed] [Google Scholar]
- 33.Wilczynski A, Wang XS, Joseph CG, Xiang Z, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, Haskell-Luevano C. Identification of Putative Agouti-Related Protein(87-132)-Melanocortin-4 Receptor Interactions by Homology Molecular Modeling and Validation Using Chimeric Peptide Ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 34.Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DB, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, Van Der Ploeg LH, Patchett AA, Nargund RP. Design and Pharmacology of N-[(3R)-1,2,3,4-Tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol- 1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a Potent, Selective, Melanocortin Subtype-4 Receptor Agonist. J Med Chem. 2002;45:4589–4593. doi: 10.1021/jm025539h. [DOI] [PubMed] [Google Scholar]
- 35.Bondebjerg J, Xiang Z, Bauzo RM, Haskell-Luevano C, Meldal M. A Solid Phase Approach to Mouse Melanocortin Receptor Agonists Derived From a Novel Thioether Cyclized Peptidomimetic Scaffold. J Am Chem Soc. 2002;124:11046–11055. doi: 10.1021/ja0123913. [DOI] [PubMed] [Google Scholar]
- 36.Ho G, MacKenzie RG. Functional Characterization of Mutations in Melanocortin-4 Receptor Associated with Human Obesity. J Biol Chem. 1999;274:35819–35822. doi: 10.1074/jbc.274.50.35816. [DOI] [PubMed] [Google Scholar]
- 37.Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Structure Activity Studies of the Melanocortin-4 Receptor by In Vitro Mutagenesis: Identification of Agouti-Related Protein (AGRP), Melanocortin Agonist and Synthetic Peptide Antagonist Interaction Determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- 38.Chen CA, Okayama H. Calcium Phosphate-Medicated Gener Transfer: A Highly Efficient Transfections System for Stably Transforming Cells with Plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 39.Chen W, Shields TS, Stork PJS, Cone RD. A Colorimetric Assay for Measuring Activation of Gs- and Gq-Coupled Signaling Pathways. Anal Biochem. 1995;226:349–354. doi: 10.1006/abio.1995.1235. [DOI] [PubMed] [Google Scholar]
- 40.Schild HO. pA, A New Scale for the Measurement of Drug Antagonism. Brit J Pharmacol. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SBH, Gantz I. Characterization of Agouti-Related Protein Binding to Melanocortin Receptors. Mol Endo. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 42.Bowen WP, Jerman JC. Nonlinear Regression Using Spreadsheets. TiPS. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- 43.Proneth B, Xiang Z, Pogozheva ID, Litherland SA, Gorbatyuk OS, Shaw AM, Millard WJ, Mosberg HI, Haskell-Luevano C. Molecular Mechanism of the Constitutive Activation of the L250Q Human Melanocortin-4 Receptor Polymorphism. Chem Biol Drug Des. 2006;67:215–229. doi: 10.1111/j.1747-0285.2006.00362.x. [DOI] [PubMed] [Google Scholar]
- 44.Haskell-Luevano C, Monck EK. Agouti-related Protein (AGRP) Functions as an Inverse Agonist at a Constitutively Active Brain Melanocortin-4 Receptor. Regulatory Peptides. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 45.Nijenhuis WA, Oosterom J, Adan RA. AGRP(83-132) Acts as an Inverse Agonist on the Human-Melanocortin-4 Receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 46.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High-Resolution NMR Structure of the Chemically-Synthesized Melanocortin Receptor Binding Domain AGRP(87-132) of the Agouti-Related Protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 47.VanLeeuwen D, Steffey ME, Donahue C, Ho G, MacKenzie RG. Cell Surface Expression of the Melanocortin-4 Receptor is Dependent on a C-Terminal Di-Isoleucine Sequence at Codons 316/317. J Biol Chem. 2003;278:15935–15940. doi: 10.1074/jbc.M211546200. [DOI] [PubMed] [Google Scholar]
- 48.Nijenhuis WA, Garner KM, van Rozen RJ, Adan RA. Poor Cell Surface Expression of Human Melanocortin-4 Receptor Mutations Associated with Obesity. J Biol Chem. 2003;278:22939–22945. doi: 10.1074/jbc.M211326200. [DOI] [PubMed] [Google Scholar]
- 49.Yeo GS, Lank EJ, Farooqi IS, Keogh J, Challis BG, O'Rahilly S. Mutations in the Human Melanocortin-4 Receptor Gene Associated with Severe Familial Obesity Disrupts Receptor Function Through Multiple Molecular Mechanisms. Hum Mol Genet. 2003;12:561–574. doi: 10.1093/hmg/ddg057. [DOI] [PubMed] [Google Scholar]
- 50.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 Receptor Mutations are a Frequent and Heterogeneous Cause of Morbid Obesity. J Clin Invest. 2000;106:253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan K, Pogozheva ID, Yeo GS, Hadaschik D, Keogh JM, Haskell-Luevano C, O'Rahilly S, Mosberg HI, Farooqi IS. Functional Characterization and Structural Modeling of Obesity Associated Mutations in the Melanocortin 4 Receptor. Endocrinology. 2009;150:114–125. doi: 10.1210/en.2008-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lubrano-Berthelier C, Cavazos M, Dubern B, Shapiro A, Stunff CL, Zhang S, Picart F, Govaerts C, Froguel P, Bougneres P, Clement K, Vaisse C. Molecular Genetics of Human Obesity-Associated MC4R Mutations. Ann N Y Acad Sci. 2003;994:49–57. doi: 10.1111/j.1749-6632.2003.tb03161.x. [DOI] [PubMed] [Google Scholar]
- 53.Proneth B, Pogozheva ID, Portillo FP, Mosberg HI, Haskell-Luevano C. Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 Modified at the Para Position of the Benzyl Side Chain (DPhe): Importance for Mouse Melanocortin-3 Receptor Agonist Versus Antagonist Activity. J Med Chem. 2008;51:5585–5593. doi: 10.1021/jm800291b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacobson P, Ukkola O, Rankinen T, Snyder EE, Leon AS, Rao DC, Skinner JS, Wilmore JH, Lonn L, Cowan GS, Jr, Sjostrom L, Bouchard C. Melanocortin 4 Receptor Sequence Variations Are Seldom a Cause of Human Obesity: The Swedish Obese Subjects, the HERITAGE Family Study, and a Memphis Cohort. J Clin Endocrinol Metab. 2002;87:4442–4446. doi: 10.1210/jc.2002-020568. [DOI] [PubMed] [Google Scholar]
- 55.Larsen LH, Echwald SM, Sorensen TI, Andersen T, Wulff BS, Pedersen O. Prevalence of Mutations and Functional Analyses of Melanocortin 4 Receptor Variants Identified Among 750 men with Juvenile-Onset Obesity. J Clin Endocrinol Metab. 2005;90:219–224. doi: 10.1210/jc.2004-0497. [DOI] [PubMed] [Google Scholar]
- 56.Tao YX, Segaloff DL. Functional Analyses of Melanocortin-4 Receptor Mutations Identified from Patients with Binge Eating Disorder and Nonobese or Obese Subjects. J Clin Endocrinol Metab. 2005;90:5632–5638. doi: 10.1210/jc.2005-0519. [DOI] [PubMed] [Google Scholar]
- 57.Lubrano-Berthelier C, Durand E, Dubern B, Shapiro A, Dazin P, Weill J, Ferron C, Froguel P, Vaisse C. Intracellular Retention is a Common Characteristic of Childhood Obesity-Associated MC4R Mutations. Hum Mol Genet. 2003;12:145–153. doi: 10.1093/hmg/ddg016. [DOI] [PubMed] [Google Scholar]
- 58.Dubern B, Clement K, Pelloux V, Froguel P, Girardet JP, Guy-Grand B, Tounian P. Mutational Analysis of Melanocortin-4 Receptor, Agouti-Related Protein, and α-Melanocyte-Stimulating Hormone Genes in Severely Obese Children. J Pediatr. 2001;139:204–209. doi: 10.1067/mpd.2001.116284. [DOI] [PubMed] [Google Scholar]
- 59.Yang YK, Dickinson C, Haskell-Luevano C, Gantz I. Molecular Basis for the Interaction of [Nle4, DPhe7] Melanocyte Stimulating Hormone with the Human Melanocortin-1 Receptor (Melanocyte α-MSH Receptor) J Biol Chem. 1997;272:23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- 60.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin Activates Anorexigenic POMC Neurons Through a Neural Network in the Arcuate Nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 61.De Lean A, Stadel JM, Lefkowitz RJ. A Ternary Complex Model Explains the Agonist-Specific Binding Properties of the Adenylate Cyclase-Coupled Beta-Adrenergic Receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 62.Lefkowitz RJ, Cotecchia S, Samama P, Costa T. Constitutive Activity of Receptors Coupled to Guanine Nucleotide Regulatory Proteins. Trends Pharmacol Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- 63.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR Engineering Yields High-Resolution Structural Insights into Beta2-Adrenergic Receptor Function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 64.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal Structure of the Human Beta2 Adrenergic G-protein-Coupled Receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 65.Wilczynski A, Wilson KR, Scott JW, Edison AS, Haskell-Luevano C. Structure-Activity Relationships of the Unique and Potent Agouti-Related Protein (AGRP)-Melanocortin Chimeric Tyr-c[β-Asp-His-DPhe-Arg-Trp-Asn-Ala-Phe-Dpr]-Tyr-NH2 Peptide Template. J Med Chem. 2005;48:3060–3075. doi: 10.1021/jm049010r. [DOI] [PubMed] [Google Scholar]
- 66.Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-Dominant Mode of Inheritance of a Melanocortin-4 Receptor Mutation in a Patient with Severe Early-Onset Obesity is Due to a Dominant-Negative Effect Caused by Receptor Dimerization. Diabetes. 2003;52:2984–8. doi: 10.2337/diabetes.52.12.2984. [DOI] [PubMed] [Google Scholar]
- 67.Tao YX, Segaloff DL. Functional Characterization of Melanocortin-4 Receptor Mutations Associated with Childhood Obesity. Endocrinology. 2003;144:4544–4551. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- 68.Holst B, Elling CE, Schwartz TW. Metal ion-mediated agonism and agonist enhancement in melanocortin MC1 and MC4 receptors. J Biol Chem. 2002;277:47662–47670. doi: 10.1074/jbc.M202103200. [DOI] [PubMed] [Google Scholar]
- 69.Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, Mosberg HI. Interactions of Human Melanocortin 4 Receptor with Nonpeptide and Peptide agonists. Biochemistry. 2005;44:11329–11341. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bromberg Y, Overton J, Vaisse C, Leibel RL, Rost B. In Silico Mutagenesis: A Case Study of the Melanocortin 4 Receptor. FASEB J. 2009;23:3059–3069. doi: 10.1096/fj.08-127530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lubrano-Berthelier C, Cavazos M, Le Stunff C, Haas K, Shapiro A, Zhang S, Bougneres P, Vaisse C. The Human MC4R Promoter: Characterization and Role in Obesity. Diabetes. 2003;52:2996–3000. doi: 10.2337/diabetes.52.12.2996. [DOI] [PubMed] [Google Scholar]
- 72.Miraglia Del Giudice E, Cirillo G, Nigro V, Santoro N, D'Urso L, Raimondo P, Cozzolino D, Scafato D, Perrone L. Low Frequency of Melanocortin-4 Receptor (MC4R) Mutations in a Mediterranean Population with Early-Onset Obesity. Int J Obes Relat Metab Disord. 2002;26:647–651. doi: 10.1038/sj.ijo.0801983. [DOI] [PubMed] [Google Scholar]
- 73.Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge Eating as a Major Phenotype of Melanocortin 4 Receptor Gene Mutations. N Engl J Med. 2003;348:1096–1103. doi: 10.1056/NEJMoa021971. [DOI] [PubMed] [Google Scholar]
- 74.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 75.Hinney A, Hohmann S, Geller F, Vogel C, Hess C, Wermter AK, Brokamp B, Goldschmidt H, Siegfried W, Remschmidt H, Schafer H, Gudermann T, Hebebrand J. Melanocortin-4 Receptor Gene: Case-Control Study and Transmission Disequilibrium Test Confirm that Functionally Relevant Mutations are Compatible with a Major Gene Effect for Extreme Obesity. J Clin Endocrinol Metab. 2003;88:4258–4267. doi: 10.1210/jc.2003-030233. [DOI] [PubMed] [Google Scholar]
- 76.Kobayashi H, Ogawa Y, Shintani M, Ebihara K, Shimodahira M, Iwakura T, Hino M, Ishihara T, Ikekubo K, Kurahachi H, Nakao K. A Novel Homozygous Missense Mutation of Melanocortin-4 Receptor (MC4R) in a Japanese Woman with Severe Obesity. Diabetes. 2002;51:243–246. doi: 10.2337/diabetes.51.1.243. [DOI] [PubMed] [Google Scholar]
- 77.Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS. Elevated Plasma Ghrelin Levels in Prader Willi Syndrome. Nat Med. 2002;8:643–644. doi: 10.1038/nm0702-643. [DOI] [PubMed] [Google Scholar]
- 78.Lee YS, Poh LK, Kek BL, Loke KY. Novel Melanocortin 4 Receptor Gene Mutations in Severely Obese Children. Clin Endocrinol (Oxf) 2007 doi: 10.1111/j.1365-2265.2007.03071.x. [DOI] [PubMed] [Google Scholar]
- 79.Buono P, Pasanisi F, Nardelli C, Ieno L, Capone S, Liguori R, Finelli C, Oriani G, Contaldo F, Sacchetti L. Six Novel Mutations in the Proopiomelanocortin and Melanocortin Receptor 4 Genes in Severely Obese Adults Living in Southern Italy. Clin Chem. 2005;51:1358–1364. doi: 10.1373/clinchem.2005.047886. [DOI] [PubMed] [Google Scholar]
- 80.Valli-Jaakola K, Lipsanen-Nyman M, Oksanen L, Hollenberg AN, Kontula K, Bjorbaek C, Schalin-Jantti C. Identification and Characterization of Melanocortin-4 Receptor Gene Mutations in Morbidly Obese Finnish Children and Adults. J Clin Endocrinol Metab. 2004;89:940–945. doi: 10.1210/jc.2003-031182. [DOI] [PubMed] [Google Scholar]
- 81.Hinney A, Bettecken T, Tarnow P, Brumm H, Reichwald K, Lichtner P, Scherag A, Nguyen TT, Schlumberger P, Rief W, Vollmert C, Illig T, Wichmann HE, Schafer H, Platzer M, Biebermann H, Meitinger T, Hebebrand J. Prevalence, Spectrum, and Functional Characterization of Melanocortin-4 Receptor Gene Mutations in a Representative Population-Based Sample and Obese Adults from Germany. J Clin Endocrinol Metab. 2006;91:1761–1769. doi: 10.1210/jc.2005-2056. [DOI] [PubMed] [Google Scholar]
- 82.Ma L, Tataranni PA, Bogardus C, Baier LJ. Melanocortin 4 Receptor Gene Variation is Associated with Severe Obesity in Pima Indians. Diabetes. 2004;53:2696–2699. doi: 10.2337/diabetes.53.10.2696. [DOI] [PubMed] [Google Scholar]
- 83.Santini F, Maffei M, Ceccarini G, Pelosini C, Scartabelli G, Rosellini V, Chiellini C, Marsili A, Lisi S, Tonacchera M, Agretti P, Chiovato L, Mammoli C, Vitti P, Pinchera A. Genetic Screening for Melanocortin-4 Receptor Mutations in a Cohort of Italian Obese Patients: Description and Functional Characterization of a Novel Mutation. J Clin Endocrinol Metab. 2004;89:904–908. doi: 10.1210/jc.2003-031175. [DOI] [PubMed] [Google Scholar]